
Abstract
Human embryonic stem (hES) cells have the potential as starting materials for a wide variety of applications in cell therapy, drug discovery and development. However, the challenge is to produce large numbers of well-characterized hES cells that are pluripotent and of high quality. This is needed to be capable of producing future cell therapies that are safe, effective, and affordable for use in routine clinical practice. A major bottleneck is the present requirement for complex culturing regimes that are very labor intensive and unscalable. hES cells have traditionally been grown on feeder layers made from inactivated mouse or human embryonic fibroblasts, in medium containing serum and other nondefined factors. This makes conditions difficult to reproduce over multiple passages. With a view to simplifying culture conditions we have tested a novel proprietary good manufacturing practice-based system that circumvents the use of feeders completely. The system consists of a matrix and a formulated medium that, in combination, demonstrate a reliable and reproducible way to culture hES cells without the use of feeders. We have been able to grow hES cells (Shef3 and Shef6) for over 20 passages, in this system, without loss of pluripotency, capacity to differentiate, or acquisition of karyotypic abnormalities. Furthermore, we have demonstrated the feasibility of propagating hES cells at clonal dilutions from single cells using this system.
Introduction
H
Traditionally, hES cells have been derived on either human or mouse embryonic fibroblast (mEF) and are subsequently maintained on these feeder layers in most research laboratories [3]. This type of culture is labor intensive and has all the disadvantages of a co-culture, a contaminating cell population that is ever present. Moreover, conditions are difficult to reproduce over several passages as there is variation between different batches of feeders, as well as being unsuitable environment for the production of clinical grade cells. For future regenerative medicine products based on hES cells, these uncertainties and lack of robustness (reproducibility) must be eliminated to produce safe, effective, and affordable therapies [4].
For these reasons, substantial efforts have been made to substitute animal and other undefined components from culture medium and substrates to standardize and simplify culture methods, as well as to make culture conditions suitable for the production of clinical-grade cells.
Several authors have reported the use of alternative culture methods that address some of these concerns. Some efforts have concentrated on the formulation of defined medium components for the culture of both the hES cells and feeders (see, eg, refs. [5 –9]), whereas others have emphasized the substitution of all matrices and medium components for totally xeno-free substances (see, eg, refs. [10 –12]). Unfortunately, not all of these novel formulations are commercially available and may be costly and difficult to reproduce in standard research laboratories. More recently, other authors have sought to test commercially available formulations that are readily accessible, and in many cases, already good manufacturing practice compliant [13,14].
With a view to simplify culturing regimes, circumvent the use of feeder layers, and move toward xeno-free culture conditions, we have tested a commercially available system using the hES lines Shef3 and Shef6. This system has 2 components: a xeno-free human-derived, good manufacturing practice- compliant matrix (CELLstart), and a defined medium formulation (StemPro) (both from Invitrogen).
We have passaged both the Shef3 and the Shef 6 cell lines continuously for more than 20 passages and have characterized the cells grown under these conditions for morphology, pluripotency, and ability to differentiate in vitro. Further, we have looked at the potential of the system to support clonal expansion of hES cells, which would be a great advantage for genetic manipulation. We pragmatically describe here the passaging regime, pitfalls, and peculiarities of this culture system.
Materials and Methods
Cell culture conditions
hES cell lines Shef3 and Shef6 were cultured in mytomycin C-inactivated mEFs in knockout Dulbecco's modified Eagle's medium (DMEM) supplemented with 20% knockout serum replacement (KSR), 2 mM L-glutamine, nonessential amino acids (all from Invitrogen), 0.1 mM β-mercaptoethanol (Sigma), and 4 ng/mL human recombinant basic fibroblast growth factor (bFGF; R&D systems) before the testing. Most hES cell lines in our laboratory are grown in IVF dishes or 6-well plates.
Manufacture's culture conditions were followed for CELLstart (Invitrogen). Briefly, the CELLstart reagent was diluted in Dulbecco's modified phosphate-buffered saline (DPBS) containing calcium and magnesium (Invitrogen), at 1:50 dilution. Tissue culture vessels (6-well plates or T25 flasks) were then coated with this reagent for 2 h at 37°C. After incubation the reagent was removed and replaced with fresh hES StemPro complete medium (Invitrogen) supplemented with 0.1 mM β-mercaptoethanol (Sigma) and 8–20 ng/mL human recombinant bFGF (R&D systems). This system can also be used totally free of animal components by substituting bovine serum albumin (BSA) with xeno-free serum replacement (Invitrogen).
Cell passaging
To each culture vessel, the fresh medium was added and cells were mechanically scrapped from the vessel using plastic Pasteur pipettes, under a dissecting microscope. No collagenase was added before scrapping. Clumps in the supernatant were then pipetted into a new vessel previously treated with CELLstart. Cells were split at a ratio of 1:2 to 1:4, depending on confluency at the time of passaging.
Enzymatic passaging was also carried out in a subset of the cells using TrypLE Express (Invitrogen). Briefly cells were washed with DPBS, treated with TrypLE Express for 3 min at 37°C, and then quenched with the complete medium. Cells were passaged at a split ratio of 1:5 into freshly prepared flasks.
Freezing and thawing
Vitrification
Cells were frozen using the open pulled straw method described previously [15]. Briefly, cell clumps were sequentially immersed in drops of 10% dimethyl sulfoxide/10% ethylene glycol for 1 min, then for 30 s in 20% dimethyl sulfoxide/20% ethylene glycol/0.5 M sucrose, and finally aspirated into straws. The straws were then flash-frozen in liquid nitrogen. For thawing, straws were quickly warmed and the cells placed into drops of the standard growth medium, washed twice, and then plated in parallel in either wells treated with CELLstart or feeders.
Slow cooling method
After enzymatic passaging using TrypLE Express, cells were centrifuged and resuspended in mFreSR (Stem Cell Technologies) medium, and frozen at −70°C in 1 mL cryovials placed in “Mr. Frosty” 1°C freezing containers (Nalgene) overnight, and then transferred to liquid nitrogen.
In vitro differentiation
Cells scrapped or ezymatically dissociated using TryplExpress, as described above, were plated into bacteriological-grade Petri dishes in embryoid body (EB) medium. The EB medium consists of knockout-DMEM, 10% KSR, 2 mM L-glutamine, and nonessential amino acids (all from Invitrogen). Cells were allowed to form EBs for either 7 or 14 days with medium changes every 3 days. At the end of the differentiation period the EBs were pooled, collected, centrifuged, and washed in PBS, after which they were centrifuged again. The dry pellets were frozen at −70°C until needed for RNA extraction.
Some EBs were further plated in 12-well plates treated with 0.1% gelatine, allowed to attach and cultured for 1–2 weeks in either EB medium, mEF medium [10% fetal bovine serum (FBS) in DMEM], or N2/B27 medium (DMEM/F12 supplemented with 1% BSA, 1 × N2 supplement and 1 × B27 supplement (both from PAA labs). At the end of the culture period the plated EBs were fixed in situ using 4% paraformaldehyde (PFA), and then stained by immunocytochemistry for markers of the 3 embryonic germ layers.
Immunocytochemistry
Cells were fixed in 4% PFA for 20 min at room temperature in the 24-well tissue culture plates where they had been grown. After a DPBS wash, cells were either permeabilized using 0.25% Triton in DPBS (as required for the antibody) for 10 min or placed straight into blocking buffer consisting of 2% FBS in DPBS (for the detection of intracellular markers, 0.25% Triton was added to the blocking buffer). After a 30 min incubation in blocking buffer, cells were incubated with antibody diluted 1:200 to 1:300 in blocking buffer, overnight at 4°C. The primary antibodies used were as follows: mouse IgG anti-Oct3/4 (Santa Cruz), rabbit polyclonal anti-Nanog (Millipore), mouse IgM anti-SSEA1, rat IgM anti-SSEA3, mouse IgM anti-TRA-1-81, and mouse IgM anti-TRA-1-60 (all kind gifts from Prof. Peter Andrews Sheffield University). After incubation with primary antibodies, the cells were washed 3 times in DPBS for 5 min in a shaking platform at room temperature. After washing, cells were incubated with secondary antibodies diluted in blocking buffer 1:500 for 1 h at room temperature. Secondary antibodies used were as follows: Alexa fluor 488 goat anti-mouse IgG, Alexa fluor 488 goat anti-rat IgM, Alexa fluor 555 goat anti-mouse IgM, and Alexa fluor 488 goat anti-rabbit IgG (All from Invitrogen). The cells were then washed in DPBS as before, and finally counterstained with DAPI and mounted in DPBS. Cells were imaged using an epifluorescence Nikon Eclipse 2000 inverted microscope, and the NIS-elements software. Isotypes controls were run in parallel.
Reverse transcriptase-polymerase chain reaction
RNA extractions were carried out using the Qiagen RNeasy mini-kit under manufacturer's instructions, with an in column DNase step. The eluted RNA was quantified using a nano-drop. First-strand cDNA was synthesized using the Ambion retroscript kit following manufacturer's instructions with 2 μg of RNA per reaction (in a total volume of 20 μL).
Polymerase chain reactions (PCRs) were carried out using Bio-Taq polymerase (Bioline) with buffer supplied with the enzyme and 2 mM MgCl2 in a Verity cycler (Applied Biosystems). For most reactions a denaturing step at 95°C for 5 min was carried out followed by 35 cycles of 95°C for 30 s, 55°C for 30 s, and 72°C for 30 s, and a final elongation step of 7 min at 72°C.
The primer pairs used were designed in different exons to avoid amplification of genomic DNA and were run against all databases to ensure sequence specificity. The primer sequences used were as follows: GPDH (forward CGACCACTTTGTCAAGCTCA and reverse AGGGGTCTACATGGCAACTG), NESTIN (forward TGCGGGCTACTGAAAAGTTC and reverse AGGAGGGTCCTGTACGTGG), β-III Tubulin (forward AACGAGGCCTCTTCTCACAA and reverse AAGGTGTTCATGATGCGGTC), Brachyury (forward GTCCACCTGCAAATCCTCAT and reverse GGGTACTGACTGGAGCTGGT), and SOX17 (forward AAGATGCTGGGCAAGTCGT and reverse AGACCTGCGCGTAGCTGTAG).
Real-time PCR
PCRs were carried out using the MESA BLUE qPCR MasterMix Plus for SYBR® Assay (Eurogentec) under manufacturer's instructions. Ready validated primer pairs from Qiagen were used as follows: β-ACTIN (QT00095431), UBC (QT00234430), GAPDH (QT01192646), POU5F (QT00210840), NANOG (QT01844808), SOX2 (QT00237601), and TERT (QT00073409). Reactions were set in triplicate in a total volume of 20 μL containing 1 μL of 1st strand cDNA prepared as described above.
All reactions were carried out in an Eppendorf Realplex cycler. Relative quantification analysis was carried out using the Pfaffl method [16].
For normalization, a geometric average of the Ct values for all 3 house-keeping genes was used. Efficiencies for each reaction were calculated by running qPCR reactions using a cDNA dilution curve.
Karyology
Cell karyotyping was carried out by TDL Genetics using standard G banding. A minimum of 20 metaphase spreads were counted per sample.
Flow cytometry
Cell surface markers
Cells were enzymatically detached from the flasks, washed in DPBS, and counted using a hemocytometer. Cells were then divided into aliquots of 106 cells. Each aliquot was then resuspended in blocking buffer (2% FBS in DPBS) and incubated on ice for 20 min. After centrifugation, cells were resuspended in antibody solution (1:10 in 2% FBS in DPBS) and incubated on ice for 1 h. After incubation, cells were washed in DPBS, and incubated with secondary antibody (diluted 1:500 in blocking buffer) for 1 h on ice. After 3 DPBS washes, cells were resuspended in DPBS and analyzed by flow cytometry using a CyAnADP and the Summit software (Beckman Coulter).
Primary antibodies used were as follows: mouse IgM anti-SSEA1, rat IgM anti-SSEA3, mouse IgM anti-TRA1-160, and mouse IgG anti-SSEA4 (kind gifts from Prof. P. Andrews. Sheffield University). Secondary antibodies were Alexa fluor 488 goat anti-mouse IgG, Alexa fluor 488 goat anti-rat IgM (Invitrogen), and FITC goat anti-mouse IgM (Sigma).
Intracellular markers
After cell counting, aliquoted cells were fixed in 2% PFA in DPBS for 20 min at room temperature. Subsequently, they were washed in DPBS, permeabilized, and blocked in 0.2% saponin in DPBS with 1% BSA for 30 min at room temperature. After centrifugation, cells were resuspended in primary antibody solution (1:200 in 0.2% saponin in DPBS with 1% BSA) and incubated at room temperature for 1 h. After 3 DPBS washes, cells were resuspended in secondary antibody solution (1:500 in 0.2% saponin in DPBS with 0.1% BSA) and incubated at room temperature for 1 h. After 3 DPBS washes, cells were resuspended in DPBS and analyzed by flow cytometry as above. Primary antibodies used were mouse IgG anti-UTF1, mouse FITC-conjugated IgG anti-SOX2, rabbit polyclonal anti-NANOG, PE-conjugated mouse IgG anti-Nestin (all from Millipore), and mouse IgG anti-OCT3/4 (Santa Cruz).
Clonal expansion
Cells were harvested using TrypLE Express as detailed above. Once detached, cells were counted and plated at limiting dilutions in CELLstart-treated plastic in complete StemPro medium. The cells were plated in 35 mm dishes at concentrations of 500, 1000, and 1500 cells/cm2 (5000, 10,000, and 15,000 cells/dish). They were then fixed and stained. In a further experiment, 8 clones were picked, expanded, and analyzed by flow cytometry for markers of pluripotency.
Results
Cell culture and passaging
The hES cell line Shef 3 [17] was derived and has been grown traditionally on inactivated mEFs under relatively low feeder density (∼12,000 cells/cm2). Under these conditions cells form small colonies that gradually grow outward pushing the fibroblasts in their wake. They have very characteristic morphology: small round cells with high nuclear-to-cytoplasm ratio. The edges of the colony contain cells that may be actively proliferating and appear larger and more elongated (Fig 1). We wanted to investigate whether these hES cells would maintain the same morphology when grown on a non-cellular substrate and whether there was a correlation between the morphology and the pluripotency of the cells.
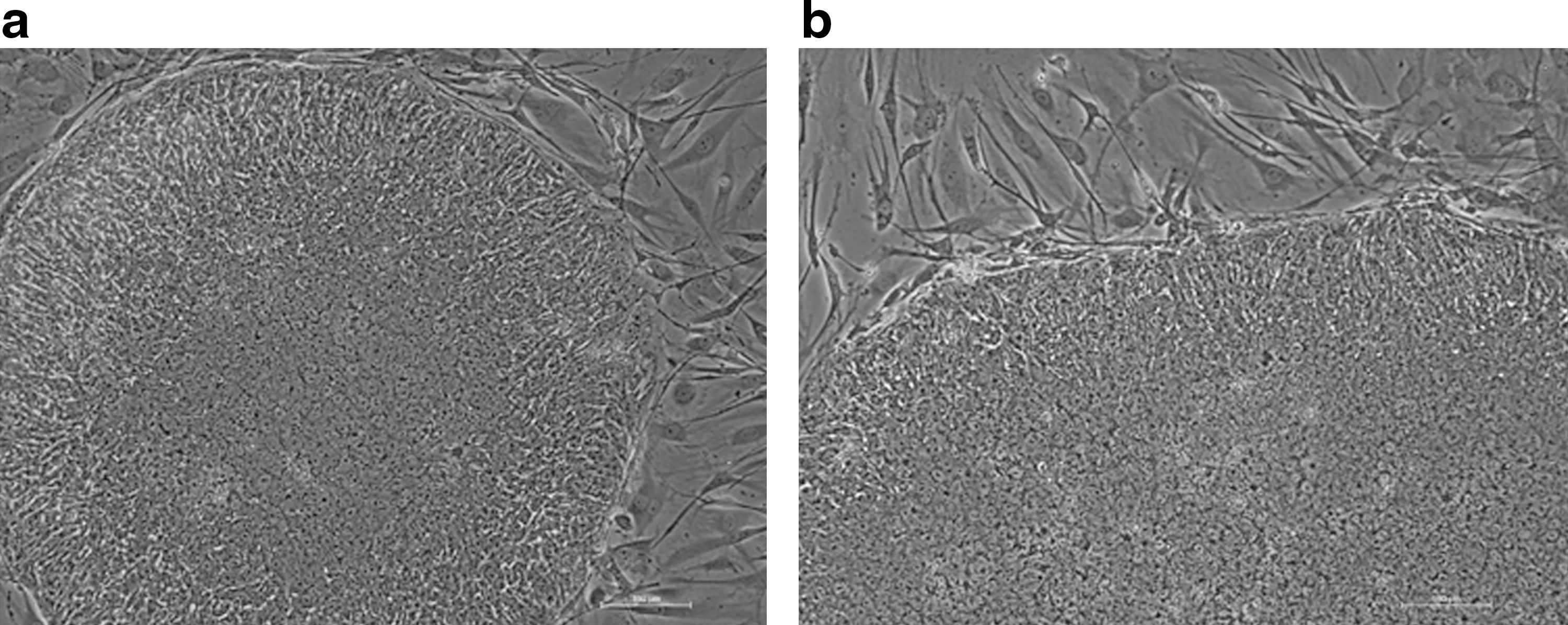
Photograph showing the classical morphology of the Shef 3 hES cell line grown under standard conditions on inactivated mouse embryonic fibroblasts.
When cells were first passaged from feeders onto the CELLstart matrix, they attached within 48–72 h, and we noted many floating clumps and single cells, which eventually attached if left undisturbed for short periods of time (2 days). When the colonies first form on the new matrix, the colonies appear larger and less compact than colonies on feeders, which could be regarded as a sign of imminent differentiation; however, the colonies compact within a few days (but may take up to 2 weeks) and show the characteristic morphology of hES cells, that is, small round cells with characteristic high nuclear-to-cytoplasm ratio (Fig. 2).
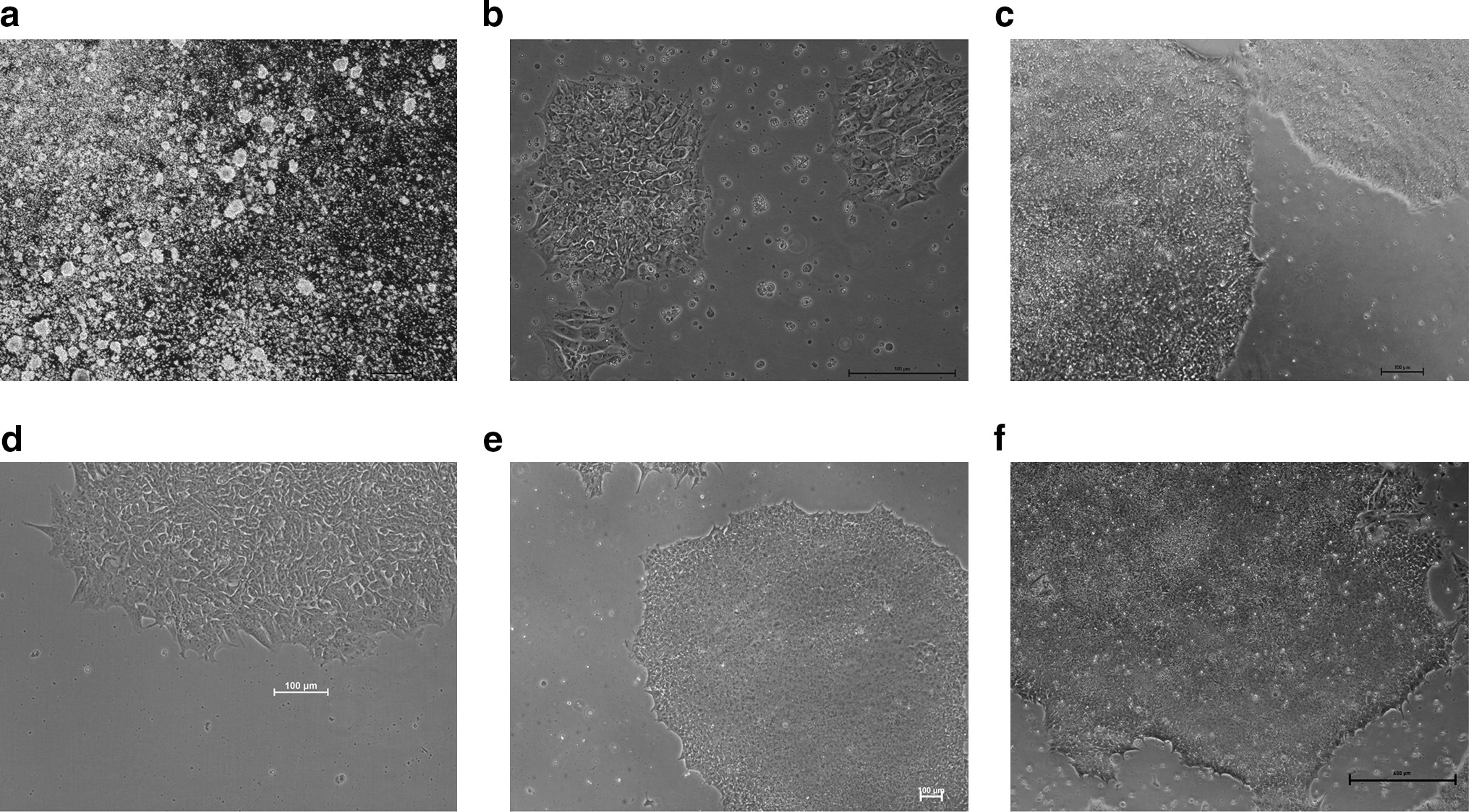
Series of phase-contrast photographs showing the different stages of growth of the Shef 3 and the Shef 6 hES cell lines in CELLstar matrix with StemPro.
With Shef3 in the first 3–4 passage, cells were split at similar time points to those grown on feeders (every 4 days), which may not be advisable as cells may be undergoing an adaptation phase and colonies compact more slowly. We found that passaging cells only when they show high compaction (high density colonies) yielded much healthier, less differentiated, and faster attaching colonies. We noted that to maintain the colony morphology we had to passage the cells less frequently and only when colonies were highly compact.
We also found that if the medium was replenished in old flasks from which cells had been scrapped, the cells would re-grow again and the colonies would re-expand faster, with classic morphologies and insignificant differentiation. We were able to re-scrape cells from these flasks at least one more time, and the matrix was still able to support growth for at least 2 weeks after initial plating.
We noted, on occasions, that passaged cells would attach as clumps and form structures on the new matrix that look similar to EBs; however, after 2 or 3 days, the cells would spread and form normal hES cell colonies (Fig. 3).
Series of phase-contrast photographs showing the EB-like structures that are sometimes formed by hES cells after mechanical passaging into new vessels coated with CELLstart. Note that the cells lose their EB-like appearance, attach, and spread on the substrate and form more classical-looking colonies, within a few days.
When adapting the Shef6 cell line, we noted that after the first 4 passages, the cells formed colonies that, instead of compacting after 3 days, would show a characteristic morphological change in the center of the colony akin to differentiation (Supplementary Fig. S1a; Supplementary Data are available online at
Culturing cells in this matrix was very straight forward, both Shef3 and Shef6 cells were able to proliferate and expand, and the morphology was equivalent to that of colonies grown on feeders, though the concentration of bFGF used had to be increased to maintain the stem properties of the Shef6 line, and the time between passages was different between cell lines. The colonies were easy to passage, even without the need for collagenase treatment, and detached cells would subsequently attach quickly to fresh matrix. This was also the case for cells that were enzymatically passaged using TrypLE Express.
We were able to adapt the Shef6 and the Shef3 cell line once more from cells that had been grown on feeders, with the second batch we have been able to grow past passage 7, we have found no loss of pluripotency markers, and the morphology of the cells is indistinguishable from that of the first batch.
Pluripotency after continuous passaging
After 8 continuous passages in CELLstart, both the Shef 3 and the Shef 6 cells still showed markers of pluripotency by immunocytochemistry. We could see expression of OCT3/4, SSEA3, TRA 1-60 (Fig. 4), TRA1-81, and NANOG (data not shown). We also stained for the differentiation marker SSEA1 and were unable to locate any cells expressing it (data not shown) except in the center of old colonies from the Shef 6 line (Supplementary Fig. S1). Flow cytometry analysis carried out on Shef 3 cells after the first freeze–thaw cycle at passages 11 and 20 after adaptation showed that there was one main cell population. When gated in this medium-sized population we found that the proportion of cells positive for all pluripotency markers tested was between 88% and 99%., whereas there were very few cells expressing either nestin (13%) or SSEA1 (1.9%). The Shef 6 cell line was tested after 14 passages in CELLstart; at this point, between 91% and 96% of cells showed markers of pluripotency, whereas only 20% and 2.7% of cells were positive for Nestin and SSEA1, respectively (Fig. 5 and Supplementary Fig. S2).
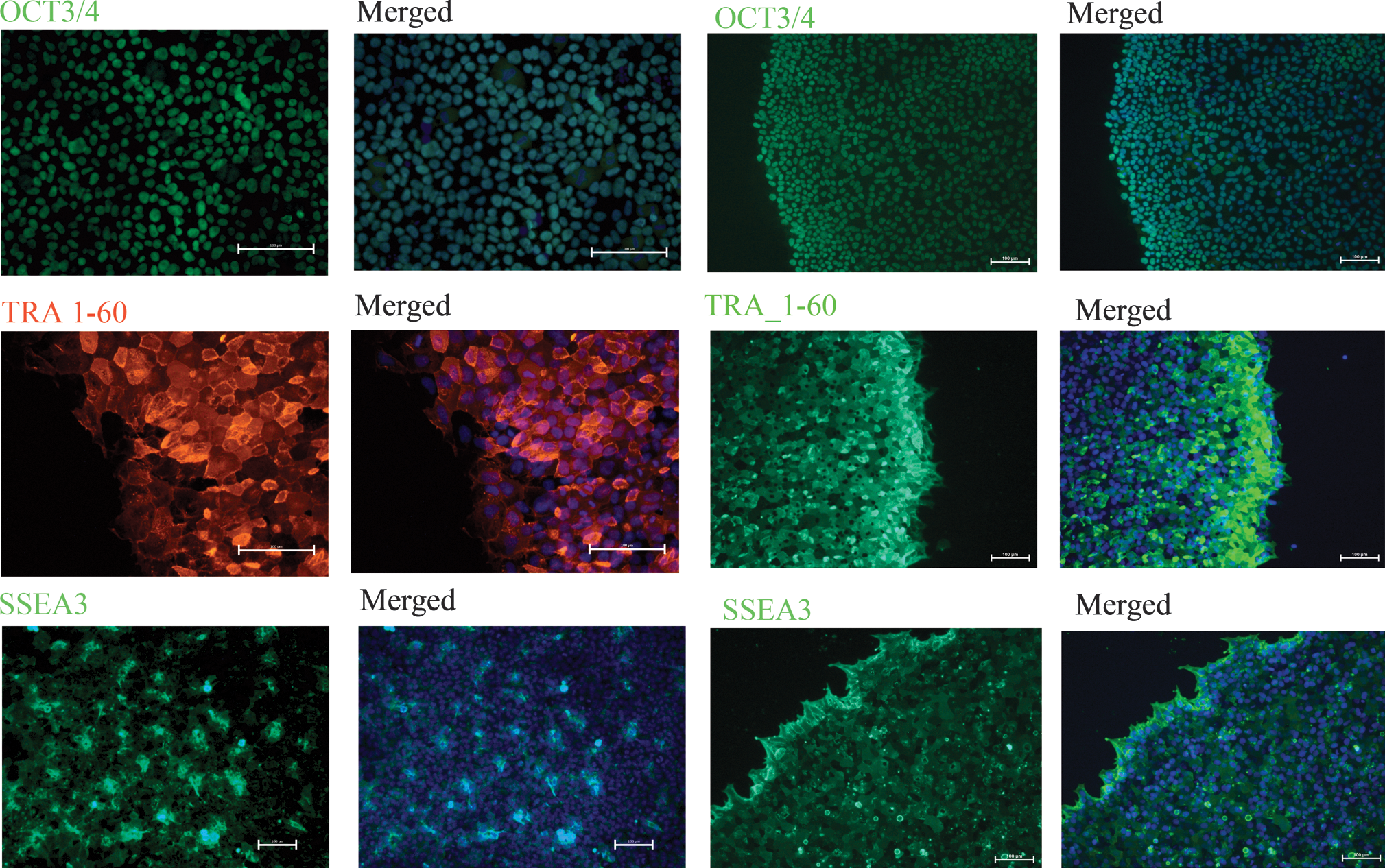
Immunocytochemistry showing both Shef 3 and Shef 6 hES cell colonies grown on CELLstart and stained for the markers OCT3/4, TRA-1-60, and SSEA3 (green). The 2 columns on the left show the Shef 3 cell line; the 2 columns on the right correspond to the Shef 6 line. In each set of columns, the panels on the left show the staining with the antibody; the merged photos are on the right panels. Magnification, 20 × ; scale bar = 100 μM for OCT3/4 and TRA-1-60. Magnification, 10 × ; scale bar = 100 μM for SSEA3.
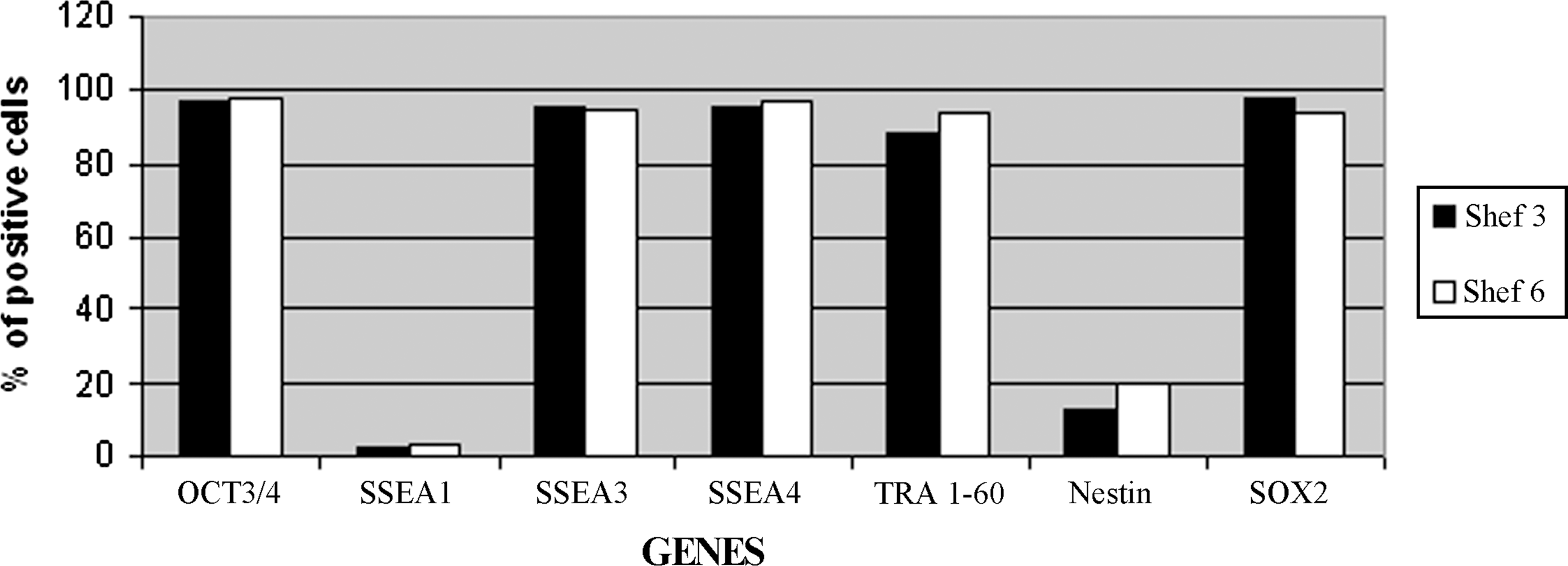
Graph representing the proportion of cells expressing different pluripotency genes as measured by FACS analysis. The Shef 3 line is represented by the solid bars and the Shef 6 line by the clear bars. Complete data for the FACS analysis are shown in Supplementary Fig. S2. FACS, fluorescence-activated cell sorting.
Quantitative RT-PCR analysis (Fig. 6) showed very little variation in the levels of expression of the pluripotency genes OCT3/4 and NANOG in either Shef 3 or Shef6, between cells grown on feeders compared to cells grown on CELLstart after 8 or 12 passages (Shef3) and 5, 8, and 15 passages (Shef 6) (P > 0.05). However, there is a statistically significant upregulation of SOX2 in Shef 3 cells grown on the CELLstart matrix compared to the original cell population grown on feeders. At present we do not know what the cause of this upregulation is, but it does not appear to affect the cells ability to differentiate. In Shef 6 there is a transient upregulation of TERT in the first 5 passages, but it returns to normal levels after 15 passages. The significance of this upregulation remains to be investigated further.

Freeze–thawing
To further investigate the characteristics of the adapted Shef 3 cell line, we carried out 2 cycles of freeze–thawing. Cells that had been grown on CELLstart for 8 passages were frozen after mechanical scrapping by either vitrification or in mFreSR. After 3 weeks in liquid nitrogen, cells were thawed in a fresh medium and plated into either CELLstart or feeders. Cells thawed into CELLStart did not settle in the first instance; however, they remained viable and grew in suspension as small aggregates, during this time the medium was added to the wells, but not exchanged. After 2–3 weeks, the cells settled, attached to the substrate, and formed large colonies with perfect hES cell morphology and very little differentiation at the center of the colonies (Fig. 7). These cells were expanded by mechanical passaging and re-analyzed for pluripotency.
Shef 3 cells after being thawed.
Cells thawed onto feeder layers in the KSR medium attached within 16–36 h and colonies formed as expected. Both sets of cells were expanded and analyzed side by side for comparison.
After further passaging the thawed cells were frozen in mFreSR, and thawed a month later. This time the cells attached quickly onto the CELLstart matrix (3–4 days) and formed classic hES cell colonies within a week.
Differentiation potential
To determine if Shef 3 cells were still capable of differentiation, we carried out EB formation by cell aggregation in nonadherent bacteriological-grade dishes. Initially, we plated cells in the StemPro medium in nonadherent dishes, but were unable to obtain any EBs, cells continued to survive in suspension, but cell aggregation did not occur; moreover, when these cells were plated back onto CELLstart matrix, they re-attached and formed classic ES cell colonies. When EB formation was carried out in the medium containing 10% KSR (in the absence of bFGF), EBs formed readily within a week to 10 days (Fig. 8). After 7 days, EBs from 3 independent dishes were pooled, RNA was extracted from them and analyzed by RT-PCR. Markers of all 3 embryonic germ layers were readily amplified from these EBs [Fig. 9 (columns 1–3)], although we noted lower levels of expression of the mesodermal marker BRACHYURY (T) and the endodermal marker FOXA2 in EBs derived from cells cultured on CELLstart versus those cultured on feeders. We cannot be sure if this is due to the use of the matrix, the absence of feeders, a slight delay in the formation of mesoderm in EBs derived from cells grown on CELLstart, or part of an adaptation process that may select against cells expressing endodermal markers.

EBs formed by Shef 3 cells grown in CELLstart.
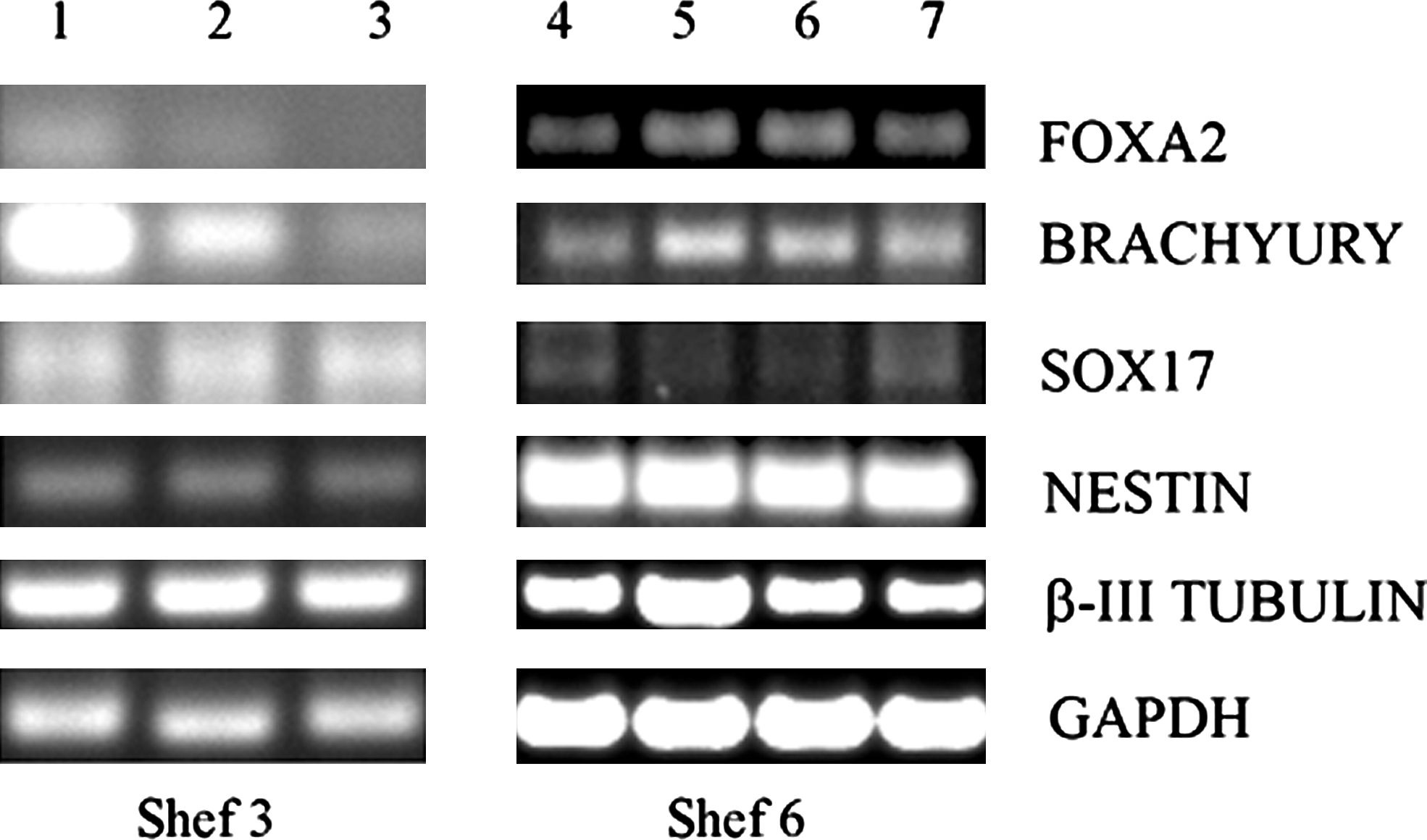
RT-polymerase chain reaction amplification of differentiation markers from EBs. Column 1: pooled EBs (7 days old) made from Shef 3 cells that had been grown in CELLstart for only 2 passages. Column 2: pooled EBs (7 days old) made from Shef 3 cells that had been grown in CELLstart for 8 passages had then been frozen and thawed onto feeders. Column 3: pooled EBs (7 days old) made from Shef 3 cells that had been grown in CELLstart for 8 passages had then been frozen and thawed back onto CELLstart. Column 4: pooled EBs (10 days old) made from Shef 6 cells grown on feeders. Column 5: pooled EBs (11 days old) made from Shef 6 cells grown on CELLstart for 19 passages. Column 6: Shef6 cells grown on feeders and differentiated in monolayers for 15 days. Column 7: Shef 6 cells grown on CELLstart for 16 passages and differentiated in monolayers for 15 days. The markers used were GAPDH (housekeeping gene), β-III TUBULIN and NESTIN (ectoderm), SOX17 and FOXA2 (endoderm), and BRACHYURY (mesoderm).
On cells cultured after the freeze–thaw cycle, EBs were readily formed in a variety of media, including StemPro, 10% KSR, and 10% FBS, in bacteriological-grade dishes. We noted that EB formation was cell concentration dependent, but the relationship was not linear. We plated a known number of cells in 35 mm bacteriological-grade dishes in 2 mL of complete StemPro medium, and assessed the capacity of EB formation. On dishes with <10,000 cells/dish, no aggregation occurred. When the number of cells was increased to 100,000, large EBs formed readily within a week. However, when the cell number was increased further to 1 or 5 million cells, the aggregation was very poor and almost no EBs were formed. This observation implies that there is a narrow window of cell number/area/volume at which EB formation is optimal for hES cells cultured in this system.
Embryoid bodies cultured for 14 days were plated into gelatinized tissue culture dishes and after 1 week were fixed and stained with antibodies for all 3 embryonic germ layers. We were able to readily identify cells expressing markers of all 3 embryonic germ layers (BRACHYURY (T), β-III-TUBULIN, NESTIN, and AFP) (data not shown).
Embryoid body formation on the Shef 6 was achieved using both the EB medium and StemPro medium without bFGF; again, the formation of EBs was dependent on the cell concentration (as for the Shef3). We were also able to differentiate these cells as monolayers by withdrawing bFGF from the medium. After 10 to 15 days, multiple types of cells could be observed on the plates. These cells were harvested and analyzed by PCR for markers of the 3 germ layers, along with the EBs [Fig. 9 (columns 4–7)].
There are some differences in the levels of expression of the different markers between cell lines; this may suggest an intrinsic difference in the competency of individual hES cell lines to differentiate into the different germ-layers, rather than a direct effect of the culturing regime.
Karyology
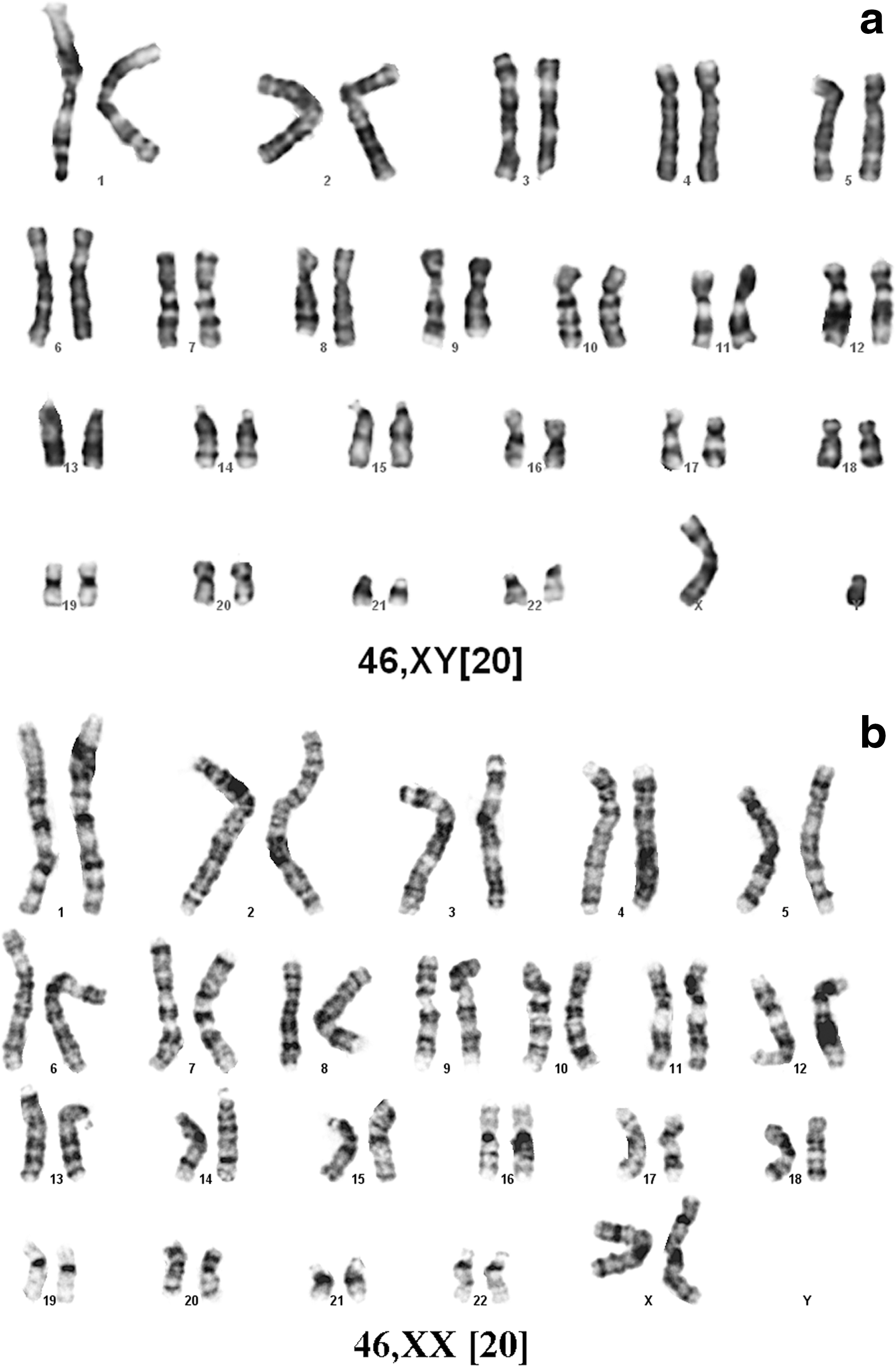
Karyology was carried out at DSL Genetics on the Shef3 cell line before culturing in CELLstart, after 8 passages in CELLstart and after 3 passages after the freeze–thaw cycle. In all cases the karyotype was normal 46 XY (Fig. 10a). For the Shef 6 line karyotyping was carried out after 8 passages in CELLstart; these cells had a normal 46,XX karyotype on all 20 cells counted.
Clonal growth
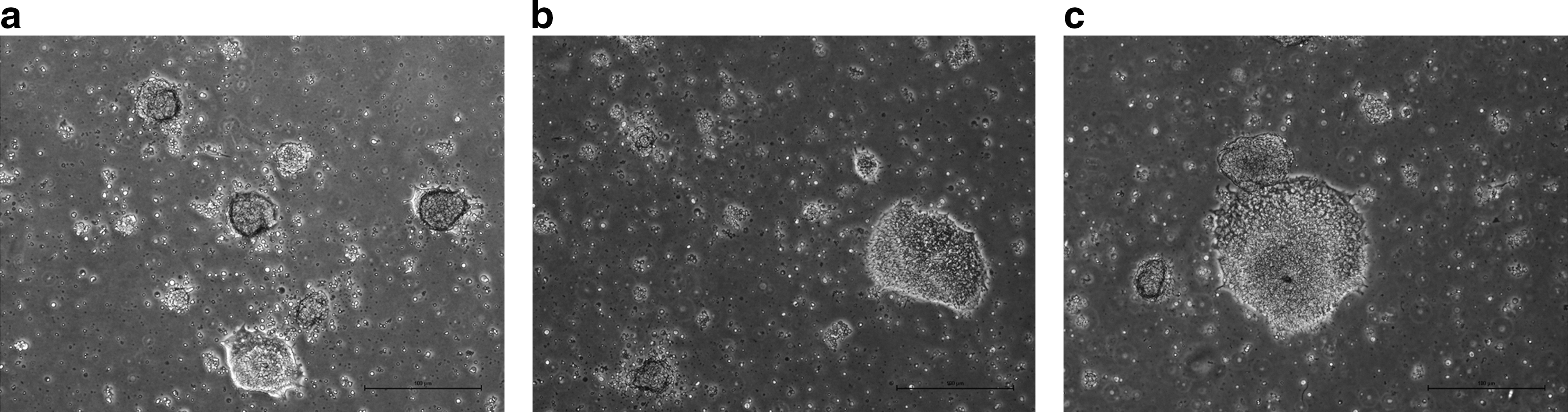
Shef3 cells were plated at limiting dilution in CELLstart and photographed using time-lapse microscopy over a 72-h period. Subsequently, cells were grown under normal conditions for a further 72 h. At the end of the growth period, cells were fixed using 4% PFA and stained for pluripotency markers.
From the time-lapse movie (Supplementary Movie S1) we can see growth from a single attached cell, and subsequent expansion of the colony; there is extensive movement of cells along the matrix as well as detachment and re-attachment after cell division.
After the total growth period we could see well-formed colonies with morphologies indistinguishable from those grown under normal conditions. To measure the plating efficiency of these cells, we stained the plates with crystal violet and counted colonies. We found that plating 500 to 1000 cells/cm2 yielded a plating efficiency of ∼0.6%. Moreover, we could still detect in these colonies co-expression of the pluripotency markers SOX2 and SSEA4, implying that colonies arising from single cells do not loose their phenotypic characteristics, though we acknowledge the possibility that karyotypic or other genetic abnormalities may have been acquired by the cells, which would require further investigation (Fig. 11). A further experiment where 8 colonies were picked and expanded showed that the cells retain normal morphology and still express high levels of pluripotency markers as shown by fluorescence-activated cell sorting analysis of the markers Nanog and SSEA3 (Supplementary Fig. S3).

Shef 3 colony grown from a single cell in CELLstart
Discussion
From our experience, growing cells using the CELLstart/StemPro medium combination was a very efficient and simple way of growing hES cells. The morphology of the colonies is very even, which makes passaging straight forward. For example, while we do use a dissecting microscope to cut colonies, it could also be achieved without any such specialized equipment, by simply scrapping cells from the flasks. Although it has been reported that colony size is crucial for maintaining pluripotency [18], we found that even very big colonies that have merged and overlap still retain pluripotency markers and are capable of in vitro differentiation. The initial adaptationwas fairly straight forward for the Shef 3 cell line though it may in part be due to the fact that Shef3 cells have been traditionally grown at relatively low feeder densities. With the Shef 6 line we noted more propensity to spontaneously differentiate when cultured on the feeder-free matrix, but we were able to resolve this by adding extra bFGF, and passaging more regularly, this propensity for differentiation was also noted on Shef 6 cells grown on feeder layers, implying that this a characteristic of this line. Our most obvious concern after the freeze–thaw cycle, and as the cells were growing and dividing fast, was that they had become karyotypically abnormal, a well-documented phenomenon [19]. However, we have found that not to be the case, as the cells have remained karyotypically normal (46XY) after 12 passages and 1 freeze–thaw cycle in the case of Shef3. For Shef6, the karyotype was also found to be a normal 46 XX, after 8 passages in CELLstart and StemPro.
Nevertheless, it is possible that the cells have undergone more subtle genetic of epigenetic changes that have selected for a population of cells which favors self-renewal over differentiation, which would in part explain the high growth rate and the downregulation of endodermal markers seen. This phenomenon of adaptation, in which cells expressing higher levels of, say, endodermal markers (bone morphogenetic proteins), are selected against, as they favor differentiation above self-renewal, has been postulated by Andrews and colleagues [20,21]. This phenomenon though appears to be independent of the mode of culturing conditions used.
The StemPro/CELLstart system, however, is especially useful for applications where feeders may interfere with the overall outcome of the experiment, such as genetic manipulation. Major stumbling blocks in the production of genetically manipulated hES cells, such as fluorescent reporters, have been the difficulty in efficiently transfecting cells, as the feeders interfere or mop up the DNA, and as it has been notoriously hard to achieve clonal selection in hES cells [22]. With this system, these 2 problems are circumvented. As cells are grown free of feeders, transfections should be as efficient as they are in other primary cells, and secondly we have shown that using CELLstart it is possible to grow cells at clonal densities. Therefore, selecting for rare transfectants should be greatly improved. Moreover, the choice of drug resistance markers that can be used for positive selection is wider, as there is no need to obtain specific feeders for each different type of drug resistance gene used.
Overall using CELLstart as a matrix for growing hES cells is a vast simplification of the current feeder culture system, it is much less labor intensive, and the quality of the cells produced is very high. Unlike other feeder-free systems we tried, in CELLstart we were able to grow cells continuously for over 20 passages without loss of pluripotency, normal karyotype, or morphology. Furthermore, we were able to grow cells at clonal densities.
We acknowledge the fact that we have not performed in vivo teratoma formation assays to assess the differentiation potential of our feeder-free adapted cells, which would confirm that the adapted cells remain pluripotent. Although this method of assessment has been traditionally used by many laboratories as the gold standard of pluripotency assays in hES cells, it is not without its problems. Not only is it an extremely expensive assay, but it also requires access to specialized facilities and in the United Kingdom there is a requirement for a Home office licence to be obtained. There are no commercial providers of this service in the United Kingdom, but even in the United States the cost is substantial with a turn around time of 15 weeks. These problems aside, the assay itself has come under criticism, as the way in which it performed and reported has not been standardized [23]. Further, there have been calls, in the induced pluripotent stem cell field, that for in vitro use of cells for research, there should be no requirement for this assay to be performed for all new cell lines created [24], as in vitro assays can show the capability of cells to differentiate into all 3 germ layers, which seems a sensible as more and more cell lines are derived. Recent publications by the International Stem cell consortium have not included in vivo assays, when assessing the suitability of various medium formulations (eg, refs. [25,26]).
Footnotes
Acknowledgments
We would like to thank Prof. Peter Andrews (Sheffield University) for the SSEA1, SSEA3, SSEA4, TRA-1-60, and TRA-180 antibodies. We would also like to thank Dr. Alaine Maxwell, David Welch, and Dr. Mahendra Rao at Invitrogen for helpful comments and suggestions. D.H. is in receipt of a UK Technology Strategy Board grant.
Author Disclosure Statement
The authors disclose no conflict of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.