
Abstract
In order for human embryonic stem cells (hESCs) to be cultured on mouse embryonic fibroblast (MEFs) feeder cells, continuous basic fibroblast growth factor (bFGF) supplementation is required. However, the role of bFGF in a culture system using human-derived feeder cells has not been evaluated until now. In this study, we propagated the widely used hESC lines, H1 and HSF6, on human placenta-derived feeder cells (HPCs) without exogenous bFGF supplementation, and were able to propagate hESCs on HPC feeders up to 50 passages. The absence of bFGF in culture media did not interrupt the undifferentiated propagation and the expression of pluripotent stem cell markers ALP, SSEA-4, TRA-60, Oct-4, Nanog, and Rex-1, as well as the formation of embryoid bodies (EBs) and their differentiation potential. In contrast, hESCs cocultured with MEF feeders could not propagate and form EBs without exogenous bFGF supplementation. Expression of bFGF and the activation of the ERK1/2-c-Fos/c-Jun pathway, which is known as the signaling pathway of bFGF, were identifiable not only in hESCs cultured in bFGF-containing media regardless of feeder cell type, but also in hESCs cocultured with HPC feeder cells in media without bFGF. These findings may support the hypothesis that HPC feeder cells enhance endogenous bFGF production and activation of the ERK1/2-c-Fos/c-Jun pathway, which suggests that HPCs have an additional advantage in their hESC propagation compared with MEF.
Introduction
B
Recent studies demonstrated that in serum-free cultures, the addition of basic fibroblast growth factor (bFGF) to culture medium is beneficial for the maintenance of the self-renewing activity of hESCs [4 –6]. bFGF, also called FGF-2, has 5 isoforms, of which 4 high-molecular-weight isoforms (22, 22.5, 24, and 34 kDa) are thought to function in an intracrine manner, and 1 low-molecular-weight isoform (18 kDa) is thought to be secreted and function in a paracrine or autocrine manner [7]. When hESCs are cultured in a feeder-free, serum-free system, bFGF alone could support the stem cell characteristics of hESCs without the contribution of other cytokines [8]. Although it was demonstrated that bFGF affects hESCs through both autocrine and intracrine mechanisms [9,10], little is known of the exact bFGF process that enables hESCs to remain undifferentiated. It is clear that bFGF is the essential cytokine for maintaining hESCs in the undifferentiated state in a xeno-free culture system. However, xeno-free culture systems have some limitations. The hESCs cultured under these conditions have lower cloning efficiencies and growth rates, and higher rates of differentiation than cells maintained on MEF feeder cells or on MEF-conditioned media [8], which still requires addition of bFGF, unless animal serum is used [11].
Human-derived feeder cell lines have been developed to solve these problems. Recently, several studies have demonstrated that human-derived feeder cell lines are quite effective [12 –18]. Because bFGF is the essential cytokine for maintaining the undifferentiated state of hESCs on MEF feeders, bFGF has been routinely added to culture medium in these studies to escape the potential risk of accidental differentiation. However, we have postulated that cell-to-cell interactions and secreted proteins from human-derived feeder cells might be different from those of animal feeders. Based on this hypothesis, we cultured the widely used hESC lines, H1 and HSF6, on human placenta-derived feeder cells (HPCs) with and without exogenous bFGF supplementation, and on MEF feeders with and without exogenous bFGF supplementation, and compared the outcomes.
Materials and Methods
Preparation of feeder cells from placenta and MEF cells
A previously reported protocol for preparation of feeder cells from human placenta was used in this study [12,19]. In brief, placental chorionic plates from healthy women who had undergone abortion at 6–8 weeks of gestation were surgically isolated, minced, and incubated in 0.25% trypsin–EDTA (GIBCO-Invitrogen, Carlsbad, CA) at 37°C for 30 min. The cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with 20% FBS (HyClone Laboratories Inc., Logan, UT), 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C, 5% CO2, and 95% humidity. The medium was changed every 3–4 days until the third passage. During culture, cell debris floating in the culture medium was removed, and HPCs were seen growing attached to the tissue culture plate. Approximately 2 weeks after inoculation, colonies of fibroblast-like cells that were attached to the plates had formed. Mesenchymal stem cells (MSCs) in HPCs showing positive expression of CD 29 and CD 44 and negative expression of CD 34 were identified by flow cytometric analysis (data not shown). In primary cultures of HPCs from 10 initial placentas, HPCs usually became thin and lost stability after 20–25 passages, and after the 13–17 passages, HPCs could not support undifferentiated growth of the hESC lines H1 and HSF6. Therefore, HPCs were cultured to the 12th passage, and harvested for hESC cultures after trypsinization and irradiation (1,500 cGy).
Reverse transcription (RT)-PCR was used to determine if HPCs were contaminated with pathogens that commonly infect the placenta before they were frozen as stock. Pathogens include cytomegalovirus, herpes simplex virus types 1 and 2, Chlamydia trachomatis, Chlamydia spp, Mycoplasma genitalium, Mycoplasma hominis, and Ureaplasma ureaticum. Published primer sequences were used [19,20]. HPC testing positive for pathogens were abandoned.
Twenty-five placentas were used for obtaining feeder cells for hESC culture in this study. The mean number of obtained feeder cells was 1.42 × 108/chorionic plate (range: 8.7 × 106/chorionic plate to 3.95 × 108/chorionic plate, 95% confidence interval: 4.5 × 107/chorionic plate to 1.98 × 108/chorionic plate). The number of available feeder cells had a tendency to be dependent upon the size of obtained placenta. Cells were frozen in cryovials at 1 × 106 cells/mL per vial. For hESC coculture, cryovials of HPCs were thawed and seeded onto 0.1% gelatin-coated 35-mm well plates at 1 × 106 cells/well (1 × 103 cells/cm2). The MEF cell line was purchased from Millipore Corp. (Bedford, MA).
MEFs were cultured in DMEM supplemented with 10% FBS to ∼90% confluence; typically, MEFs could not be propagated after 5–7 passages and could not support undifferentiated growth of hESCs after the 4–5 passages. Therefore, MEFs were cultured to the third passage and stocked for ESC coculture. Before transfer to plates, MEFs were prepared by incubation with 10 μg/mL mitomycin for 90 min. The 2 feeder cell lines (HPC, MEF) were allowed to attach for 24 h before hESC seeding.
Human ESC culture
The H1 hESC line (passage (p) 29) was purchased from the National Stem Cell Bank and initially cocultured with MEFs according to the instructions of the provider. The HSF6 hESC line (p63), which was originally purchased from the National Stem Cell Bank, and kindly provided by Dr. Jong Hoon Kim (Laboratory of Stem Cell Biology, College of Life Sciences and Biotechnology, Korea University), was cocultured with MEFs. After the H1 and HSF6 lines were maintained on MEFs for 10 passages, colonies of each line were transferred onto freshly prepared HPC and MEF feeders and were propagated on each feeder with or without bFGF (Fig. 1). bFGF was added to culture medium at a concentration of 4 ng/mL. The other culture conditions of each experimental group were the same as follows: DMEM/F-12 supplemented with 20% knockout serum replacement (GIBCO), 0.1 mM β-mercaptoethanol, and 1% penicillin–streptomycin (Sigma, St. Louis, MO) were used for coculture of hESCs and feeder cells. The culture medium was exchanged every 48 h. hESCs were passaged weekly by mechanical dissociation, seeded at each passage at a density of 4× 104 cells per plate (the area of each plate is 9.62 cm2 and thus about 4158 cells/cm2 was seeded), and then cultured at 37°C, 5% CO2, and 95% humidity. During culture, ESC colonies were counted at every passage during the first 5 passages, and then every 5 passages. A grid hemocytometer were used to count colonies, and each colony counted consisted of 100–280 ESCs.

The study scheme. Samples are grouped according to hESC lines, the feeder cell type, and presence or absence of exogenous bFGF: H1/MEF/±bFGF, HSF6/HPC/±bFGF, H1/HPC/±bFGF, HSF6/HPC/±bFGF. Abbreviations: ESC, embryonic stem cell; MEF, mouse embryonic fibroblast; HPC, human placenta-derived feeder cell; bFGF, basic fibroblast growth factor; +bFGF, supplemented with exogenous bFGF; –bFGF, not supplemented with exogenous bFGF.
Characterization of hESCs
To identify the undifferentiated state of hESCs, morphology, expression of stem cell markers, and differentiation capacity were examined. Cell morphology was observed each day using an inverted microscope, and expression of stem cell markers were tested by immunostaining for alkaline phosphatase (ALP), Oct-4, stage-specific embryonic antigen (SSEA)-4, tumor-rejection antigen (TRA)-60, and by RT-PCR for Oct-4, Nanog, and Rex-1. Characterization of cultured hESCs was performed every 10 passages (Fig. 1). The primers used in RT-PCR were as follows: Oct-4, 577 bp: CGACCATCTGCCGCTTTGAG (forward), CCCCCTGTCCCCCATTCCTA (reverse); Nanog, 149 bp: AAAGAATCTTCACCTATGCC (forward), GAAGGAAGAGGAGAGACAGT (reverse); and Rex-1, 306 bp: CAGATCCTAAACAGCTCGCAGAAT (forward), GCGTACGCAAATTAAAGTCCAGA (reverse). Cocultures used for immunocytochemistry were established in 6-well plates. Prior to analysis, adherent cell layers were fixed by addition of 10% formalin (15 min) at room temperature, permeabilized with 0.1% Triton X-100/PBS for 10 min, and incubated with primary antibodies overnight at 4°C. Primary antibodies for SSEA-4 were purchased from Hybridoma Bank (Hybridoma Bank, Iowa City, IA) and other antibodies were purchased from Chemicon (Chemicon, Temecula, CA). ALP activity was detected using a commercial kit (Sigma).
RT-PCR for stem cell markers was also performed. Total RNA was prepared using a QIAGEN RNeasy kit (Qiagen-Hilden, Germany), and RT was performed with 500 ng of total RNA primed with random hexamers, using avian myeloblastosis virus (AMV) reverse transcriptase (Roche Molecular Biochemicals, Germany). After PCR was performed, products were analyzed on a 1.5% agarose gel stained with ethidium bromide.
Karyotype analysis
To evaluate chromosomal stability, karyotype analysis was performed of cultured hESCs every 10 passages (Fig. 1). ESCs were incubated with 0.1 μg/mL colcemid for 3–4 h, trypsinized, and then incubated in 0.075 M KCl for 20 min at 37°C. After fixation with 3:1 methanol/acetic acid, the karyo-types of ESCs were analyzed at 550-band resolution.
Formation of embryoid bodies and RT-PCR analysis for lineage-specific markers
Embryoid body (EB) formation was induced from cultured hESCs every 10 passages to determine hESC differentiation potential during long-term in vitro maintenance (Fig. 1). The hESC colonies were harvested at each designated point in time in order to induce EB formation. They were transferred to EB culture medium: DMEM/F-12 supplemented with 20% serum replacement without bFGF. The number of EBs formed was counted, and the percentage of formed EBs per seeded hESC colony was calculated. To detect the presence of 3 germ layers within the formed EB, RT-PCR analysis of EBs was performed on Day 21 every 10 passages. The primers are as follows: Sox-1, 132 bp: CACAACTCGGAGATCAGCAA (forward), GGTACTTGTAATCCGGGTGC (reverse); Brachyury, 111 bp: AATTGGTCCAGCCTTGGAAT (forward), CGTTGCTCACAGACCACA (reverse); and AFP, 675 bp: AGAACCTGTCACAAGCTGTG (forward), GACAGAAGCTGAGGATGTC (reverse).
RT/RQ-PCR analysis and enzyme-linked immunosorbent assay for bFGF
To identify whether feeder cells (MEF and HPC) and cultured hESCs could express and secrete bFGF, RT/real-time quantitative (RQ)-PCR analysis was performed on feeder cells and hESCs every 10 passages and enzyme-linked immunosorbent assay (ELISA) was used to analyze supernatants. Primers for bFGF are GGCTTCTTCCTGCGCATCCAGCATCCA (forward) and GCTCTTAGCAGACATTGGAAGA (reverse). RQ-PCR of bFGF expression in H1 and HSF6 hESCs was performed every 10 passages. At each measurement the results were adjusted with the amount of housekeeping gene (GAPDH) expression (value of bFGF/value of GAPDH) and, after setting the value of the control group (hESCs cultured on MEF feeder with exogenous bFGF supplementation) as 1, the proportional values of HPC feeder groups (hESCs cultured on HPC feeder with or without exogenous bFGF supplementation) were compared with the control. The mean of these calculated values in HPC feeder group were compared with the control.
Before culture media were changed, that is, after the culture media were conditioned for 48 h, 1 μL each of supernatants were collected from all experimental groups during the first 10 passages. The bFGF concentration of each supernatant was measured using a commercially available bFGF ELISA kit (R&D Systems, Germany) according to manufacturer's instructions. Each measurement was performed twice, and the mean concentration of bFGF was determined.
Western blot, densitometry, and RT-PCR analysis for bFGF signaling pathway
To identify the bFGF signaling pathway in hESCs, we performed immunoblot assays for phosphorylated extracellular signal-regulated kinase (pERK), and RT-PCR for c-Fos, and c-Jun expression. The primers of c-Fos and c-Jun are as follows: c-Fos, 236 bp: GAATAAGATGGCTGCAGCCAAATGCCGCAA (forward), CAGTCAGATCAAGGGAAGCCACAGACATCT (reverse) and c-Jun, 325 bp: GGAAACGACCTTCTATGACGATGCCCTCAA (forward), GAACCCCTCCTGCTCATCTGTCACGTTCTT (reverse). For western blot analysis, hESCs from both H1 and HSF6 were harvested by gently using a glass pipette, washed with phosphate-buffered saline (PBS), and lysed in 100 μL of sodium dodecyl sulfate (SDS) sample buffer (12 mM Tris–HCl, pH 6.8, 0.4% SDS, 5% glycerol, 0.02% bromophenol blue, 0.288 mM 2-mercaptoethanol). Samples were subjected to SDS-polyacrylamide gel electrophoresis (PAGE) and electrotransferred onto polyvinylidene fluoride (PVDF) membranes (Millipore Corp., Bedford, MA). The membranes were soaked for 2 h at room temperature in blocking buffer (PBS, 0.5% Tween-20 containing 5% nonfat dry milk) and then incubated overnight at 4°C with appropriate primary antibody. Proteins were detected by horseradish peroxidase (HRP)-conjugated secondary antibody and an enhanced chemiluminescence (ECL) reagent (Amersham Pharmacia Biotech, Piscataway, NJ). Antibodies to ERK1/2, pERK1/2 (Thr202/Tyr204) were purchased from Cell Signaling Technology (Beverly, MA). Anti-c-Fos and anti-c-Jun were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). ImageJ software was used to conduct the densitometry analysis.
Statistical analysis
Each hESC group (H1/MEF/±bFGF, H1/HPC/±bFGF, HSF6/MEF/±bFGF, HSF6/HPC/±bFGF) consisted of 5 culture dishes; values for each group were the mean of the 5 dishes. Experiments were repeated 10 times, and results were verified by 2 other specialists. All quantitative data were presented as mean ± SEM. Results were considered significant when the P value was <0.05. The overall study scheme is shown in Figure 1.
Results
Comparison of colony counts
In the MEF feeder groups with exogenous bFGF, the mean numbers of colonies were 147 ± 17 per plate (15.3 ± 1.8 colonies/cm2) in H1 hESCs cultured on MEF feeder with bFGF supplementation (H1/MEF/+bFGF group) and 150 ± 13 per plate (15.6 ± 1.4 colonies/cm2) in HSF6 hESCs cultured on MEF feeder with bFGF supplementation (HSF6/MEF/+bFGF group) over 50 passages, and it was possible to propagate hESCs in both H1/MEF/+bFGF and HSF6/MEF/+bFGF groups beyond 50 passages (Fig. 2A and 2B). However, in the MEF feeder groups without exogenous bFGF, the mean numbers of colonies were 71 ± 13 per plate (7.4 ± 1.4 colonies/cm2) in H1 hESCs cultured on MEF feeder without bFGF supplementation (H1/MEF/–bFGF group) and 56 ± 11 per plate (5.8 ± 1.1 colonies/cm2) in HSF6 hESCs cultured on MEF feeder without bFGF supplementation (HSF6/MEF/–bFGF) and the colonies could not be maintained beyond 10 passages (Fig. 2A and 2B). There were significant differences in the mean colony counts of H1 and HSF6 according to the presence or absence of exogenous bFGF (P < 0.001 in H1 and HSF6, respectively) (Fig. 2A).
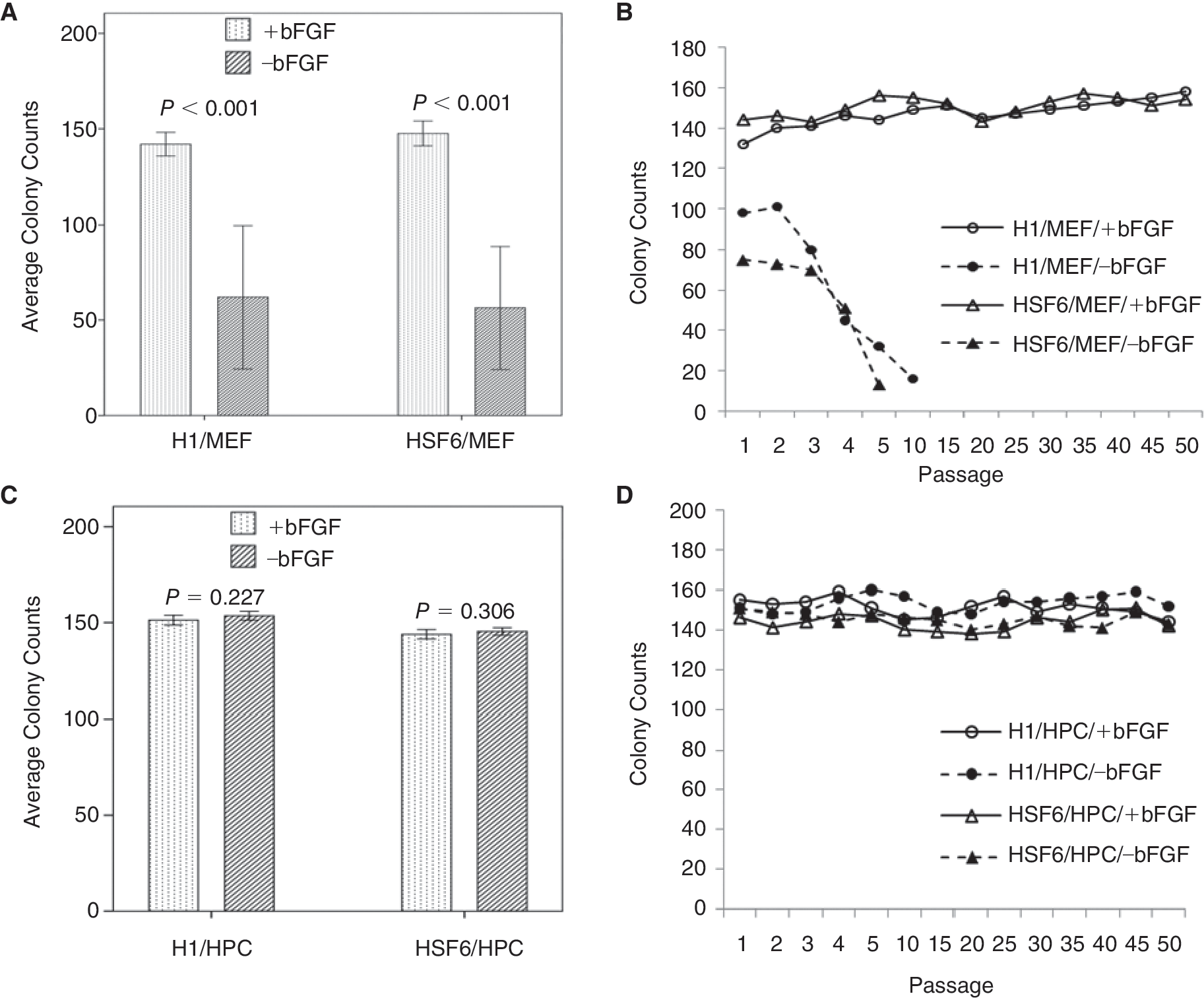
The results of colony counts of human embryonic stem cells (hESCs) on each feeder cell type over passages. (
In the HPC feeder groups with exogenous bFGF, the mean numbers of colonies were 151 ± 11 per plate (15.7 ± 1.1 colonies/cm2) in H1 hESCs cultured on HPC feeder with bFGF supplementation (H1/HPC/+bFGF group) and 143 ± 13 per plate (14.9 ± 1.4 colonies/cm2) in HSF6 hESCs cultured on HPC feeder with bFGF supplementation (HSF6/HPC/+bFGF group) over the 50 passages (Fig. 2C). In the HPC feeder groups without exogenous bFGF, the mean numbers of colonies were 153 ± 10 per plate (15.9 ± 1.0 colonies/cm2) in H1 hESCs cultured on HPC feeder without bFGF supplementation (H1/HPC/–bFGF group) and 145 ± 9 (15.1 ± 0.9 colonies/cm2) in HSF6 hESCs cultured on HPC feeder without bFGF supplementation (HSF6/HPC/–bFGF group) (Fig. 2C). In the HPC feeder groups, colonies of hESC could be propagated beyond 50 passages regardless of the presence or absence of exogenous bFGF (Fig. 2D). There were no significant differences in the mean colony counts of H1 and HSF6 according to the presence or absence of exogenous bFGF (P = 0.227 in H1 and P = 0.306 in HSF6). H1 and HSF6 colonies could be propagated on HPC feeders without exogenous bFGF as efficiently as on HPC feeders with exogenous bFGF.
Undifferentiated state and chromosomal stability of hESCs during in vitro culture
Exogenous bFGF was required to maintain the colonies of hESCs (H1 and HSF6) on MEF feeders. Without exogenous bFGF, the colonies of hESCs became thin and detached from feeder cells at passage 8 or 9 (Fig. 3A, 3B, 3I, and 3J). However, on HPC feeders, hESC colonies were well maintained regardless of the presence or absence of exogenous bFGF, and morphologically there were no differences between exogenous bFGF-supplemented groups (+bFGF) and exogenous bFGF-unsupplemented groups (−bFGF) (Fig. 3C, 3D, 3K, and 3L). On MEF feeders, hESC colonies strongly expressed ALP only in the presence of exogenous bFGF (Fig. 3E, 3F, 3M, and 3N). In contrast, hESC colonies strongly expressed ALP on HPC feeders regardless of exogenous bFGF (Fig. 3G, 3H, 3O, and 3P). Immunocytochemical staining for stem cell markers such as Oct-4, SSEA-4, and TRA-60 showed well-conserved stem cell markers on MEF feeders with exogenous bFGF (data not shown). On HPC feeders, highly expressed stem cell markers were identified regardless of the presence or absence of exogenous bFGF over long-term maintenance (Fig. 4). Stem cell characteristics were also evaluated every 10 passages by RT-PCR for Oct-4, Nanog, and Rex-1. Expression of these markers was identified in hESCs on MEF feeders with bFGF. On HPC feeders, similar levels of expression of these stem cell markers were identified in hESCs cultured with and without exogenous bFGF (data not shown). Therefore, the absence of exogenous bFGF did not influence the expression of stem cell genes in hESCs cocultured with HPC feeders. Determination of the hESC karyotype every 10 passages did not identify karyotypic abnormalities in H1 and HSF6 lines cocultured with MEFs plus exogenous bFGF (H1/MEF/+bFGF, HSF6/MEF/+bFGF) and in all the HPC groups (H1/HPC/±bFGF, HSF6/HPC/±bFGF) (data not shown).
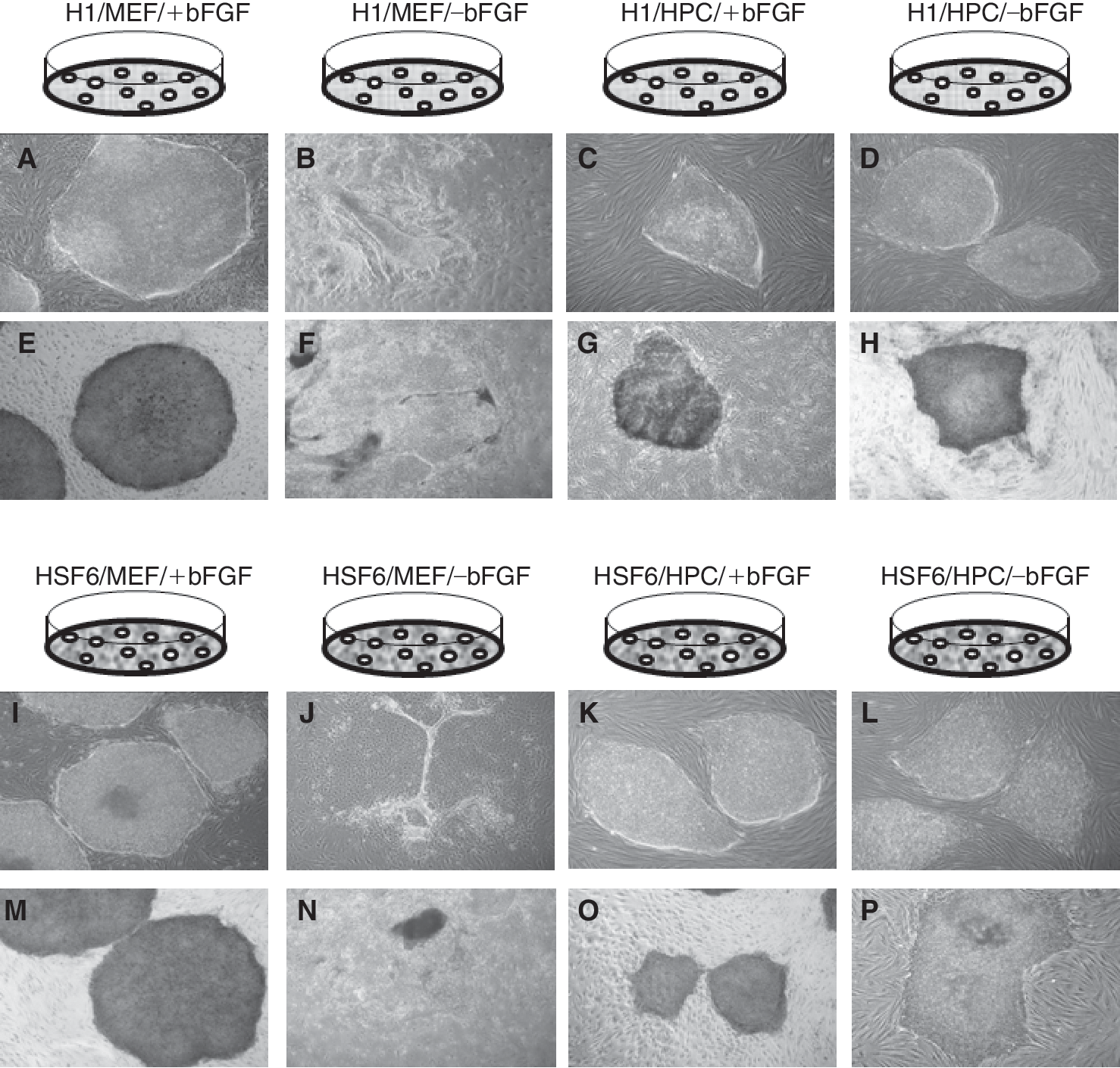
. (
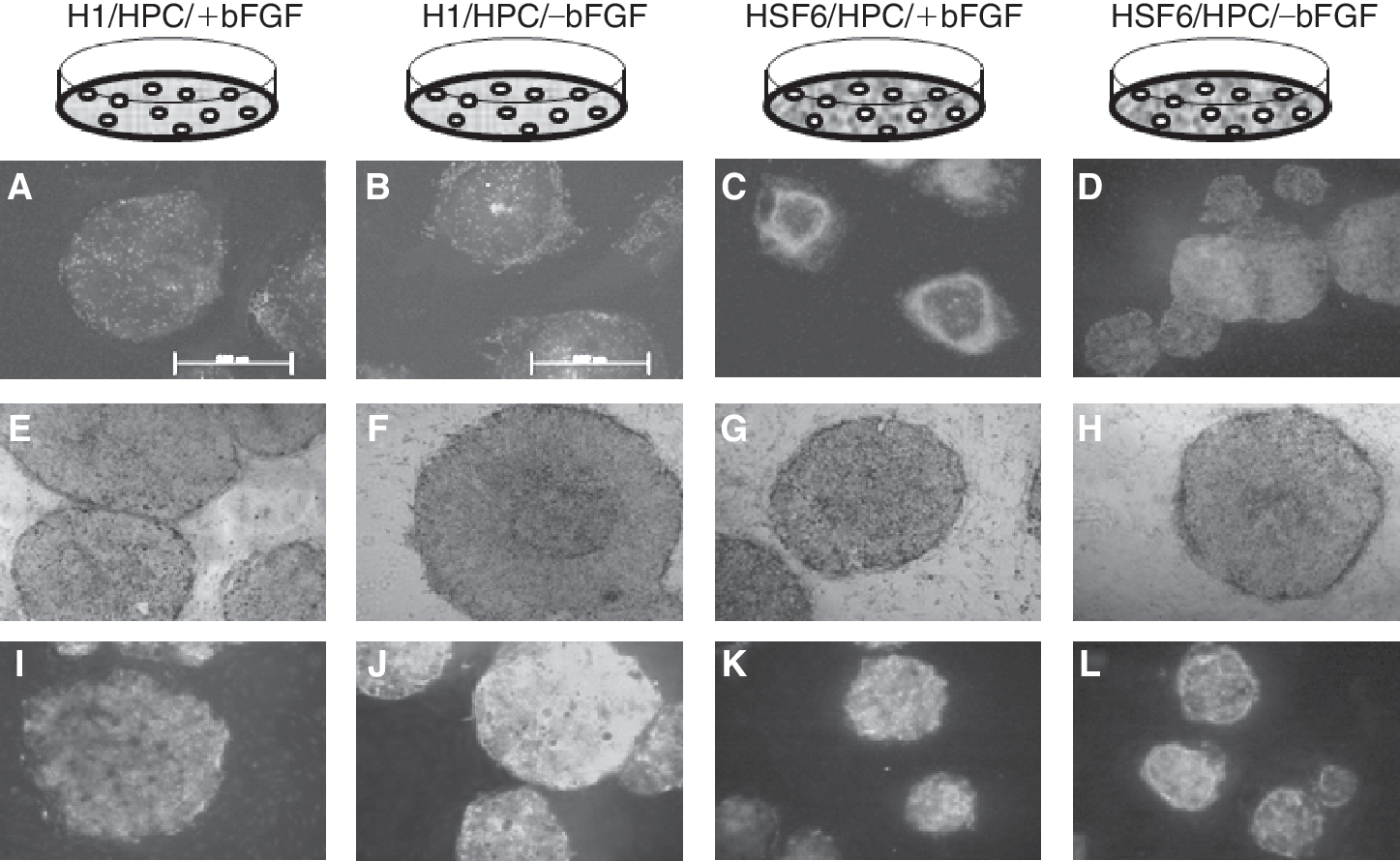
Immunostaining results for stem cell markers in the human placenta-derived feeder cell (HPC) groups. (
EB formation and identification of lineage-specific markers
Embryoid body formation was induced from cultured hESCs every 10 passages. The efficiency of EB formation was calculated by the percentage of formed EBs per seeded ESC colony. The mean percentages of formed EBs from hESCs cocultured with MEF feeders were 79.1% ± 3.2% in the H1/MEF/+bFGF group and 82.7% ± 2.5% in the HSF6/MEF/+bFGF group. The mean percentages of formed EBs from hESCs cocultured with HPC feeders were 75.3% ± 5.6% in the H1/HPC/+bFGF group, 77.1% ± 2.6% in the H1/HPC/–bFGF group, 81.6% ± 1.6% in the HSF6/HPC/+bFGF group, and 80.6% ± 3.6% in the HSF6/HPC/–bFGF group. In the HPC feeder groups, there were no significant differences in EB formation efficiency between +bFGF and –bFGF groups. In other words, the absence of exogenous bFGF did not influence EB formation efficiency in hESCs cocultured with HPCs. The presence of 3 germ layers within formed EBs was evaluated and identified by RT-PCR every 10 passages. Expression of Sox-1, Brachyury, and AFP was demonstrated within formed EBs from all HPC groups, indicating the presence of ectoderm, mesoderm, and endoderm (Fig. 5).
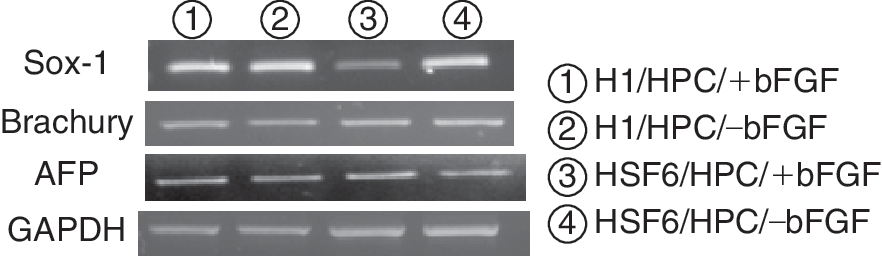
RT-PCR analysis for lineage-specific markers in embryoid bodies (EBs) derived from human embryonic stem cells (hESCs) cultured on human placenta-derived feeder cells (HPCs). All 3 germ-layer-specific markers are well identified in EBs derived from hESCs on HPCs without exogenous basic fibroblast growth factor (bFGF) (lanes (2) and (4)).
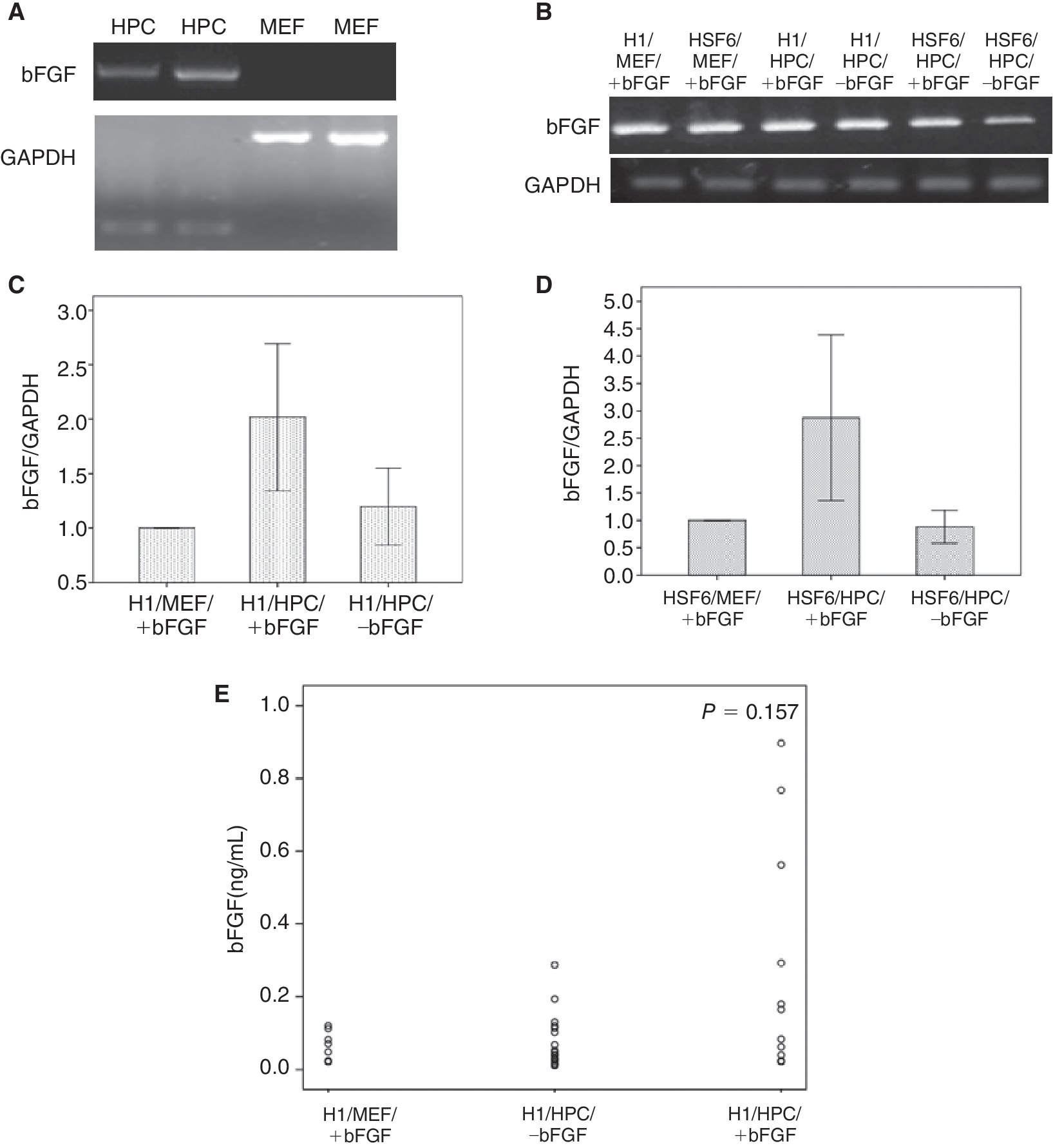
Expression and release of bFGF in both hESCs and HPCs
As mentioned previously, exogenous bFGF was required to maintain the undifferentiated state of hESCs on MEF feeders, but on HPC feeders, hESCs could be propagated in the undifferentiated state without exogenous bFGF. To investigate the biologic differences between MEF and HPC feeders, which could impact the growth and differentiation of hESCs, RT-PCR was performed to detect bFGF expression in feeder cells. The results showed that HPCs expressed bFGF but MEFs did not (Fig. 6A). In hESCs on MEF feeders plus exogenous bFGF supplementation (H1/MEF/+bFGF, HSF6/MEF/+bFGF), bFGF was also identified by RT-PCR analysis. Moreover, all hESCs cocultured with HPCs expressed bFGF regardless of the presence or absence of exogenous bFGF (Fig. 6B). RQ-PCR analysis was performed every 10 passages to compare the degree of bFGF expression within cultured hESCs. It showed that the mean bFGF expression of hESCs in the H1/HPC/+bFGF group was significantly (× 2.01 ± 0.24) higher compared with the expression of hESCs in the H1/MEF/+bFGF group (P = 0.008), and the mean bFGF expression of hESCs in H1/HPC/–bFGF group was not significantly different compared with that of hESCs in the H1/MEF/+bFGF group (P = 0.690) (Fig. 6C). In other words, H1 hESCs cultured on HPC feeder without exogenous bFGF supplementation expressed as much bFGF as H1 hESCs cultured by standard method using MEF feeder with exogenous bFGF supplementation. These trends were also observed in HSF6 cells (Fig. 6D). For quantitation of secreted bFGF, ELISAs were performed on H1 supernatants. Supernatants obtained before transfer were assayed, that is, supernatants conditioned with H1 on feeder cells for 48 h. The mean concentration was 0.07 ± 0.15 ng/mL in the H1/MEF/+bFGF group, 0.14 ± 0.06 ng/mL in the H1/HPC/–bFGF group, and 0.28 ± 0.09 ng/mL in the H1/HPC/+bFGF group. However, there were no significant differences between these groups (P = 0.157). The high mean concentration of H1/HPC/±bFGF is considered to be caused by some extraordinarily high values of outliers (Fig. 6E).
(
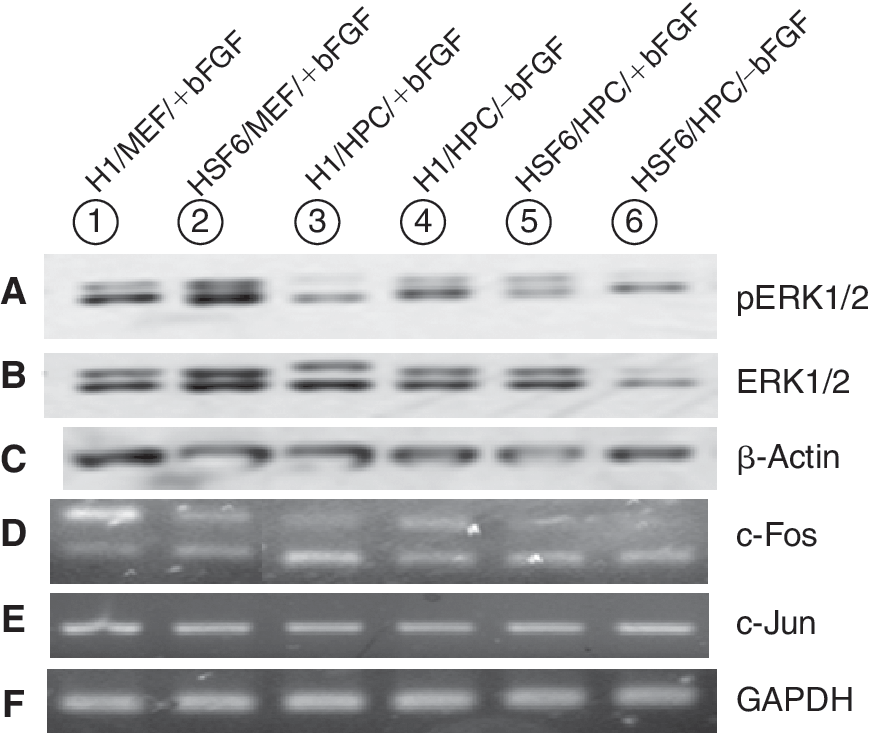
Activation of ERK1/2 in hESCs by bFGF
It was recently reported that bFGF activated the MEK/ERK1/2 pathway in hESCs [9,21]. Western blot analysis for pERK1/2 identified it in hESCs cultured with exogenous bFGF (H1/MEF/+bFGF, HSF6/MEF/+bFGF, H1/HPC/+bFGF, HSF6/HPC/+bFGF) (Fig. 7A and 7B). Interestingly, this pathway was also activated in hESCs cocultured with HPC feeders without exogenous bFGF (H1/HPC/–bFGF, HSF6/HPC/–bFGF) (Fig. 7A and 7B). Because pERK is the activated form of ERK, we thought relative strength of pERK to ERK could reflect the degree of activation of this pathway. We performed densitometric analysis of the western blot result for pERK and ERK and calculated the ratio, value of pERK/value of ERK (pERK/ERK). We tried to find any difference in pERK/ERK according to cell line, feeder type, and the presence or absence of the bFGF supplementation but the statistical analysis showed there were no significant differences according to cell lines, feeder type, and the presence or absence of bFGF supplementation (data not shown).
The results immunoblotting (
Induction of early transcription factors: c-Fos, c-Jun
The c-Fos gene is known to be induced by ERK activation of bFGF in hESCs [21]. c-Jun is also known to form early transcription factors in combination with c-Fos [22]. We evaluated the induction of these genes by RT-PCR. In hESCs cultured with exogenous bFGF (H1/MEF/+bFGF, HSF6/MEF/+bFGF, H1/HPC/+bFGF, HSF6/HPC/+bFGF), induction of these early transcription factors was identified (Fig. 7D and 7E). Moreover, in hESCs cocultured with HPC feeders minus exogenous bFGF, induction of these genes was also identified (Fig. 7D and 7E).
Discussion
It is clear that bFGF has a role in maintaining the undifferentiated state of hESCs, and bFGF is considered to be an essential growth factor, especially in hESC cultures using MEF feeder cells. Recently various human-derived feeder cells were introduced to hESC cultures to overcome the problems of animal feeder cells. It is expected that the secreted proteins and cell-to-cell interactions in human-derived feeder cells are relatively different from those in MEF feeder cells, although it has not been definitively demonstrated. Therefore, we performed this study to re-evaluate the role of bFGF in culture systems using human-derived feeder cells.
The most important finding of this study was that in hESC cultures using HPC feeders, the absence of exogenous bFGF did not influence the outcome of long-term culture, thereby providing additional merit. Therefore, medium supplementation with exogenous bFGF is not thought to be necessary when using HPC feeder cells for hESC culture.
We demonstrated that HPC expressed bFGF but MEF did not. Therefore based on the hypothesis that HPCs might secrete sufficient bFGF to sustain the undifferentiated state of hESCs, we performed ELISA of culture supernatants. ELISA results showed that the mean bFGF concentration of supernatants from H1 cells cocultured with HPCs without exogenous bFGF supplementation (H1/HPC/–bFGF group) was not significantly different from that of supernatants from H1 cells cocultured with MEFs with exogenous bFGF supplementation (H1/MEF/+bFGF group), which is the standard method for hESC culture (Fig. 6E). In other word, supernatant bFGF of H1/HPC/–bFGF group was enough to sustain the undifferentiated state of H1 hESCs despite absence of exogenous bFGF supplementation. Additionally, we also identified the endogenous bFGF synthesis within hESCs of HPC feeder group without exogenous bFGF supplementation (Fig. 7B). A recent study demonstrated that bFGF knockdown within hESCs cultured on MEF feeder induce differentiation of hESCs despite the exogenous bFGF supplementation and therefore endogenous bFGF within hESCs has a crucial role in maintaining the pluripotency of hESCs [23]. In our RQ-PCR analysis, bFGF in hESCs cultured on HPC feeder without exogenous bFGF supplementation was expressed similarly in hESCs cultured on MEF feeder with exogenous bFGF supplementation (Fig. 6C and 6D). Hence, endogenous expression of bFGF might also have an important role to proliferate undifferentiated hESCs cultured on HPC feeder. Further additional studies will be warranted to verify this hypothesis, and the study to search the other mechanisms such as cell-to-cell interactions also have to be tried, because little is known about the exact mechanism for this phenomenon.
With regard to the bFGF signaling pathway, activation of the ERK1/2-c-Fos/c-Jun pathway was identified. This is consistent with a previous study [21] that was investigated in hESCs on MEF feeders plus exogenous bFGF. However, our study demonstrated that this pathway is also activated in hESCs cocultured with HPCs without exogenous bFGF. Therefore, taking into consideration the recent data showing that autocrine bFGF signaling is involved in the maintenance of undifferentiated hESCs [9], it can be postulated that HPCs might support hESCs so that sufficient bFGF is expressed to sustain the pluripotency of hESCs over long-term culture, and then this endogenous bFGF might be secreted and act on the ERK1/2-c-Fos/c-Jun pathway via an autocrine/paracrine route. Actually recent studies suggested that an autocrine FGF signal is involved in the maintenance of proliferating hESCs in the undifferentiated state [9,23]. We demonstrated that the degree of ERK activation is significantly different among the study groups. However, we could not determine whether these differences could impact the outcomes of long-term culture. Additional long-term follow-up is required for that.
In order for hESCs to be a source for cell replacement therapy, mass production of undifferentiated hESCs may be required. To date, culture systems using feeder cells best achieves this purpose. There are some advantages in using HPCs for feeder cells. First, because they are human-derived, the concerns about animal feeder cells are minimized. Second, placentas can be attained without additional damage to donors. Third, because the HPC feeder cell culture system does not require continuous bFGF supplementation, the expense and degree of labor can be considerably reduced.
In conclusion, the undifferentiated state of hESCs can be well maintained on HPCs without supplemental bFGF over long-term culture, and it has been shown that continuous supplementation with exogenous bFGF does not provide additional merit for culturing hESCs on HPC feeder cells. To our knowledge, this is the first report evaluating the role of bFGF in coculturing hESCs with human-derived feeder cells.
Footnotes
Acknowledgments
This research was supported by a grant (SC-2240) from the Stem Cell Research Center of the 21st Century Frontier Research Program, funded by the Ministry of Science and Technology, Republic of Korea.
Author Disclosure Statement
All authors indicated no potential conflicts of interests and no competing financial interests exist.