
Abstract
The in vitro generation of neural cells from human embryonic stem cells is a powerful tool to acquire better knowledge of the cellular and molecular events involved in early human neural and brain development under physiological and pathological conditions. Prenatal alcohol exposure can induce important anomalies in the developing brain, the embryogenesis being an important critical period for the craniofacial defects and mental disabilities associated with fetal alcohol syndrome. Here, we report the generation of neural progenitors (NPs) from human embryonic stem cells. Neuroepithelial progenitors display the morphological and functional characteristics of their embryonic counterparts and the proper timing of neurons and glia cells generation. Immunocytochemical and real time (RT)-polymerase chain reaction analyses reveal that cells appeared as clusters during neuroepithelial cell proliferation and that the genes associated with the neuroectodermal (Pax-6) and the endodermic (α-fetoprotein) lineages decreased in parallel to the upregulation of the genes of NPs (nestin and Tuj1), followed by their differentiation into neurons (MAP-2+, GABA+), oligodendrocytes [galactocerebroside (GalC+)], and astrocytes (GFAP+). We further demonstrate, for the first time, that human NPs express the endocannabinoid receptors (CB1 and CB2) and the enzymes involved in endocannabinoids synthesis (NAPE-PLD) and degradation (FAAH). Using this in vitro culture, we demonstrate that ethanol exposure impairs NPs survival, affects the differentiation of NPs into neurons and astrocytes, disrupts the actin cytoskeleton, and affects the expression of different genes associated with neural differentiation. The results provide new insights into the effects of ethanol on human embryogenesis and neuroprogenitors and offer an opportunity to delineate potential therapeutic strategies to restore early ethanol-induced brain damage.
Introduction
I
Experimental studies have demonstrated that central nervous system development originates from the embryonic dorsal ectoderm with the formation of the neural tube. This structure is composed of germinal cells that originate most of the initial neurons and glial cells and culminate with the complex processes involved in the formation of the different brain regions (regionalization and morphogenesis) [2,3]. By using hESCs, the results obtained in the last decade have provided evidence that neuroectodermal cells from hESCs are capable of forming structures known as “neural rosettes,” which mimic the neural tube and can differentiate into various region-specific neuronal and glial subtypes in response to appropriate developmental cues [4 –7]. These cellular models allow the systematic functional evaluation to explore functional neural development conditions [4,8] and to decipher not only how these physiological traits are regulated but also how these processes are altered by pathological conditions, such as developmental disorders or exposure to neuroteratogens.
Ethanol is a well-recognized teratogen, and defects found in children of mothers who consumed alcohol during pregnancy are one important brain developmental disorder. Ethanol disrupts the developing brain; and alcohol consumption during pregnancy can produce a wide range of irreversible cognitive, behavioral, structural, and physical anomalies [9] and is one of the leading preventable causes of birth defects and neurobehavioral disorders [10]. In the most severe cases, these anomalies make up a pattern of malformations known as fetal alcohol syndrome (FAS). Children and animals prenatally exposed to alcohol have a smaller brain, a thinner cerebral cortex, and a significantly reduced total cell number [11,12]. Experimental evidence demonstrates that ethanol interferes with many ontogenic phases of brain development, affecting crucial processes such as neuronal migration, neurogenesis, and gliogenesis [13]. However, an important critical period of ethanol-induced teratogenesis and neurodevelopmental defects occurs during embryogenesis, as heavy ethanol consumption during this period can lead to the craniofacial defects and mental disabilities associated with FAS [14,15]. We have demonstrated that NP cells are important targets of ethanol neuroteratogesis, because chronic ethanol exposure during rat embryogenesis reduces the telencephalic radial glia progenitor pool and affects its transformation into neurons and astrocytes [12]. However, whether these effects also occur in human precursor cells is presently unknown.
In this study, we have used a derivation of neural human NP cells from hESCs and an in vitro protocol for neural induction, proliferation, and differentiation, which mirror early forebrain neural development [16]. Using this in vitro model of human NPs, we herein report, for the first time, that both endocannabinoid receptors (CB1 and CB2) and the enzymes involved in their synthesis (NAPE-PLD) and degradation (FAAH) of endocannabinoids (EC) are expressed in NPs during neural proliferation and differentiation. We further demonstrate that ethanol, at physiological relevant concentrations, impairs neural cell survival, alters the gene expression pattern, and affects the differentiation of NPs into neurons and astrocytes. These results could provide new insights into the effects of ethanol on early human embryogenesis.
Materials and Methods
Cell culture of hES cell-derived NPs
hESCs, the H9 line (WiCell Inc.), were cultured on commercially available human foreskin fibroblast (American Type Culture Collection), inactivated by mitomycin C in ES medium containing knockout-DMEM (Invitrogen), 20% serum replacement (Invitrogen), L-glutamin (Invitrogen), penicillin/streptomycin (Invitrogen), β-mercaptoethanol (Sigma), and basic fibroblast growth factor (bFGF; Invitrogen) at 37°C in 5% CO2 in a humidified atmosphere. The hESC colonies were replated, according to their size, after a 6- to 7-day period, by passaging small pieces by mechanical dissection. To obtain embryoid bodies (EBs), the hESC colonies were transferred to low attachment plates and maintained in the same medium for 6 days. For neural induction, after 6 days in culture, EBs were scattered and transferred to adherent conditions on Matrigel-coated plates and cultured with neural induction medium, which contained DMEM/F12 and glutamax:neurobasal (1:1), B27 supplement (Invitrogen), N2 supplement (Invitrogen), 2 mM glutamax (Invitrogen), and penicillin/streptomycin. The cells from attached floating formations started to radially spread after 5 days. Rosettes were cut and desegregated to obtain single cells. Cells were plated on polyornithine/laminin plates (350,000 cells/2 mL plate) and cultured with neural proliferating medium (NPM) containing bFGF (20 ng/mL), DMEM/F12, glutamax:neurobasal (1:1), B27 supplement, N2 supplement, 2 mM glutamax, and penicillin/streptomycin. Neuroprogenitors proliferated on the plates until they became confluent and were passaged onto polyornithine/laminin plates with a fresh medium (NPM) in accordance with their number. On day 21, the NPM was replaced with a neural differentiating medium (NDM) containing neurobasal medium, B27, 2 mM glutamax, penicillin/streptomycin, and 10 ng/mL brain-derived growth factor (BDNF). Cells were maintained for an additional 15 days in NDM.
For the alcohol treatment, ethanol was added to the proliferating or differentiating media at a final concentration of 25 or 50 mM. To prevent evaporation, culture plates were placed in sealed containers that also contained an open dish with a solution of 25 or 50 mM ethanol at the bottom. This procedure completely avoids evaporation after 48 h. Ethanol-containing medium was freshly prepared before use from 99% Ethanol (Merk, Co.), and the ethanol concentration in the fresh and culture-exposed medium was measured by a spectrophotometric method (Sigma-Aldrich). The culture media with or without ethanol were changed every 2 days. The alcohol concentrations used were in the range of the blood alcohol levels found in alcoholics (30–100 mM) [17].
Cell proliferation assay
For the cell proliferation assay, BrdU incorporation was assessed using a commercial kit (Roche Diagnostics, Cat No. 11810740001) and following the manufacturer's instructions. Quantification of the number of BrdU-incorporated cells was carried out by flow cytometry (Cytomics FC500; Beckman Coulter).
Apoptosis quantification and caspase-3 activity
To analyze apoptosis, cells were labeled with R-phycoerythrin-conjugated annexin-V in combination with the cell-impermeant DNA fluorophore 7-amino-actinomycin D (7-AAD) (both from Molecular Probes). The assay was performed according to the manufacturer's protocol. Cells were analyzed by Flow Cytometry (Cytomics FC500; Beckman Coulter).
Caspase-3 activity was measured by a colorimetric assay kit (Sigma-Aldrich), using acetyl-DEVD-p-nitroanilide (Ac-DEVD-pNA) as a substrate. Activity was calculated according to the manufacturer's instructions and expressed as pmol pNA/min/mL.
RNA isolation, reverse-transcription polymerase chain reaction, and quantification of mRNA levels
RNA was isolated from undifferentiated, differentiating, and differentiated cells at the established times using Tri Reagent (Sigma) according to the manufacturer's instructions. The amount of purified RNA was estimated by measuring absorbance at 260 nm. Its purity was assessed by the 260/280 nm ratio. RNA integrity was examined by agarose gel electrophoresis. The RNA (1 μg) of each sample was reverse transcribed using the Transcriptor First Strand cDNA Synthesis kit (Roche Diagnostics). Diluted cDNA was amplified with a rapid thermal cycler (LightCycler Instrument; Roche Diagnostics) in 20 μL of LightCycler 480 SYBR Green I Master (Roche Diagnostics) and 0.5 μM of each oligonucleotide. The sequences of both the forward and reverse primers used in this study are shown in Table 1. Each sample was quantified for each gene in triplicate in at least 6 independent cultures. In parallel, we analyzed the mRNA levels of the human housekeeping GAPDH as an internal control for normalization. To ensure that treatments did not alter the GAPDH mRNA levels, the Ct values were compared between groups. No significant differences were found in the GAPDH levels. Polymerase chain reaction (PCR) amplicons were confirmed to be specific by size and the melting curve analysis. The real-time monitoring of the PCR, the precise quantification of the products in the exponential phase of the amplification, and the melting curve analysis were all performed with the LightCycler 480 quantification software, as recommended by the manufacturer.
Immunofluorescence
Cells growing in laminin and polyornithine-coated glass coverslips (12 mm) on 4-well plates were used for the immunofluorescence studies. Cells were fixed with paraformaldehyde (4% in PBS) for 20 min, permeabilized with 0.2% Triton X-100 for 5 min, as previously described [12]. After washing with PBS, fixed cells were incubated with 5% bovine serum albumin in PBS for 30 min to block nonspecific antibody binding. Cells were then incubated with the primary antibodies: mouse anti-GFAP (Sigma), rabbit anti-nestin (Chemicon), mouse anti-nestin (Abcam), rabbit anti-MAP-2 (Chemicon), mouse anti-Tuj1 (Abcam), rabbit anti-GABA-A (Chemicon), rabbit anti-glutamate (Sigma-Aldrich.), mouse anti-Pax-6 (Abcam), mouse anti-A2B5 (Sigma-Aldrich), rabbit anti-Olig1 (Chemicon), and rabbit anti-Gal C (Abcam). All the antibody incubations were performed in 0.1% Triton-1% bovine serum albumin in PBS for 1 h at room temperature. Appropriate secondary antibodies (Jackson Immunoresearch) were conjugated with fluorophores and used thereafter. For actin filaments staining, cells were incubated for 10 min with phalloidin-TRICT (Sigma-Aldrich), as previously described [18]. Nuclei were stained by incubating in DNA-binding dye Hoeschst 33342 (Molecular Probes). Micrographs were digitally recorded with a Zeiss microscope (Axioskop 2), and morphological measurements were taken with the Metamorph-Offline software (Universal Imaging). At least 10 fields of at least 3 different cultures were taken.
Statistical analyses
Statistical analyses were assessed by one-way analysis of variance. Following a significant F value, a post hoc analysis (Student–Newman–Keuls) was performed. P < 0.05 was considered statistically significant.
Results
Characterization of hESC-derived NP cultures
Figure 1A shows a scheme of the protocol used to obtain NPs from hESCs (H9). Specifically, hESCs were allowed to form EBs (Fig. 1B) and were then transferred to adherent conditions and scattered on Matrigel-coated plates, thus allowing them to differentiate into neuroectodermal structures known as neural rosettes. These rosettes are neural tube-like structures containing high levels of neuroprogenitors cells [4,19] (Fig. 1B). After the dissociation of the rosettes, cells were plated under adherent conditions and cultured in a serum-free proliferating medium (NPM) containing bFGF. Under these conditions, NPs proliferate and form spherical aggregates or clusters (Fig. 1B). On days 7–8, the aggregates were dissociated, and cells were split and re-plated with fresh media. This process was repeated up to 3 times (1/week). During the 3-week proliferation period, cells initially adopted a rosette-like structure. Then as the cell number increased, they formed aggregates or clusters displaying a radial glia array with long filament projections emerging from the central area to the peripheral area (Figs. 1B and 4D). For terminal differentiation, the clusters were dissociated on day 21, and cells were cultured in NDM, containing BDNF, for an additional 15 days. Under these conditions, cells were allowed to differentiate into mature neurons, astrocytes, and oligodendrocytes.
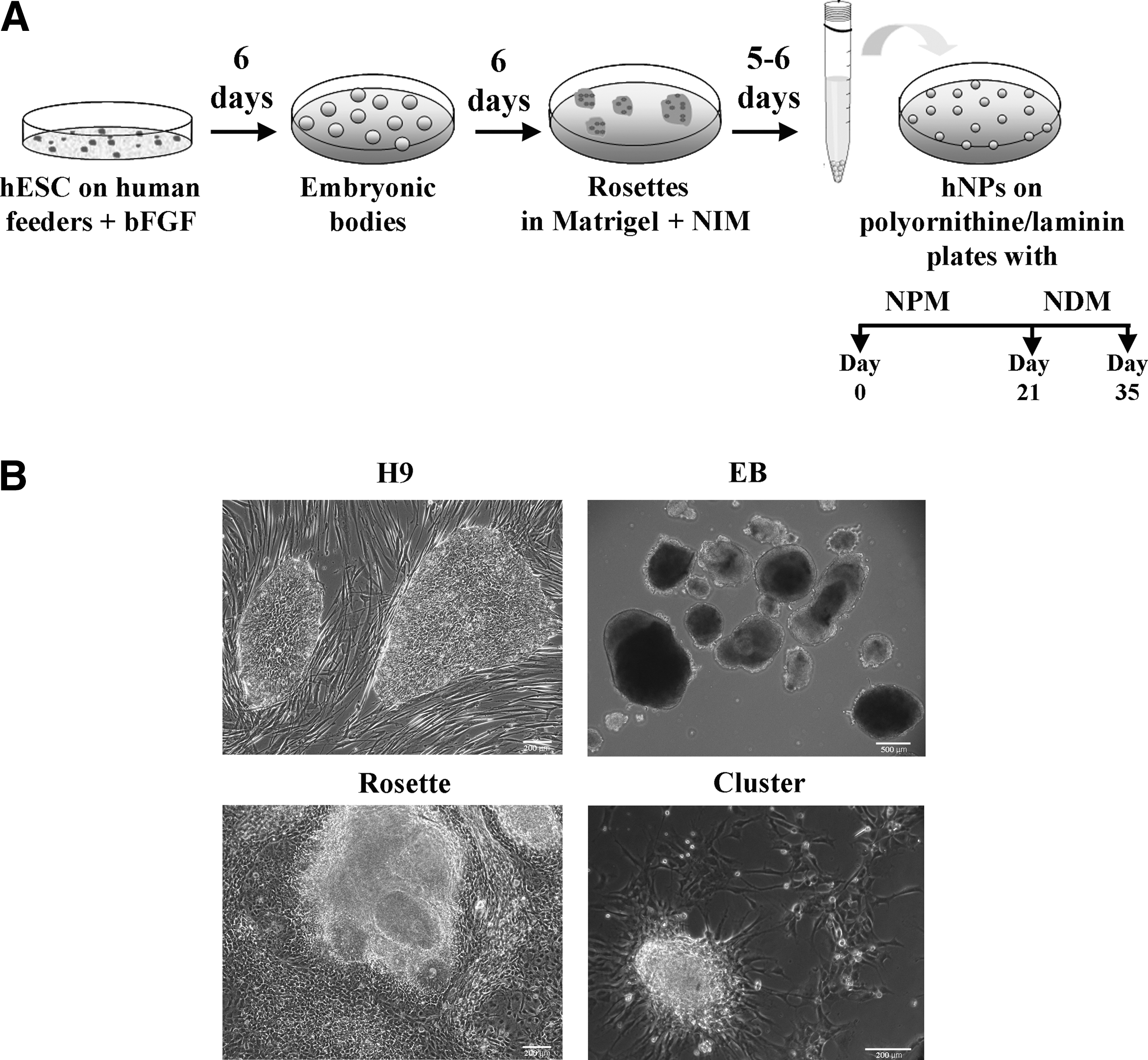
Neural differentiation from human embryonic stem cells (hESCs).
The quantitative real-time-polymerase chain reaction (RT-PCR) analysis indicates that the cultured cells expressed Nanog (a hESC-specific marker), Sox2 (a neural stem cell marker), Pax-6 (a neuroectodermal marker), and α-fetoprotein (AFP, an endodermic marker) in the first week of the NPs culture; but the expression of these genes markedly decreased or disappeared (eg, AFP) during the 3 weeks of NPs proliferation (Fig. 2A). The lack of expression of Brachyury (data not shown) confirmed no mesodermal contamination. Further, the expression of the neural-related genes revealed that, although the neural precursors nestin and Tuj1 were high during cell proliferation and then decreased during cell differentiation (Fig. 2B), the levels of MAP-2 and GFAP were undetectable, or very low, during cell proliferation, but were markedly upregulated in cell differentiation (Fig. 2B). Similarly, the levels of the oligodendrocyte precursor Olig1 increased during the culture time, reaching maximal values on day 28 (Fig. 2B). We also assessed the expression of CD133 (prominin-1), a marker of neural stem/precursor cells [20] that can give rise to neurons and glial cells in vivo [21]. Under our experimental conditions, CD133 levels slowly increased during the proliferation period, being upregulated when undifferentiated NPs become committed to differentiation. Under our experimental conditions, these results indicate that the NPs contained a relatively uniform population of forebrain progenitors that can be differentiated from hESCs and that the temporal pattern of neurogenesis/gliogenesis from the in vitro-produced human NPs was preserved in our culture system.
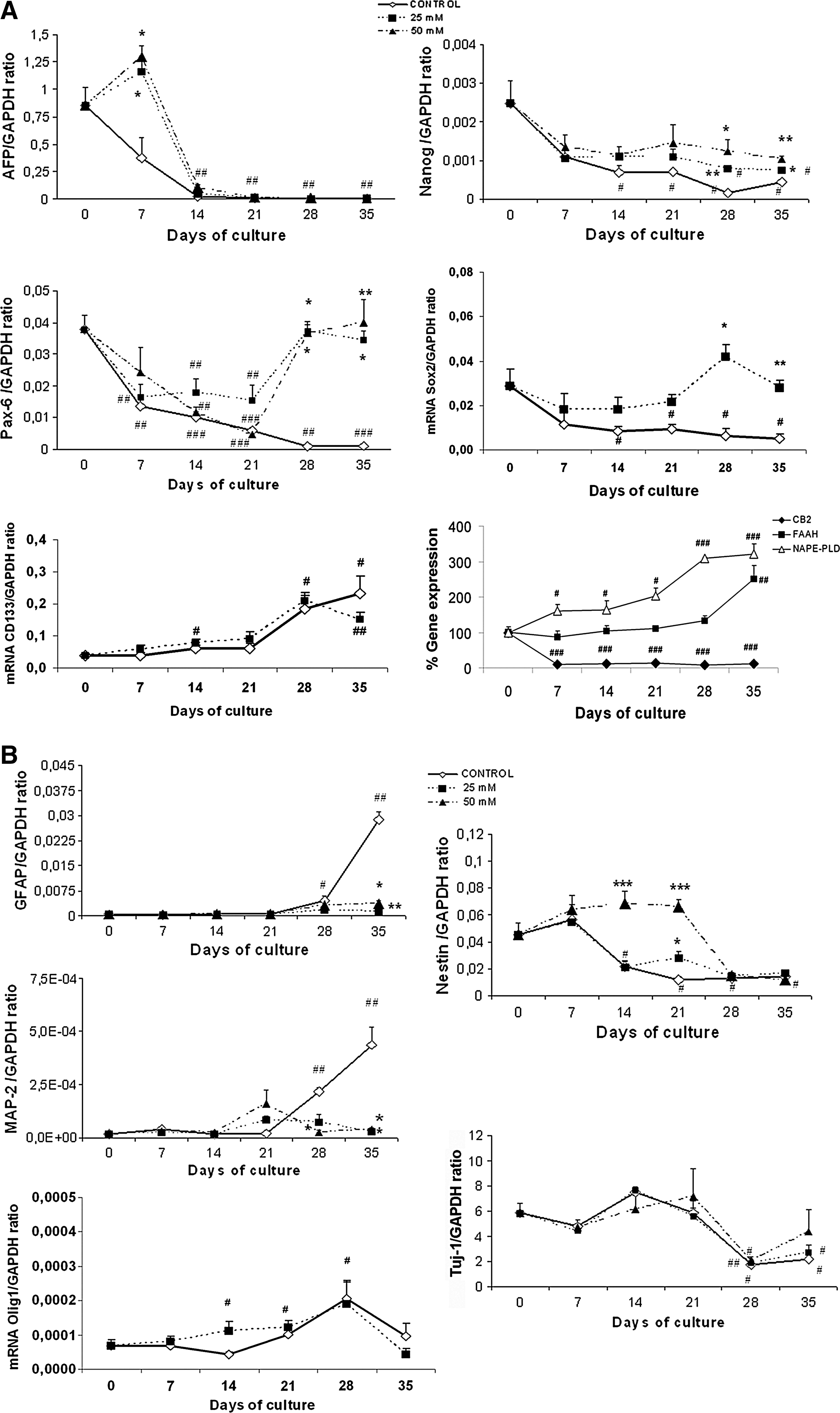
Real-time polymerase chain reaction analysis of the gene expression for different markers determined at the established times in NPs proliferation and differentiation: effects of ethanol (25 and 50 mM).
Since recent studies with rodents have demonstrated the role of the EC system in the regulation of NP proliferation [22 –24], we, therefore, wondered whether the EC system was also expressed in human NPs. For this purpose, the expression of the transcripts of the EC receptors (CB1 and CB2), along with the enzymes involved in the EC synthesis (NAPE-PLD) and degradation (FAAH), was assessed. As Fig. 2A illustrates, CB2 decreased from day 0 to 7 and was then maintained with a very low expression during cell proliferation and differentiation. No significant changes were noted in the CB1 expression (data not shown). Conversely, a progressive increase in NAPE-PLD was observed throughout the culture time, which reached maximal levels during differentiation. The FAAH expression also significantly increased on day 35, when the maximal NAPE-PLD expression was observed. These results suggest that EC are present during the early stage of human neural development and may play a role during neural differentiation into neurons and astrocytes.
Ethanol impairs human neuroprecursors survival
Our previous studies have demonstrated that in utero alcohol exposure impairs the cell proliferation and self-renewal capacity of rat neural stem cells [12]. Therefore, to evaluate whether human NPs are sensitive to alcohol effects, we added ethanol (25 or 50 mM) to the medium. Then, we evaluated the potential actions of ethanol on NPs proliferation and differentiation. One of the initial effects observed in the ethanol-treated cells was the reduced size of the aggregates or clusters formed during cell proliferation (Fig. 3A). Indeed, the number of NPs in the clusters lowered in a dose-dependent manner, although the values were only statistical significant in the 50 mM-treated cells (P < 0.01, Fig. 3B). To investigate whether the reduction in the number of cells occurred by impairment in cell proliferation and/or by increase in cell death, we first incorporated BrdU into the control and the ethanol-treated cells. As expected, the percentage of BrdU incorporation into the control NPs was higher during cell proliferation than during their differentiation (Fig. 3C). The data in Fig. 3C also reveal that ethanol treatment slightly reduced the incorporation of BrdU into NPs, but this reduction was not statistically significant.
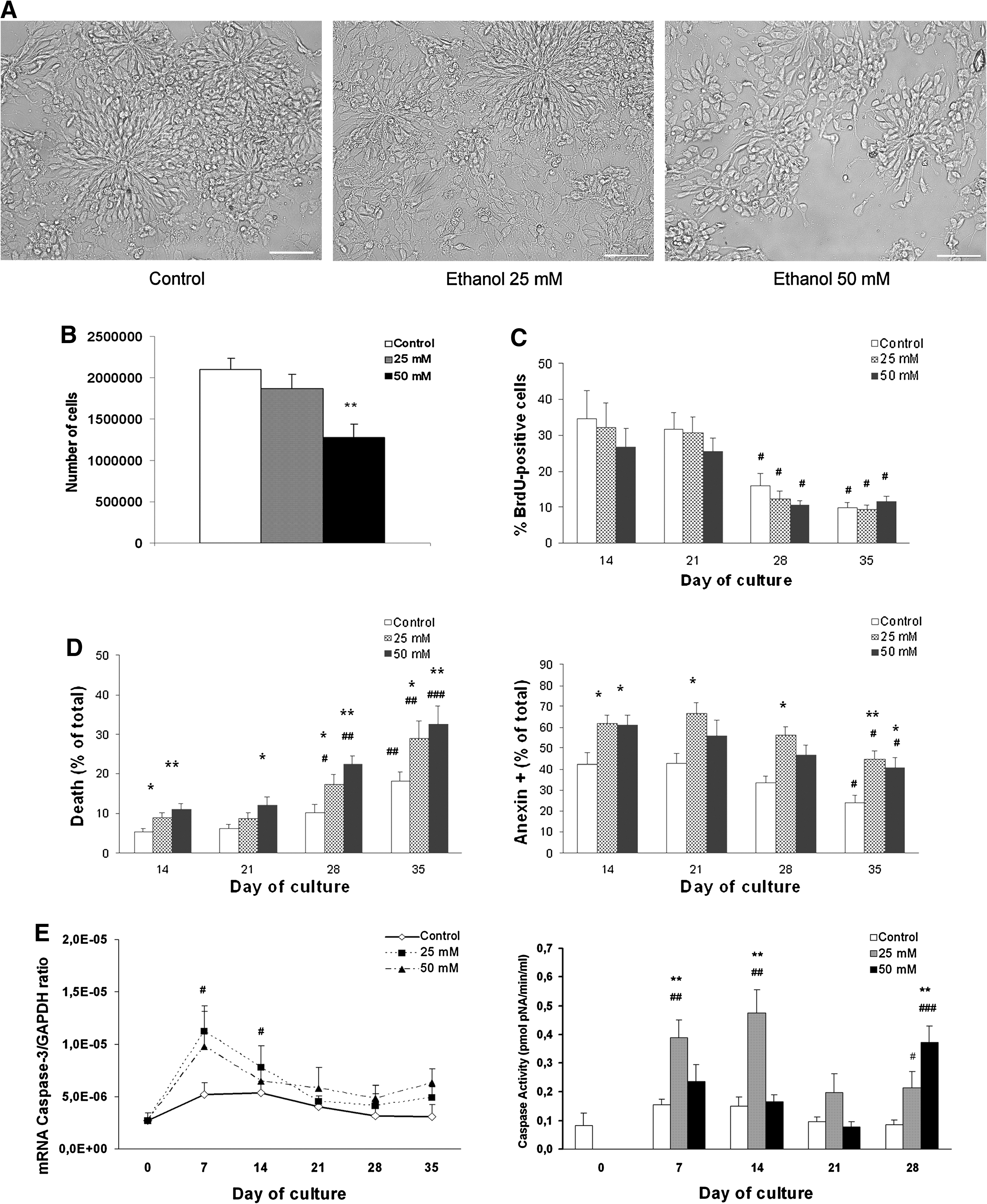
Effects of ethanol on the proliferation and survival of neuroprecursors. NPs were cultured in the presence and absence of ethanol (25 mM or 50 mM).
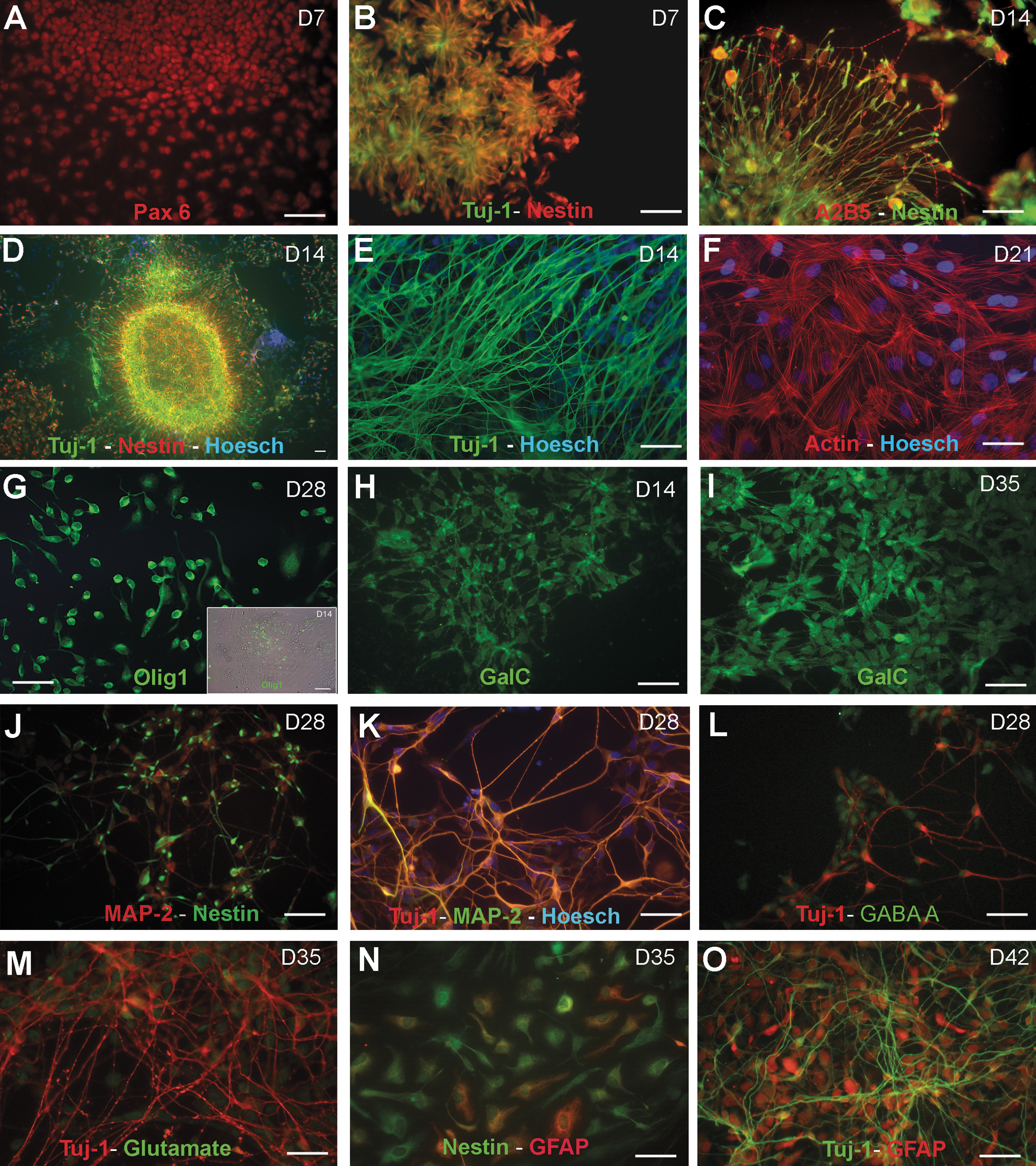
Immunocytochemical characterization of hESC-derived neural precursors. Human NPs cultured with NPM expressed Pax-6
We then analyzed the potential effects of ethanol on cell survival. For this purpose, cells were labeled with annexin V-PE, a marker of the early stage of apoptosis [25]. In addition, the annexin V analysis was simultaneously performed with a dye exclusion stain, such as 7-AAD, to distinguish necrotic cells from apoptotic cells [25,26]. Flow cytometry analysis revealed that ethanol significantly increased the annexin V+ population or the number of apoptotic cells (Fig. 3D). Moreover, the data in Fig. 3 also show that ethanol at 25 mM mainly increases the annexin V+ cells, whereas ethanol at higher concentrations, such as 50 mM, also triggers necrotic cell death, as shown by the increased population of annexin V+ and 7-AAD+. Confirming these data, an increase in the nuclear fragmentation from the alcohol-treated cells was also observed in the immunofluorescence studies (see Fig. 5E, M). According to these data, ethanol at 25 mM significantly upregulated the caspase-3 activity, whereas a higher ethanol concentration (eg, 50 mM) increased caspase-3 only during the cell differentiation stage (Fig. 3E). Likewise, ethanol also upregulated the mRNA levels of caspase-3 (Fig. 3E). These results suggest that ethanol reduced the number of NPs by increasing cell death.
Morphological alterations in ethanol-exposed neural progenitors. Ethanol exposure during neural progenitor proliferation (days 7–21) alters Pax-6 expression
Ethanol affects the expression of human NPs and impairs their differentiation into mature neural cells
To gain additional insight into the potential actions of ethanol on NPs during their in vitro proliferation and differentiation, we determined the expression levels of different gene markers using a quantitative RT-PCR analysis at the established times. The data shown in Fig. 2A illustrate how ethanol at 25 and 50 mM significantly increased the gene expression of AFP on day 7 of the proliferating stage, suggesting that ethanol affects the loss of endodermal markers toward neural precursors. Further, the levels of the transcription factors Nanog, Sox2, and Pax-6 were downregulated during NP differentiation; whereas ethanol treatment abolished the downregulation of Nanog and Sox2 and markedly increased the gene expression of Pax-6 (Fig. 2A). Analysis by RT-PCR also demonstrates that although ethanol treatment significantly increased the expression levels of the neural precursor, nestin, it reduced the upregulation of MAP-2 (a marker of mature neurons) and GFAP (an astrocytes marker) observed during cell differentiation in the control NPs (Fig. 2B). No significant changes in the mRNA-Olig1 levels were observed in NPs treated with ethanol. These results suggest that ethanol impairs the transformation of hESC into NPs and into mature neural cells, causing an upregulation of neural precursors (eg, nestin) and decreasing the expression of the genes of mature neurons and astrocytes. Finally, ethanol exposure did not significantly alter the expression of the endocannabinoid system (data not shown).
Morphological characterization of the human neural precursor culture and its differentiation: effects of ethanol
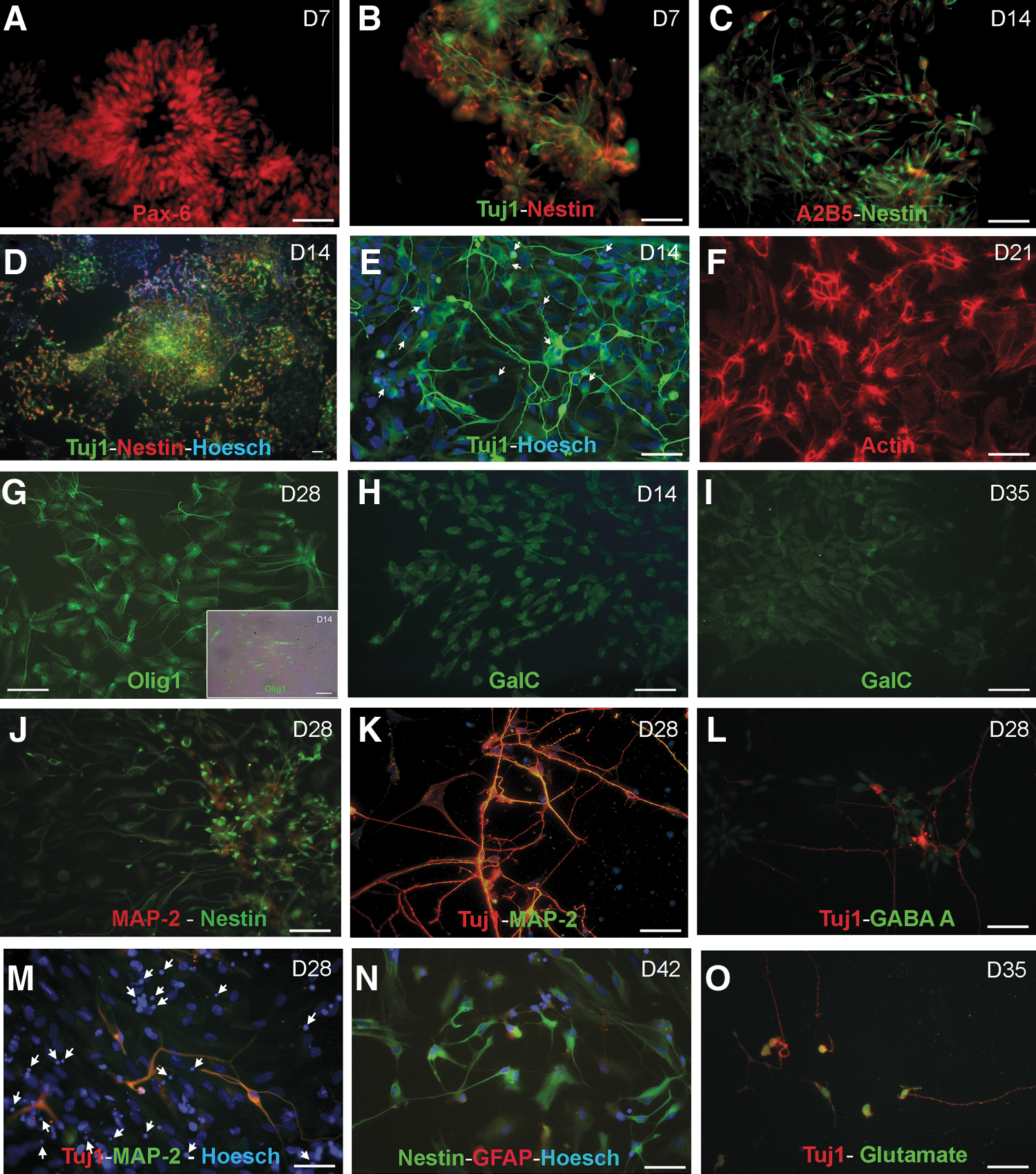
The immunocytochemical analysis of the NP colonies demonstrates that on days 7–14 after plating, the majority of NPs form rosette-like pattern structures (Figs. 3A and 4B) and spherical clusters (Fig. 4D). The spherical structures were mainly composed of a central population of small tightly packed cells forming round structures along with a flat epithelial-like peripheral population (A2B5+) that migrated outward from the cluster (Figs. 3A and 4C). The double immunostaining performed for nestin and Tuj1 reveals that 76% ± 15% of the epithelial-like peripheral and central cells were positive for nestin on day 7, whereas 34% ± 12% of the cells with a unipolar and bipolar morphology at the cell periphery expressed Tuj1 (Fig. 4D). At this stage, most cells expressed Pax-6 (Fig. 4A). On day 14, the nestin+ cells showing a radial glia (RG) morphology decreased (43% ± 10%), whereas the Tuj1+ cells displaying dendritic arborizations markedly increased (58% ± 13%) (Fig. 4E). On days 14 and 21, 41% ± 17%, respectively, of the cells expressed nestin; whereas 60% ± 14% of them are Tuj1+. We were unable to detect the glial fibrillary acidic protein (GFAP) and the MAP-2-positive cells during cell proliferation. However, we were able to detect the oligodendrocyte precursor marker, Olig1, which was predominantly expressed in the nuclei of NPs during cell proliferation (Fig. 4G). All of these findings agree with the results obtained by RT-PCR (Fig. 2).
The immunocytochemical analysis also reveals that the cells which were initially organized as aggregates displayed more complex dendritic arborizations and were stained not only with nestin and MAP-2 but also with Tuj1 and MAP-2 during cell differentiation (day 28) (Fig. 4J, K). Some Tuj1-positive neurons were also immunoreactive for GABA-A+ (Fig. 4L) and glutamate (Fig. 4M). In addition, the specific cell-surface marker for oligodendrocytes, galactocerebroside (GalC), was also detected; and its immunoreactivity increased throughout the culture time, thus confirming the oligodendroglial identity of our NPs culture (Fig. 4H, I). During prolonged maintenance, specifically on day 35, about 15% ± 9% of the cells were immunoreactive for the astroglial marker GFAP (Fig. 4N). On day 42, the percentage of astrocytes (GFAP+) increased (20% ± 12%). and some of them appeared at the bottom of Tuj1-positive neurons (Fig. 4O).
Ethanol exposure markedly affects the morphology profile of NPs cells. Thus, Fig. 5 shows how ethanol alters the morphology of the rosette-like structures (Fig. 5A–C) and reduces the number of the nestin- and Tuj1-positive cells forming the spherical aggregates during the proliferation stage (days 7–21) (Fig. 5D). Despite the number of cells being significantly reduced by ethanol, the percentage of cells expressing nestin and Tuj1 at the different proliferation times was similar to that of the controls. Further, a striking effect of ethanol noted was the alteration in cell morphology and cytoskeletal changes. Those cells immunoreactive for nestin (type VI, intermediate filament) and Tuj1 (class III β-tubulin isotype) displayed shorter processes and abnormal filaments (Fig. 5D, E, K, L, M, O). In addition, the actin filaments were organized into stress fibers as bundles running throughout the cytoplasm in the control cells (Fig. 4F), whereas ethanol exposure markedly disrupted actin organization by changing the stress fibers into actin rings at the cell periphery (Fig. 5F). These effects were more marked in the NPs exposed to 50 mM than in those exposed to 25 mM. Further, ethanol brought about a reduction in the percentage of cells expressing MAP-2 during cell differentiation. Thus, 21% ± 10% of the control cells expressed MAP-2, whereas only 15% ± 7% of the alcohol-exposed (50 mM) cells were MAP-2+ on culture day 28 (Fig. 5K). Similarly, the immunoreactive GABA-A and glutamate neurons also slightly reduced in the alcohol-treated NPs (Fig. 5L, O). Likewise, 20% ± 12% of the control cells were immunoreactive for GFAP on day 42, whereas only 5% of the ethanol-treated cells (25 and 50 mM) were GFAP+ (Fig. 5N). Finally, ethanol also impaired oligodendrocytes precursors by reducing the number of both the Olig1+ cells (Fig. 5G) and the GalC-positive processes (Fig 5H, I) and by affecting Olig1 + cell distribution.
Taken together, our findings demonstrate that ethanol both affects the proliferation of NP cells and markedly reduces their differentiation into mature neurons, astrocytes, and oligodendrocytes. The results also indicate the disruption of several cytoskeletal proteins by ethanol and suggest that the changes noted in the cytoskeletal proteins might underlie alterations in the cell shape and functions of the ethanol-exposed NPs [27].
Discussion
In this study, we used an in vitro culture that efficiently directs the differentiation of hESC toward NPs and the generation of radial glia, neurons, and astrocytes in a temporal pattern, which is capable of reproducing the early stages of human brain development. This culture protocol maintains progenitors with forebrain specifications [16] in absence of extrinsic factor and in the presence of FGF2 for NPs proliferation [28] and BDNF for their differentiation [29]. Using this in vitro culture model, we have investigated the actions of ethanol on NP proliferation and/or differentiation, as early human embryogenesis is considered a critical period of ethanol-induced teratogenesis and for the craniofacial and neurodevelopmental defects associated with FAS [9,13].
Although our culture system has some limitations, it mostly reproduces the early stages of human brain development. Indeed, neurulation is the first event in organogenesis, and it begins from ESCs during the early events in embryonic development [3,30,31]. Previous studies have demonstrated that hESC can form in vitro neural tube-like rosettes after 2 weeks of differentiation [19], which temporally correlates with the formation of the neural groove by 18 days in vivo and its subsequent rostrocaudal delineation by day 20 [32]. Therefore, this stage is approximately equivalent to the third week of gestation if we consider that hESC are derived from 1-week-old embryos [5]. Regarding neurogenesis, recent evidence in developing human embryos has shown that the earliest born neurons are observed within the forebrain by days 31–35 [33] and that these early neurons precede local cortical neurogenesis. The present in vitro culture of the NPs derived from hESC is in concurrence with the above findings. We found that rosettes are formed on days 15–17. Then, in the presence of FGF2, epithelial cells, radial glia, and immature neurons (Tuj1+) were observed during the maximal proliferation period of NPs (days 18–39 of hESC or days 1–21 of NPs proliferation), followed by the appearance of early neurons (Tuj1+ and MAP-2+) (days 39–48 or days 21–30 NPs differentiation) and postmitotic neurons (days 50–53, day 35 NP proliferation), which mirrors the early phase of neural development in a human embryo. Further, gliogenesis and the GFAP protein expression were observed after neurogenesis on days 30–40 of NPs differentiation (days 48–58 of hESC). In humans, this protein starts expressing in neocortex after 6–7 weeks of human gestation [34]. Interestingly, NPs can also differentiate into oligodendrocytes, as demonstrated by not only the expression of Olig1 during the proliferation and differentiation but also the presence of GalC expressing cells during the differentiation stage, thus confirming the oligodendroglial identity of our NPs culture. The immunocytochemical and RT-PCR data also reveal a temporal decrease in the expression of the markers for the endoderm (AFP), ectoderm (Pax-6), and the neural stem cell marker Sox2 in parallel to the upregulation of NP markers, such as Tuj1 and nestin, in the NPs proliferation stage. Then, NPs are converted into neurons and astrocytes during their differentiation, as revealed by the upregulation of the protein and gene expression of MAP-2, GABA, and glutamatergic neurons as well as GFAP. These results indicate that, under our conditions, hESC-derived NPs can reproduce the processes that occur during the early development of the nervous system and can be used as a tool to study both the cellular and molecular mechanisms during early forebrain development. Indeed, by using this in vitro culture, we report for the first time that EC receptors (CB1 and CB2) are expressed in NPs, although their expression is low during cell proliferation and differentiation. However, our results support the potential role of ECs in the early stages of human neural development, as the gene expression of both the EC synthesis enzyme, NAPE-PLD, and degradation, FAAH, markedly increases during NPs differentiation. Although further studies are needed to ascertain their role in NPs, recent studies have demonstrated their participation in the proliferation and differentiation of mice neural stem cells [35]. Indeed, the genetic ablation or the pharmacological blockade of CB1 receptors induces deficits in NPs proliferation and axon fasciculation [36], and the selective pharmacological activation of the CB2 receptor in vitro promotes NP cell proliferation and neurosphere generation, an action that is impaired in CB2-deficient cells [37].
Using the in vitro culture of hESC-derived NPs, we also assessed the sensitivity of the early human embryogenesis to the neuroteratogenic effects of ethanol. Both binge drinking and chronic alcohol abuse in the early human embryogenesis stage and before pregnancy recognition (corresponding to the first 4–6 weeks of human gestation) are associated with a greater incidence of craniofacial defects and mental disabilities [38,39]. Likewise, mice exposed to ethanol on embryonic day 7 (E7) or E8 [40], and macaques exposed to ethanol on E19 or E20 [41], also exhibited FAS-associated facial dysmorphia and brain anomalies. In this study, we have attempted to reproduce the first weeks of human gestation to assess the effects of ethanol on embryogenesis. For this purpose, we used NPs in culture exposed to ethanol for 5–6 weeks at concentrations that can be reached in the blood of alcoholics [17]. We demonstrate that ethanol induces cell death and reduces the number of proliferating human NPs. Thus, ethanol at 25 mM mainly induces apoptosis, as revealed by increases in annexin+ cells and caspase-3 activity; whereas we observed that higher ethanol concentrations, that is, 50 mM, promote cell death by necrosis. In agreement with our results, a recent study has demonstrated that ethanol alters the cell cycle and induces apoptosis during early mice neurulation [42]. Conversely, other studies report that NPs from murine [43] or fetal human brain (obtained from fetal tissues corresponding to gestation weeks 14–17) [44] are resistant to ethanol-associated apoptosis. However, in both studies, the authors used neuroprogenitors isolated from the second trimester-equivalent period of gestation. Differences in the sensitivity to ethanol of either neuroprogenitors isolated from distinct developmental stage or brain regions might explain these discrepancies. In fact, we have previously demonstrated that alcohol exposure during late rat embryogenesis in vivo (E12) does not induce apoptosis in NPs but reduces the proliferation of the radial glial progenitor pool, impairs their self-renewal capacity of thelencephalic multipotent progenitor cells, and affects the generation of neurons and astrocytes [12]. In agreement with these results, our immunocytochemical and RT-PCR data reveal that in vitro ethanol exposure impairs the transformation of NPs into neurons (eg, MAP-2) and astrocytes (eg, GFAP).
The present findings also show that the derivation of NPs from hESCs toward neural cells is affected by ethanol. Indeed, ethanol treatment alters the disappearance of AFP (an endodermal marker) during NPs proliferation and also upregulates the transcription factors Nanog, Sox2, and Pax-6 during their differentiation. Nanog and Sox2 are expressed in ESCs and are key factors in maintaining pluripotency [45 –48]. An ethanol-induced upregulation of their expression during NPs differentiation might result in a maintained undifferentiating stage, thus avoiding their differentiation into mature neural cells. Further, alterations in Pax-6 have been shown to result in defects in neural stem and progenitor cells, causing forebrain anomalies in both humans and experimental animals [49]. The Pax-6 mutant mice show defects in neural stem and progenitor cell proliferation and result in microcephaly and abnormal development in the subventricular zone [50,51]. Moreover, the overexpression of Pax-6 also leads to the depletion of the stem cell pool by driving stem cells to a basal progenitor fate, which leads to an overproduction of early-born, deep-layer cortical neurons, causing progenitors apoptosis [52,53] and resulting in forebrain anomalies [54]. By considering the similarities of the later alteration and those induced by ethanol in NPs in culture, it is feasible that an ethanol-induced upregulation of Pax-6 could underlie some of the defects in neuronal differentiation along with the upregulation in immature neurons (Tuj1) and ethanol-induced NPs apoptosis. Changes in Pax-6 may also play a role in the forebrain anomalies observed in animals and children prenatally exposed to ethanol [9,12].
Notch signaling also plays a critical role in maintaining the progenitor pool, controlling cell renewal, and regulating cell differentiation and cell fate determination. Our preliminary results indicate that the expression of Notch decreases in those NPs exposed to 25 mM ethanol at the initiation of astrogliogenesis (day 28, data not shown). According to these findings, our previous results have shown that in utero ethanol exposure decreases not only the levels of activated Notch1 and FGF receptor 2 but also astrogliogenesis [12]. Since one prominent role of Notch is to promote astrocytic fate [55], an ethanol-induced decrease in Notch activation at the onset of gliogenesis might account for the reduction in the GFAP expression noted in NPs differentiation.
Finally, our findings demonstrate that ethanol affects cell morphology and impairs several cytoskeletal proteins, including nestin, Tuj1, and actin. Interestingly, the actin reorganization (eg, loss of stress fibers and actin ring formation) observed in NPs was similar to that described in ethanol-exposed astrocytes [18,56] and that these effects were associated with loss of focal adhesions, activation of the RhoA/ROCK-I/MLC pathway, and apoptosis by anoikis in astrocytes [18]. Therefore, ethanol-induced actin cytoskeletal reorganization may not only underlie disruptions in cell morphology, synaptic formation, and plasticity [27] but also participate in ethanol-induced NPs apoptosis.
In conclusion, using NPs derived from hESC in culture, we demonstrate that ethanol exposure during early embryogenesis impairs NPs survival, affects the differentiation of NPs into neurons and astrocytes, disrupts several cytoskeleton components, and affects the expression of the different genes associated with neural differentiation. Our results provide new insights into the effects of ethanol on early human embryogenesis and offer an opportunity to delineate potential therapeutic strategies to restore the early brain damage induced by ethanol.
Footnotes
Acknowledgments
This work has been supported by the PNSD, Spanish Ministry of Health, the RTA Network (G03/005), the Carlos III Institute, the Direc. Gen. of Drugdependence (GV), and Regerenative-Medicine grant (Generakitat Valenciana and Carlos III Institute).
Author Disclosure Statement
No competing financial interests exist.