
Abstract
The development of culture processes for hematopoietic progenitors could lead to the development of a complementary source of platelets for therapeutic purposes. However, functional characterization of culture-derived platelets remains limited, which raises some uncertainties about the quality of platelets produced in vitro. The aim of this study was to define the proportion of functional platelets produced in cord blood CD34+ cell cultures. Toward this, the morphological and functional properties of culture-derived platelet-like particles (PLPs) were critically compared to that of blood platelets. Flow cytometry combined with transmission electron microscopy analyses revealed that PLPs formed a more heterogeneous population of platelets at a different stage of maturation than blood platelets. The majority of PLPs harbored the fibrinogen receptor αIIbβ3, but a significant proportion failed to maintain glycoprotein (GP)Ibα surface expression, a component of the vWF receptor essential for platelet functions. Importantly, GPIbα extracellular expression correlated closely with platelet function, as the GPIIb+ GPIbα+ PLP subfraction responded normally to agonist stimulation as evidenced by α-granule release, adhesion, spreading, and aggregation. In contrast, the GPIIb+ GPIbα− subfraction was unresponsive in most functional assays and appeared to be metabolically inactive. The present study confirms that functional platelets can be generated in cord blood CD34+ cell cultures, though these are highly susceptible to ectodomain shedding of receptors associated with loss of function. Optimization of culture conditions to prevent these deleterious effects and to homogenize PLPs is necessary to improve the quality and yields of culture-derived platelets before they can be recognized as a suitable complementary source for therapeutic purposes.
Introduction
M
In practice, current culture techniques yield limited level of hematopoietic stem cells and Mk expansion, whereas other systems are incompatible with large-scale platelet production. Matsunaga et al. [6] published a 3-step culture system of cord blood (CB) CD34+ cells using a combination of modified stromal cell line and cytokine cocktails. This strategy provided massive cell expansion and generated sufficient PLPs to enable their function to be tested. However, the major drawbacks were the inability to scale-up their culture protocol, the long duration of the protocol (33 days), and the lack of specificity in the expansion phase that resulted in very low Mk purity. Recently, Sullenbarger et al. [8] reported a promising 3-dimensional hydrogel culture bioreactor system that produced between 10 and 20 αIIb+ [glycoprotein IIb (GPIIb)] PLPs per starting CD34+ cells. However, the majority of the PLPs produced appeared already activated.
Most studies published on culture-derived PLPs estimate platelet production mainly on size and on αIIb integrin expression. GPIIb is indeed a specific marker for Mks and platelets and is also essential for platelet function since it forms with β3, the main fibrinogen receptor. However, expression of GPIIb as sole platelet marker is questionable given that platelets express many other receptors that play major roles in the hemostatic process. For instance, the GPIbα of the GPIb-V-IX complex is the main receptor facilitating platelet recruitment and adhesion to thrombi under arterial flow conditions [11]. In addition, platelet responsiveness is controlled in part by members of the ADAM family of metalloproteinases, which mediates the proteolysis of GPIb, GPV, and GPVI at their juxtamembrane region, leading to ectodomain shedding of these receptors [12 –15]. High rate of receptor shedding was also observed in stored and culture-derived platelets [3,14,15], suggesting that the integrity of the metalloproteinase-sensitive receptors should be carefully monitored. Taken together, these observations suggest that a more thorough analysis needs to be done to define the functional properties of the PLPs produced ex vivo.
We recently optimized a simple 14-day 2-step culture protocol for the expansion and differentiation of CB CD34+ cells toward Mk lineage in liquid culture [7]. In this study, we scaled-up and adapted this protocol to allow platelet production in a resource-efficient process, and then developed a reliable purification method to comprehensively study the morphology and function of the PLPs. We present a critical analysis of 2 major distinct GPIIb+ PLPs sub-populations, the first one that co-expressed GPIbα and the second one that didn't.
Materials and Methods
Production of PLPs from human CB CD34+ cells
Human CB cell collection, cryopreservation, and CD34+ cell enrichment were previously described [7,16]. CD34-enriched cells were maintained in a serum-free medium as described previously [7], supplemented with human cytokines all purchased at Feldan Bio. The Mks culture protocol is summarized in Fig. 2A. In brief, 4 × 104 CB CD34+ cells/mL were first grown in the presence of the cytokine cocktail-optimized Mk production (OMP) (Table 1). On day 4, an equal volume of the fresh medium supplemented with the cocktail BS1 (Table 1) [17] was added to the culture. Cell density was adjusted to 3–4 × 105 cells/mL on day 7 and on day 10 with the BS1 medium, and 25 μM GM6001 or its negative control analog (Calbiochem EMD) was added in selected cultures at day 12 [3]. GM6001 was added at day 12 following preliminary experiments showing no beneficial effect on GPIbα expression when added at earlier time (data not shown). Cultures were maintained in a humidified atmosphere (10% CO2, 20% O2) at 39°C and 37°C for the first and last 7 days of culture, respectively, to increase Mk and PLP yields [9,18]. Viable nucleated cells were manually counted as described [7]. Analyses of the cultures and PLPs were performed at day 14.
OMPC, optimized Mk progenitor expansion cocktail; OMP, optimized Mk production; TPO, thrombopoietin; SCF, stem cell factor.
Purification of blood platelet and culture-derived PLPs
Control platelet-rich plasma (PRP) was prepared from the blood of healthy volunteers as described [19]. Purification by centrifugation was carried out by performing a slow spin (80g, 10 min) to remove Mk cells. PLP-rich supernatants and PRPs were washed in CGS buffer (10 mM sodium citrate, 30 mM D-Glucose, and 120 mM NaCl, adjusted to pH 6.5 with citric acid) in presence of 1 μM PGE1 (Sigma) and then resuspended in Tyrode-Hepes buffer (140 mM NaCl, 3 mM KCl, 1 mM MgCl2, 16.62 mM NaHCO3, 5.5 mM glucose, and 10 mM Hepes, pH 7.4). For gel filtration, a 10-mL column was loaded with 2B-CL sepharose (Amersham Biosciences, GE Healthcare). PLP-rich supernatants were spun (1,000g, 10 min) in the presence of 1 μM PGE1, suspended in 2 mL of the BS1 medium, and run through the column. PLPs were eluted with Tyrode-Hepes buffer supplemented with 0.5% bovine serum albumin (BSA; Fisher Scientific) and collected at a selected time of elution based on previous tests done with PRPs. For BSA (bovine serum albumin, fraction V (Invitrogen)) gradient, Mks cultures and blood platelets in CGS buffer were spun (80g, 15 min) over a BSA gradient (2% to 12% prepared in phosphate-buffered saline [PBS]). PLPs and platelets were collected in the 0% to 5% BSA layers and mixed with an equal volume of 1% BSA in PBS. Purified platelets were spun (1,000g, 10 min) in the presence of 1 μM PGE1 and then suspended in the BS1 medium supplemented with 1% heat inactivated human serum (BS1-serum medium) or Tyrode-Hepes buffer as indicated. Purified platelets and PLPs were maintained in a humidified atmosphere at 37°C until functional analyses were performed.
Flow cytometry analyses
Flow cytometry data were collected with a FACS-Calibur (Becton Dickinson) and were analyzed with FCS Express version 3. Cells and PLPs were stained with an APC-conjugated anti-CD41a (APC-CD41a and GPIIb) and FITC-conjugated anti-CD42b (FITC-CD42b and GPIbα) MoAbs (BD Pharmingen), PE-conjugated anti-CD62p (p-selectin) MoAb (Immunotech; Beckman Coulter Co.), and propidium iodide (PI, 5 μg/mL) and were analyzed as previously described [7]. GPV expression was monitored on unpurified PI-stained PLPs using an unconjugated CD42d MoAb (Sanguin plesmanlaan) counterstained with an Alexa Fluor 488 goat anti-mouse IgG (Invitrogen). For α-granule release stimulation, blood platelets and PLPs were incubated with or without 1 U/mL thrombin (Sigma) or 50 μM ADP (ChronoLog Corporation) for 10 min at 37°C before staining. For viability assays, blood platelets and PLPs were incubated with calcein AM (Molecular Probes, Invitrogen) for 30 min at 37°C, washed in 1% BSA in PBS supplemented with 1 μm PGE1, and resuspended the in BS1-serum medium before immunostaining with PerCP-CD41a or APC-CD42b MoAbs. PI-based ploidy analysis was done on GPIbα+ cells as previously described [7].
Electron microscopy
Purified blood platelets and PLPs in Tyrode-Hepes buffer were stabilized for 15 min by adding an equal volume of 0.5% paraformaldehyde. Platelets were then fixed with 4% paraformaldehyde and 2.5% glutaraldehyde in sodium cacodylate buffer 0.1M, pH 7.3, for 24 h at 4°C, postfixed with 1% osmium tetroxide, dehydrated, and embedded in epon. About 60–80 nM sections were stained with 3% uranyl acetate and 0.1% lead citrate, and examined with a JEM-1230 transmission electron microscope operating at 80 kV (JEOL). Platelet diameter was measured using software ImageJ 1.38 × .
Immunostaining of adherent platelets for fluorescence microscopy
Platelets suspended in the BS1-serum medium were plated for 1 h on 100 μg/mL fibrinogen-coated coverslip in the absence or presence of 1 U/mL thrombin, then rinsed twice with PBS, and fixed with 3.7% formaldehyde for 20 min at 37°C. Platelets were then rinsed twice, treated for 5 min with 0.2% Triton X-100, rinsed twice, and blocked at 37°C for 30 min in 5% BSA in PBS (PBS-BSA). Mouse anti-CD42b MoAb was used 1:50 in PBS-BSA for 1 h at 37°C. Platelets were washed with 0.05% Tween-PBS and incubated with Alexa 555–conjugated (A555) goat anti-mouse IgG (Molecular Probes) 1:1,000 and A488 phalloidin (Lonza) 1:50 in PBS-BSA to stain GPIbα and actin filament (F-actin), respectively. Platelets were extensively washed, mounted in Prolong Gold (Molecular Probes), and observed with a TE-2000-s inverted microscope (Nikon) by using a 100 × oil, 1.25 NA objective or with a FV100 point scanning confocal microscope (Olympus) driven by fluoview software version 2.0.3 using a 63 × 1.4 NA oil objective.
Light transmittance aggregometry
The aggregometer (Chrono-Log Corporation 540 VS) driven by AGGRO/LINK® software version 5.2.1 was filled with 250 μL of PRP or with 250 μL of a mixture of 450 × 106 purified PLPs/mL suspended in filtered poor-platelet plasma. PRP and PLPs were incubated at 37°C under constant stirring for 5 min, and then ristocetin (1.25 mg/mL) was added. Light transmission was recorded continuously from the beginning of the experiment until 15 min after the addition of the agonist.
Fibrinogen coated micro well aggregation assay
For aggregate formation in the microwell, PLPs or blood platelets in the BS1-serum medium were plated into fibrinogen-coated 96 wells (100 μg/mL; Calbiochem) for 5 min, gently washed with PBS, and incubated in the absence or presence of fibrinogen (300 μg/mL) and ADP (20 μM) or with thrombin (1 U/mL) under vigorous shacking for 15 min. In some experiments, PLPs were loaded with 2 μg/mL calcein AM and mixed with nonstained blood platelets before the assay. Platelet aggregates were fixed and stained with A594 phalloidin (Molecular Probes) as described above and observed directly in the plate by using the LWD 40 × , 0.55 NA objective of the TE-2000-s microscope. All epifluorescent microscopy images were captured as 12-bit TIFF files with a cooled monochrome CCD camera (Retiga-1300; Qimaging) driven by Simple PCI software (Compix Inc, Imaging Systems) version 5. Photoshop CS2 software version 9.0.2 was used for brightness/contrast adjustment and image cropping.
Statistical analysis
Comparisons were analyzed using the Student's t-test, Mann–Whitney test, or analysis of variance followed by Tukey's multiple comparison test. Results were regarded as significant when P values were below 0.05.
Results
Production and purification of PLPs
We previously reported that production of Mks and PLPs from CB CD34+ cells can be achieved with a 2-phase culture protocol [7]. This included an expansion phase of 5 days with cytokine cocktails such as S3F6 (Table 1) followed by a differentiation phase of 9 days with the cocktail BS1 [7]. The first objective was to scale-up this protocol in a resource-efficient manner. Hence, to reduce cytokine use, we tested 2 cytokine cocktails that use 85% less cytokine than S3F6 for the first phase of culture (day 0–6). The optimized Mk progenitor expansion cocktail (OMPC, Table 1) was recently optimized by statistical design of experiments for the expansion of CB Mk progenitors. Although OMPC improves Mk production (manuscript in preparation), the Mk production model suggested that lower concentration of thrombopoietin (TPO) and stem cell factor (SCF) would provide similar level of Mk production. Hence, we developed the cocktail OMP that uses the lower concentration of TPO and SCF (Table 1). Mks produced in the OMP or OMPC cultures showed similar morphology (Wright-Giemsa staining, data not shown) and reached similar level of ploidy distribution (Fig. 1A). However, OMP generally increased the production of total (GPIIb+) and mature (GPIIb+ GPIbα+) Mks and PLPs at day 14 (Fig. 1B), though the gains remained below statistical significance.
Next, we investigated the temporal production of Mks and PLPs in the 2-phase OMP-based culture. Proplatelets formation generally started at day 9 (Fig. 1E). Total GPIIb+ Mk and mature GPIbα+ Mk productions reached a plateau around day 14, as no significant gains were obtained for either cell populations at day 16 (Fig. 1D). Finally, production of GPIbα+ PLP peaked at day 14 of culture (Fig. 1E), with or without pretreatment of the cultures with the metalloproteinase inhibitor GM6001 (see below).
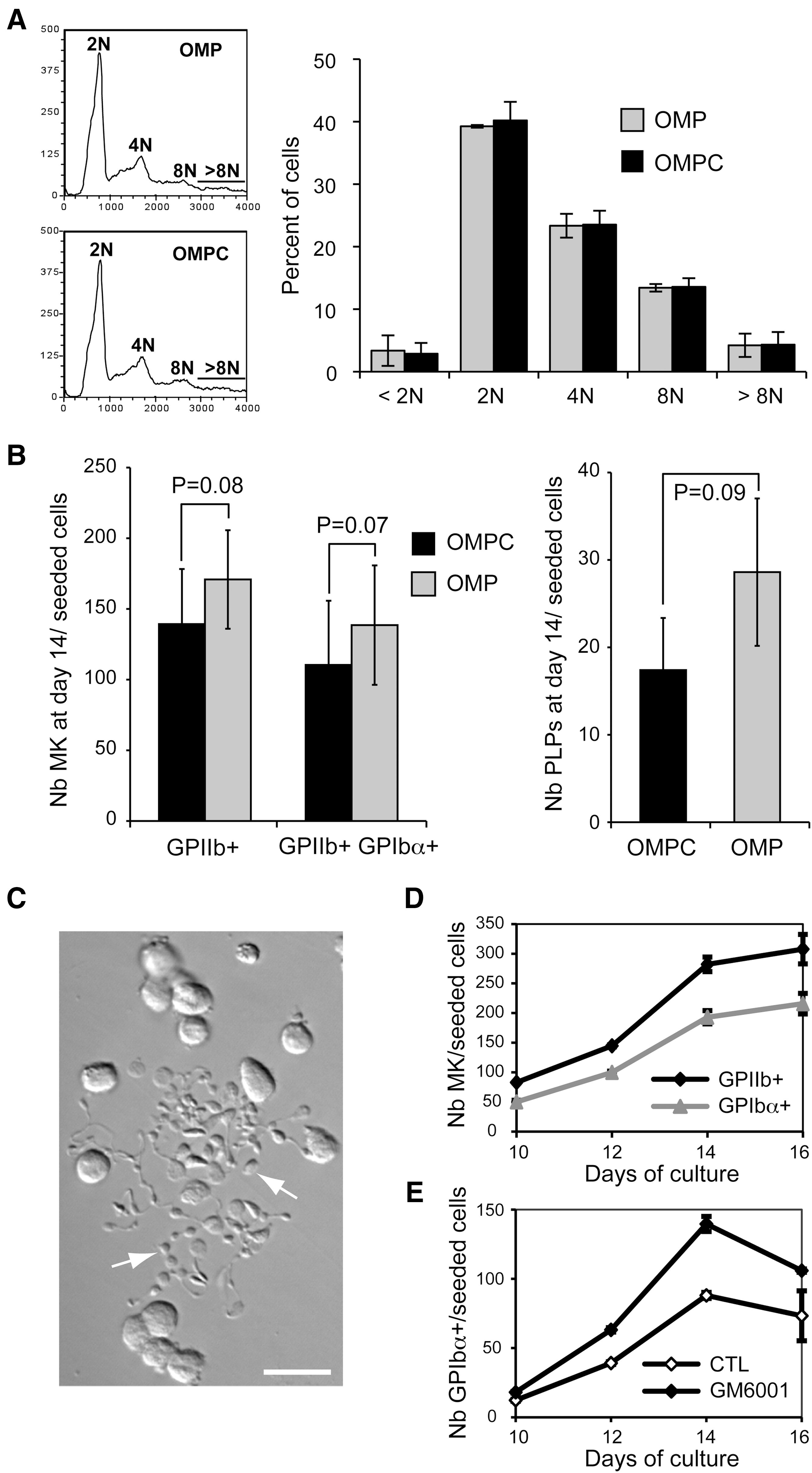
Production of Mks and PLPs in the 2-phase OMP-based culture protocol. Cord blood CD34+-enriched cells were cultured with the cytokine cocktails OMP or OMPC for the first 6 days of culture, and then with the BS1 cocktail from day 6 to day 16.
Since, OMP was as efficient as OMPC to produce PLPs despite its 65% reduction in cytokine content compared to OMPC, we used the combination of OMP and BS1 for the large-scale production (up to 180 mL) of PLPs for this work. In addition, to further reduce media and cytokine consumption, we modified the protocol by removing the washing step between the 2 phases as schematized in Fig. 2A. Under this scheme, Mk purity (% GPIIb+) averaged 67% ± 15% at day 14 (Fig. 3A).
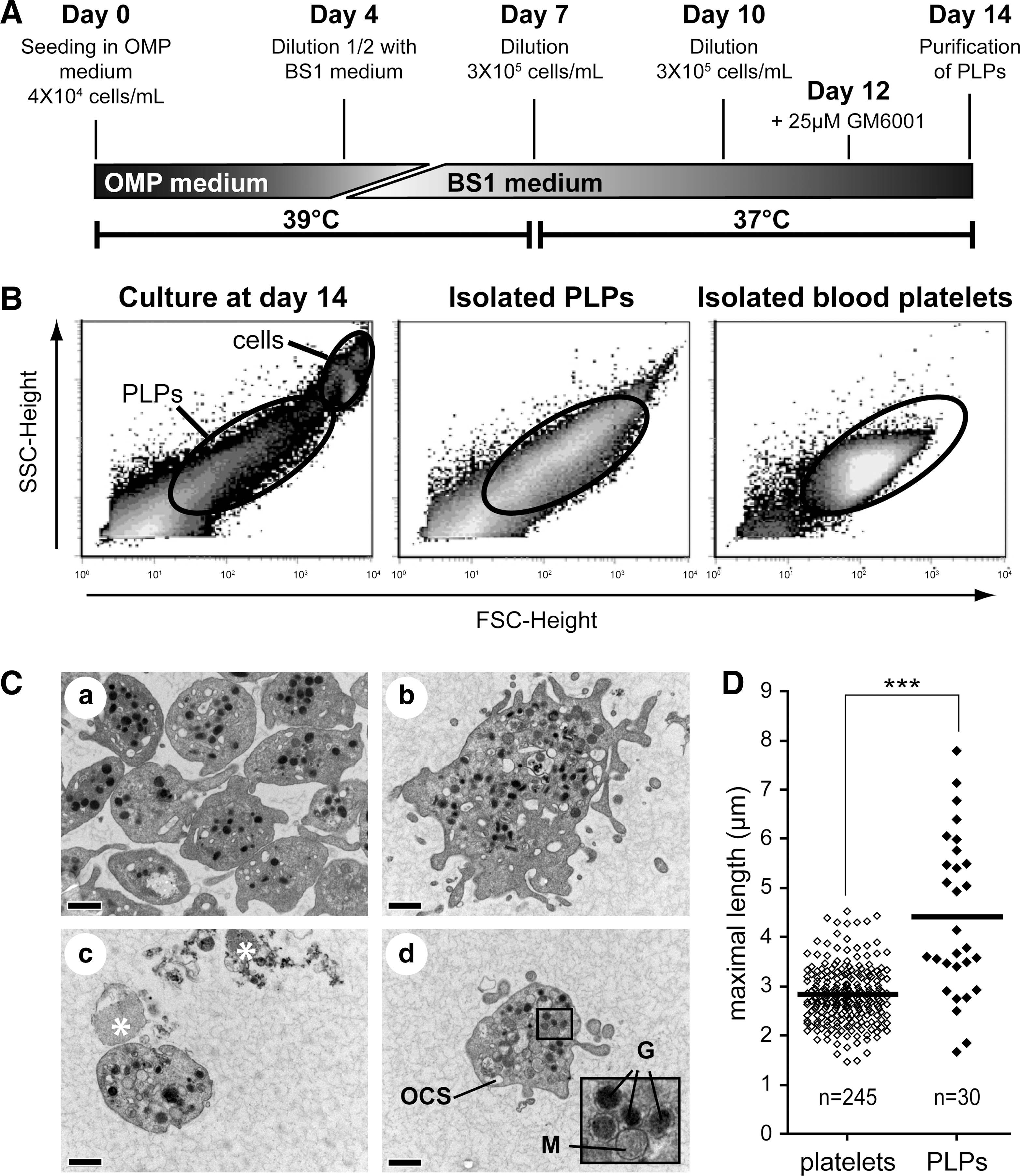
Mk cultures release a heterogeneous population of PLPs.
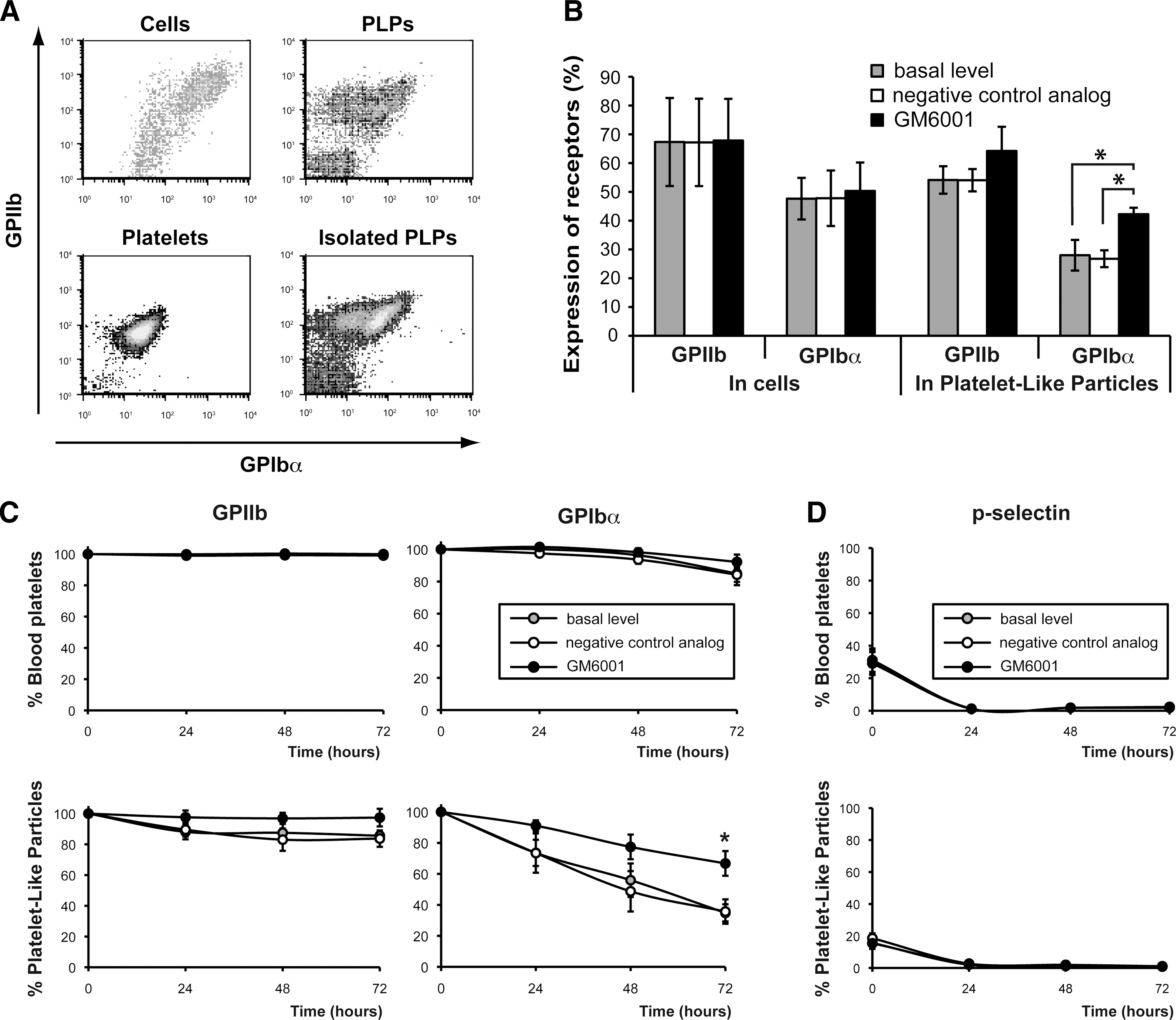
Metalloproteinase inhibition increases the stability of the GPIbα receptors on PLPs.
Next, 3 techniques were then tested to purify the PLPs from Mks and debris before their characterization; (i) centrifugation, (ii) gel filtration, and (iii) BSA gradient. Although simple, the first technique was found highly inefficient at purifying the PLPs (Table 2), while the gel filtration was found unreliable since cell debris often caused obstruction of the column. Finally, centrifugation over a BSA gradient was selected based on its reliability and excellent PLP recovery at high purity (Table 2). The production of PLPs varied among cultures, but generally 4–18 million PLPs (mean of 12 millions, N = 22) were purified per 100,000 starting CB CD34+ cells.
Mean ± standard deviation shown.
Number of independent experiments.
BSA, bovine serum albumin; PLP, platelet-like particle.
Morphology of PLPs
The forward and side scatter flow cytometry profiles of PLPs indicated that these formed a heterogeneous population compared to blood platelet (Fig. 2B, PLPs gate). Examination by transmission electron microscopy confirmed that some PLPs were of same size (Fig. 2C, panel c, d) to blood platelets (Fig. 2C panel a), whereas others were much bigger (Fig. 2C, panel b). Distribution of the measured diameters of the platelets from the transmission electron microscopy images confirmed that PLPs were heterogeneous in size with a mean of 4.4 ± 1.6 μM in length as compared to 2.8 ± 0.6 μM for normal platelet (Fig. 2D, P < 0.0001). Importantly, a significant proportion of PLPs shared morphological features with blood platelets (Fig. 2C); they contained mitochondria, alpha and dense granules, and open canalicular systems (see inset for high magnification of organelles, Fig. 2C, panel d). However, numerous ghosts presumed as degranulated platelets or cell fragments were also observed (see stars on Fig. 2C, panel c). Taken together, these analyses suggest that the current culture condition produces platelets at various maturation stages and confirmed the presence of normal-looking platelets in PLPs produced from CB CD34+ -derived Mks.
Expression and stability of the GPIIb and GPIbα receptors
Given the instability of some platelet receptors ex vivo, we examined the presence on the cell surface of GPIIb and GPIbα, 2 important receptors for platelet function specific to Mks and platelets. Surprisingly, the proportion of PLPs positive for expression of extracellular GPIbα (28%) was consistently reduced compared to the levels observed on the Mks (Fig. 3B, gray columns). In contrast, no significant differences were observed for GPIIb. Since the broad-range metalloproteinase inhibitor GM6001 was recently shown to prevent cleavage of GPIbα and the formation of its extracellular fragment (glycocalicin) in culture-derived platelets [3] and in stored PRPs [14,15], we added GM6001 (optimal dose of 25 μM; Supplementary Fig. S1, available online at
GPV is another receptor recently shown to be sensitive to metalloproteinases cleavage on murine PLPs [3]. Extracellular expression of GPV in PLPs was investigated to determine if this also occurred in human PLPs, and whether GM6001 could prevent loss of GPV. The proportion of GPV+-PLPs in unpurified total PLPs was similar to that seen for GPIbα (Table 3). Pretreatment with GM6001 significantly increased the proportion of GPV+ -PLPs (1.3-fold, Table 3), and that of GPIbα+ PLPs (1.6-fold, Table 3) as previously observed with purified PLPs (Fig. 3B). Based on the positive effect of GM6001 on expression of GPIbα and GPV, all of the remaining experiments were performed on PLPs produced in presence of GM6001 added at day 12.
Mean ± standard error of the mean shown, N = 3. Results from flow cytometry analyses of single stained (GPV or GPIbα+) PI-negative PLPs.
P < 0.01.
P < 0.001. P values calculated using the paired Student's t-test.
GPV, glycoprotein V; PI, propidium iodide.
Despite the addition of GM6001, 71% of the GPIIb+ PLPs produced in culture remained GPIbα−, suggesting perhaps that our culture conditions facilitate GPIbα downregulation. To clarify this point, purified blood platelets and PLPs were stored in the BS1-serum medium supplemented or not with GM6001 or with its inactive analog for up to 3 days. The evaluation of cell surface level of receptors by flow cytometry revealed that GPIIb level was stable in both PLPs and normal platelets in all conditions tested. Cell surface GPIbα expression was also stable in blood platelets whereas (Fig. 3C), it was markedly reduced on PLPs in basal condition and in the presence of the negative control analog. Consequently, up to 70% of the PLPs lost extracellular GPIbα expression after 72 h of storage (Fig. 3C), whereas only 20% of the PLPs lost extracellular GPIbα expression after 72 h of storage when GM6001 was present (Fig. 3C, see star). This result demonstrates that metalloproteinase-induced receptor shedding is one of the main mechanisms responsible for the low level of GPIbα receptor on PLP surface.
As GPIbα can also be eliminated from the platelet surface following internalization due to platelet activation [20], we examined the basal activation state of isolated platelets by measuring cell surface expression of the α-granule marker p-selectin (CD62p). A basal activation level in purified blood platelets (30%) was found almost identical to that of PLPs (20%). A resting period of 24 h following purification was, however, sufficient to shutdown p-selectin presentation in both platelet preparations (Fig. 3D). Thus, despite the similar level of basal activation, PLPs appeared more susceptible to GPIbα receptor shedding than blood platelets.
Function and vitality of PLPs
According to the cytometry data presented above, 2 distinct populations of PLPs were identified in the culture; the first one composed of GPIIb+ GPIbα+ particles and the second one regrouped GPIIb+ GPIbα− particles. To investigate the functionality of these PLP populations, we first evaluated their potential to induce α-granules release following agonist stimulation. Stimulation with thrombin was performed after a 24 h-resting period in the BS1-serum medium in the presence of GM6001 since this was found to restore normal basal level of p-selectin expression due to p-selectin shedding with the preservation of GPIbα receptor. The vast majority of GPIIb+ GPIbα+ PLPs responded strongly to thrombin (Fig. 4A, B). In contrast, the majority of GPIIb+ GPIbα− PLPs failed to present p-selectin following thrombin stimulation (Fig. 4A, B), whereas 95% of blood platelets upregulated p-selectin presentation after agonist activation (Fig. 4A, B). Similar results were obtained when ADP was used as an agonist (data not shown), suggesting that platelet function was seriously compromised in GPIIb+ GPIbα− PLPs.
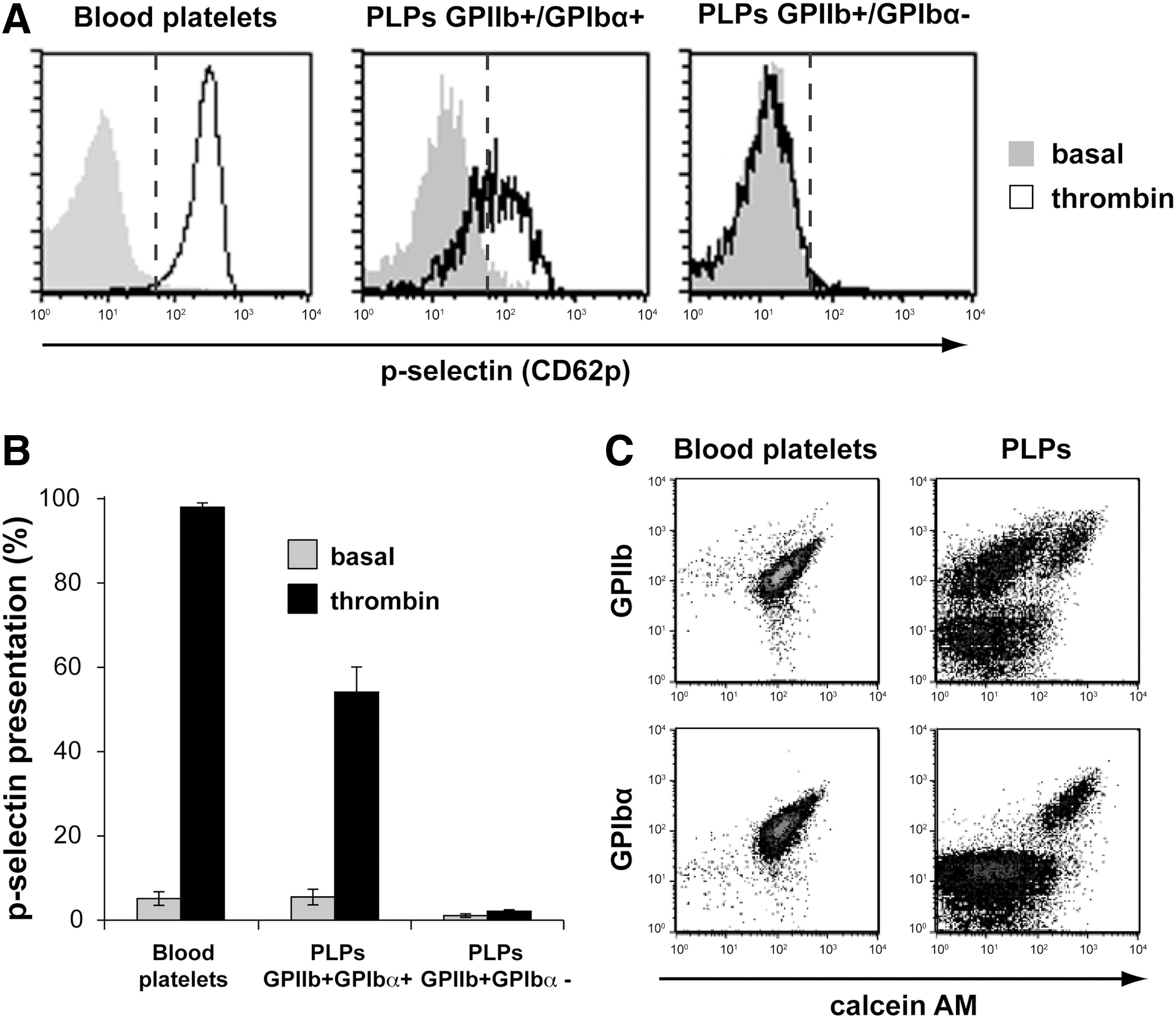
Thrombin stimulates α-granule release from GPIbα+ PLPs. Purified blood platelets and PLPs were incubated in the absence (fill gray) or presence (black line) of 1 U/mL thrombin for 10 min at 37°C before extracellular immunostaining of GPIIb, GPIbα, and p-selectin.
Next, blood platelets and PLPs were stained with calcein AM before GPIIb or GPIbα immunolabeling to establish the cell vitality of each population. Calcein AM ester is nonfluorescent until hydrolyzed by intracellular esterase present in viable cells and platelets. The vast majority of GPIIb+ or GPIbα+ blood platelets were positive for fluorescent calcein (Fig. 4C, left panels). In contrast, an important proportion (∼60%) of the GPIIb+ PLPs remained nonfluorescent (Fig. 4C, right panels), whereas all GPIbα+ PLPs were positive for fluorescent calcein. These results suggest that the majority GPIbα− particles are in fact metabolically inactive and imply that the absence of GPIbα is a marker for a more global defect. Thus, viable and perhaps functional PLPs appeared to be enriched in the GPIIb+ GPIbα+ PLP subpopulation.
Integrin αIIbβ3 bidirectional signaling in viable PLPs
The integrin αIIbβ3 (GPIIb/IIIa) drives bidirectional signaling, which supports the adhesion and aggregation activities of platelets. To investigate this process in PLPs, we evaluated the capacity of platelets to undergo normal shape change and spreading following adhesion to immobilized fibrinogen. Note that the previous cytometry analyses confirmed that all GPIIb+ PLPs also co-expressed GPIIIa (Supplementary Fig. S2, available online at
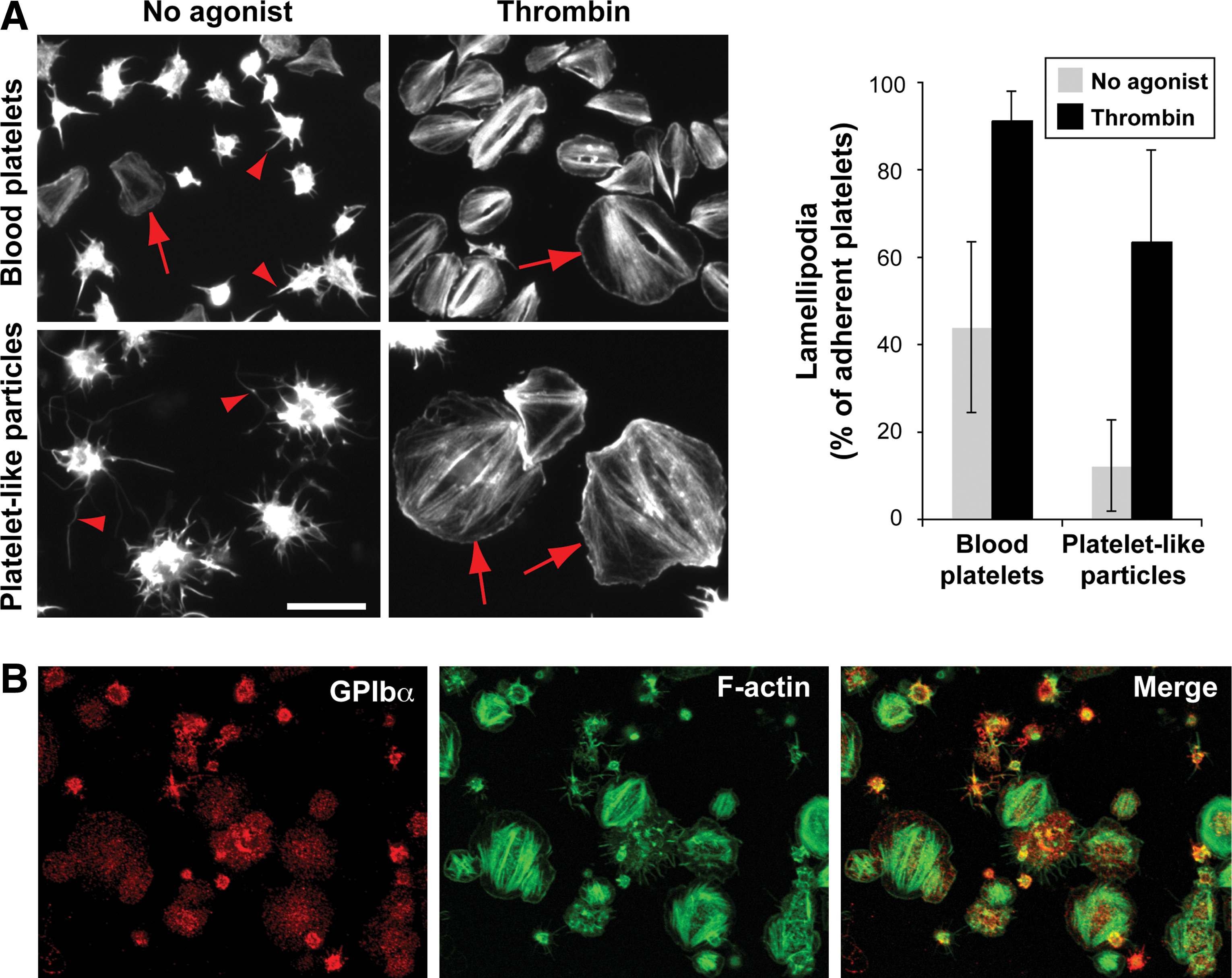
αIIbβ3 bidirectional signal response of culture-derived platelets.
Aggregation potential of culture-derived platelets
The capacity of PLPs to aggregate was first evaluated using light transmittance aggregometry. After stimulation with ristocetin, no difference in light transmittance was recorded for PLPs (Fig. 6A, right graph), whereas the control PRPs presented a typical shape change followed by a rapid aggregation response after ristocetin stimulation (Fig. 6A, left graph). Similar results were obtained when ADP was used as agonist (data not shown). Nevertheless, small aggregates were visible in the PLPs test tube after stimulation, but this differ from the larger unique aggregate observed with blood platelets, suggesting that some of the PLPs particles have the capacity to aggregate (data not shown).
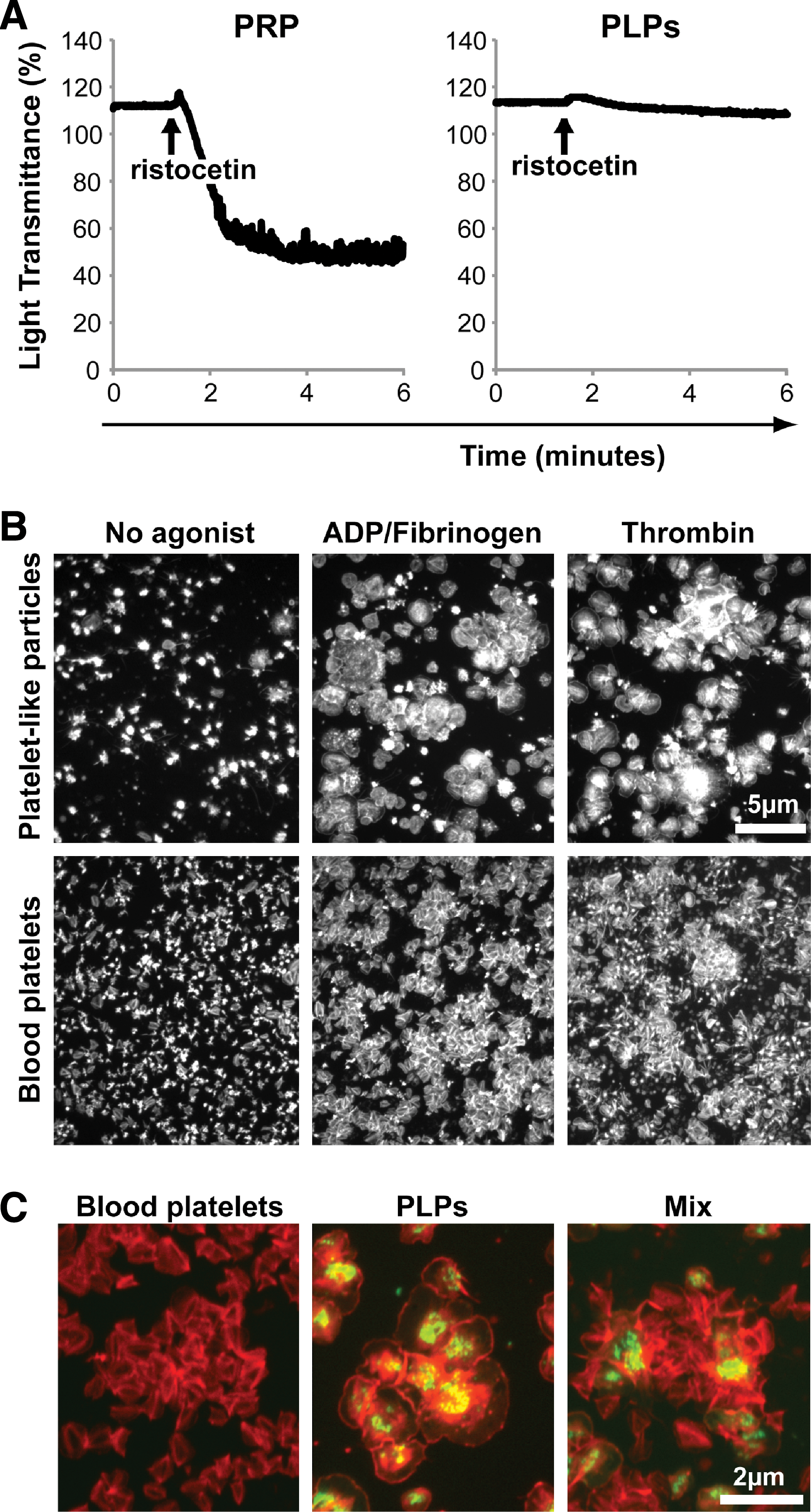
GPIbα+ PLPs form aggregates in response to agonist stimulation.
Since the previous results suggested that GPIbα− PLPs are metabolically compromised and nonfunctional and that GPIbα+ represented only 30% of all PLPs, the aggregation response of GPIbα+ PLPs may have been masked by the greater amount of GPIbα− PLPs. Hence, we designed an assay to evaluate the aggregation properties of GPIIb+ GPIbα+ PLPs. Briefly, to enrich the GPIbα+ PLPs, PLPs were first allowed to adhere to immobilized fibrinogen for 5 min, washed with PBS, and incubated in the absence or presence of soluble agonists under agitation for 15 min. GPIbα immunostaining on fixed aggregates confirmed that GPIIb+ GPIbα− PLPs were washed away with debris (data not shown). F-actin staining revealed that normal platelets and PLPs were dispersed and adopted a star shape in absence of agonists (Fig. 6B, first column). However, PLPs and blood platelets efficiently formed large aggregates following stimulation with fibrinogen and ADP or thrombin (Fig. 6B, second and third column).
PLPs were also loaded with calcein AM and mixed or not with nonstained blood platelets before aggregation assays. F-actin staining of fixed aggregates (Fig. 6C) showed that healthy PLPs and blood platelets could interact and form large aggregate together (Fig. 6C, right panel). Taken together, these results confirmed that functional platelets can be produced ex vivo, and that these are for most part restricted to the GPIIb+ GPIbα+ fraction of culture-derived PLPs.
Discussion
In this study, we produced human PLPs from the differentiation of CB CD34+ cell in liquid culture using an adapted scaled-up version of our previously described 2-phase protocol [7]. However, the main objective was rather to define carefully the properties of culture-derived platelets by comparing their morphological, phenotypic, and functional properties to that of blood platelets. Our results demonstrate that platelets with functional properties similar to that of normal platelets are restricted to PLPs that maintain co-expression of extracellular αIIbβ3 integrin (GPIIb/IIIa) and GPIbα, which represented 30% of the PLPs purified from our culture system.
Our results confirm to some extent those reported by others that human PLPs produced ex vivo could undergo α-granule release and/or aggregation [4 –6]. However, there are some differences in the efficiency reported between our work and theirs, which are probably due to differences in the purification method, origin of the cells, and culture medium composition used that may affect the PLPs.
Many studies published to date have relied on forward and side scatter properties combined with GPIIb expression to follow or measure platelets yields [2,6,8,21], whereas others have included receptors resistant to ectodomain shedding such as GPIX [5]. Two populations of PLPs were observed in these studies, GPIIb+ and GPIIb−, identified as platelets and cell debris and/or microparticles, respectively. Similar observations were also made in this work. Moreover, our study clearly distinguished major 2 subpopulations of GPIIb+ PLPs based on the presence of GPIbα at the platelet surface. Functional assays established that platelets that fail to maintain extracellular GPIbα expression have several functional abnormalities, including defects in α-granule release, adhesion, shape change, and aggregation in response to thrombin or ADP activation. The functional assays presented herein were performed with PLPs issued from cultures supplemented with GM6001 to improve the proportion of GPIbα+ PLPs. However, PLPs issued from cultures without GM6001 followed the same tendency (data not shown). These results indicate that monitoring of the GPIbα receptor is a better approach to identify functional platelets from ex vivo culture system.
Another significant finding of this work is that the high instability of the GPIbα receptor on the surface of PLPs produced ex vivo compared to blood platelets. Various cellular and proteolytic mechanisms might be responsible for the loss of GPIbα from the platelet surface. It may be either internalized following platelet activation [20] or shed due to cleavage by the metalloproteinase TACE/ADAM 17 [13]. A potential role of the calcium-dependent protease calpain has also been reported [22]. Our results suggest that metalloproteinase participate to the loss of GPIbα in culture-derived PLPs. Indeed, addition of the broad-range metalloproteinase inhibitor GM6001 at the end of the culture and during storage reduced GPIbα shedding considerably. This result is corroborated by other studies showing that metalloproteinase inhibition improves the function of in vitro-aged or -injured mouse platelets [23] and that of embryonic stem cell (ESC)-derived mouse platelets [3]. In addition, Nishikii et al. recently demonstrated that shedding of GPV and GPVI on the surface of PLPs produced from murine ESC can also be prevented by addition of GM6001 [3]. Our results confirmed that shedding of GPV on human PLPs can be prevented by GM6001. Whether GPVI is also shed by metalloproteinase on human PLPs remains to be determined, but our results suggest that a significant proportion of GPIbα+ PLPs retain extracellular GPVI expression as these responded normally to collagen.
Loss of cell surface GPIbα expression was not the sole defect of GPIbα-deficient PLPs. The failure of GPIbα− PLPs to remove the ester group from calcein AM indicates that these PLPs are metabolically severely compromised. This observation, together with their lack of proper response to various agonist stimulations, strongly suggests that these PLPs are in a senescent state or nonviable. In stored platelets, shedding of GPIbα and GPV receptors is part of the so-called platelet storage lesions (PSL) that have been shown to correlate with reduced responses in functional assays, increased apoptosis, and reduced post transfusion recovery [24]. The present study suggests that culture-derived platelets experience defects highly reminiscent of PSL. Optimization of culture process to prevent premature ectodomain shedding of receptors and senescence of platelets is clearly needed to improve the quality of PLPs produced ex vivo.
In the last decade, many efforts were made to understand and overcome the deleterious effects of PSL. Some of these findings could be transposed to current culture conditions to improve PLPs quality. As an example, activated clotting factors, cellular debris, and proteolytic enzymes found in the suspension plasma can adversely affect platelets during storage [25]. We can expect that cellular debris and metabolic waste, which accumulate in culture with time, can negatively affect culture-derived platelets and favor their premature aging and senescence. With this in mind, preliminary data suggest that washing and complete media change several days before platelet purification can improve both PLPs' quantity and quality in terms of GPIbα expression (A. Robert, unpublished results). Secondly, agitation is vital for maintenance of platelet quality during storage. Recent findings indicated that prolonged periods without agitation induce superoxide anion generation and depolarization of mitochondrial membrane potential along with a presentation of apoptotic markers [26]. The authors suggested that superoxide anion generation could be caused by local area of hypoxia when platelets are maintained stationary. Hence, gentle agitation or proper oxygenation of cultures may benefit the quality of PLPs produced ex vivo [27]. Finally, another study demonstrated that oxidative stress activates ADAM17 and induces receptor shedding in a p38-dependant manner [28]. p38 MAPK signaling inhibition prevented receptor shedding and phosphatidylserine exposure in stored human platelet, and increased post transfusion recovery [29]. Thus, it would certainly be interesting to test the effect of p38 inhibitors on PLPs viability and function.
Another issue raised by our study and corroborated by others [2 –4,8,30] is that PLPs constitute a very heterogeneous population of platelets at varying stage of maturation, suggesting that thrombopoiesis in culture occurred in an asynchronous manner. It would certainly be interesting to determine whether immature platelets and proplatelet fragments could be safely transfused so that the final stages of platelet maturation could be completed in the circulation. Live imaging with multiphoton intravital microscopy has recently shown that proplatelets extend from mature Mk through endothelial cells into blood vessel, supporting that platelet release and maturation is completed in part under sheer stress condition into the blood flow [31]. As proplatelets formation was generally observed by day 9 of culture, proplatelet fragments could be prepared from CB-derived MK cultures at earlier time to reduce lesions associated with ex vivo conditions. Another advantage of this proposal is that the final stages of thrombopoiesis would actually occur in vivo under normal shear stress conditions, which may significantly improve the yields of platelets production [32].
In summary, the present study demonstrates the importance of carefully monitoring the stability of important cell surface receptors on ex vivo-derived platelets. The optimization of culture process to prevent premature ectodomain shedding of receptors and senescence of platelets is clearly needed to improve the quality of culture-derived platelets before they can be considered as a useful complementary source of platelets for clinical application.
Footnotes
Acknowledgments
Amélie Robert owns a fellowship from the Natural Sciences and Engineering Research Council of Canada (NSERC). The authors wish to thank Nancy Trottier and Pier-Ann Gagné for their valuable technical help.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.