
Abstract
Endothelial progenitor cells (EPCs) may be recruited from the bone marrow to sites of tissue regeneration to sustain neovascularization and reendothelialization after acute vascular injury. This feature makes them particularly suitable for cell-based therapy. In mature endothelium, store-operated Ca2+ entry (SOCE) is activated following emptying of inositol-1,4,5-trisphosphate–sensitive stores, and controls a wide number of functions, including proliferation, nitric oxide synthesis, and vascular permeability. The present work aimed at investigating SOCE expression in EPCs harvested from both peripheral blood (PB-EPCs) and umbilical cord blood (UCB-EPCs) by employing both Ca2+ imaging and molecular biology techniques. SOCE was induced upon either pharmacological (ie, cyclopiazonic acid) or physiological (ie, ATP) depletion of the intracellular Ca2+ pool. Further, store-dependent Ca2+ entry was inhibited by the SOCE inhibitor, N-(4-[3,5-bis(trifluoromethyl)-1H-pyrazol-1-yl]phenyl)-4-methyl-1,2,3-thiadiazole-5-carboxamide (BTP-2). Real-time reverse transcription–polymerase chain reaction and western blot analyses showed that both PB-EPCs and UCB-EPCs express all the molecular candidates to mediate SOCE in differentiated cells, including TRPC1, TRPC4, Orai1, and Stim1. Moreover, pharmacological maneuvers demonstrated that, as well as in differentiated endothelial cells, the signal transduction pathway leading to depletion of the intracellular Ca2+ pool impinged on the phospholipase C/inositol-1,4,5-trisphosphate pathway. Finally, blockage of SOCE with BTP-2 impaired PB-EPC proliferation. These findings provide the first evidence that EPCs express SOCE, which might thus be regarded as a novel target to enhance the regenerative outcome of cell-based therapy.
Introduction
E
The finding that circulating EPCs traffic to sites of neovascularization, proliferate, and differentiate into ECs is consistent with the notion of adult vasculogenesis [4]. More specifically, EPCs are mobilized from bone marrow in response to local tissue ischemia or damage to replace dysfunctional ECs at the onset of severe cardiovascular disorders, such as atherosclerosis and arterial stenosis secondary to injury [5,6]. As a consequence, EPCs play a pivotal role in restoring the supply of oxygen and nutrients to infarcted myocardium [7], ischemic limbs [8], and growing tumors [9]. Their regenerative potential renders EPCs particularly amenable for cell-based therapy, as discussed in several recent reviews [1,5,10 –12]. However, because of their paucity in samples collected from peripheral blood (PB), which may be too low to exert beneficial effects, and their uncertain differentiation fate, which may be harmful for the recipient, unveiling the signal transduction pathway regulating EPC behavior is essential to improve the outcome of cell-based therapy [10,11]. As well as PB-derived EPCs (PB-EPCs), the therapeutic potential of umbilical cord blood–derived EPCs (UCB-EPCs) has been shown in a recent study, in which the authors demonstrated that transplantation of expanded UCB-EPCs favored neovascularization in a mouse ischemic hind-limb model [13]. Similar to their peripheral counterparts, the cellular mechanisms driving UCB-EPC function are yet to be elucidated.
An increase in intracellular Ca2+ concentration ([Ca2+]i) is the key signal in the regulation of a myriad of functions in mature ECs [14 –18]. The elevation in [Ca2+]i displayed by macrovascular ECs upon stimulation is generally biphasic and consists in an initial rise that rapidly decays to a plateau level of lower amplitude until agonist removal [18]. In more detail, ligand binding to Gq/11 protein–coupled receptors (GPCRs) results in the activation of phospholipase C-beta (PLC-β), which, in turn, cleaves phosphatidylinositol-4,5-bisphosphate to inositol 1,4,5-trisphosphate (InsP3) and diacylglycerol (DAG) [16,18]. DAG engages both protein kinase C and nonselective cation channels on the cellular membrane, whereas InsP3 rapidly diffuses within the cytosol to bind to and activate the InsP3 receptors (InsP3Rs) in the endoplasmic reticulum (ER). InsP3Rs, in turn, serve as Ca2+-permeable channels to release luminal stored Ca2+ and shape the initial Ca2+ transient [16,18]. In ECs, as well as in most other nonexcitable (ie, not endowed with voltage-gated ion channels) cells, ER depletion causes the activation of Ca2+-permeable ion channels in the plasma membrane, a mechanism that has been termed store-operated Ca2+ entry (SOCE), and sustains the following plateau phase [19,20]. The tight link between SOCE and ER Ca2+ content is underscored by the observation that inhibitors of the sarco-ER Ca2+-ATPase (SERCA), such as cyclopiazonic acid (CPA), activate Ca2+ entry by preventing Ca2+-reuptake into the stores, a maneuver leading to ER depletion [21 –23]. SOCE contributes to the global increase in [Ca2+]i, which triggers the release of a number of vasoactive/inflammatory mediators, such as nitric oxide, prostacyclin, von Willebrand factor, platelet activating factor, tissue plasminogen activator, and tissue factor pathway inhibitor [14]. Moreover, SOCE controls the genetic program underlying EC proliferation by engaging Ca2+-sensitive transcription factors, including the nuclear factor of activated T-cells (NFAT) and the nuclear factor kappaB (NF-kB) [19,24,25]. Finally, store-dependent Ca2+ inflow governs the paracellular permeability of endothelial monolayers by triggering cytoskeleton reorganization (through myosin light chain–dependent contraction) and consequent disassembly of VE-cadherins at adherens junctions [14]. Unveiling the molecular nature of SOCE in mature ECs has engendered remarkable controversy [26]. Several lines of evidence hinted at members of the canonical transient receptor potential (TRPC) family of cation channels as prime candidates for SOCE in vascular endothelium [18]. Indeed, store-dependent Ca2+ inflow was strongly dampened in cells lacking either TRPC1 [27] or TRPC4 [28,29]. Conversely, a recent report proposed the stromal interaction molecule-1 (Stim1)/Orai1 pathway to mediate SOCE in ECs [19]. More specifically, Stim1 is a transmembrane ER Ca2+ sensor that translocates in close proximity to the plasma membrane upon store depletion and activates Orai1 Ca2+ channel to gate SOCE [22]. Although EPCs incorporate into neovessels to recapitulate endothelial functions following tissue injury or ischemia [4], at the present time, it is unknown whether these cells are endowed with such a critical signaling pathway, that is, store-dependent Ca2+ inflow, before differentiating into mature ECs.
The aim of this article was to investigate SOCE in EPCs harvested from both PB and UCB. We provide the first evidence that SOCE occurs also in EPCs upon emptying of InsP3-sensitive intracellular Ca2+ stores. Further, we show that EPCs express all the molecular candidates to mediate SOCE in mature ECs, such as TRPC1, TRPC4, Stim1, and Orai1. Finally, we demonstrate that SOCE inhibition blocks EPC proliferation. These findings gain crucial insights into the molecular mechanisms that govern EPC signaling and provide novel putative targets for genetic manipulation aimed at enhancing EPC response to angiogenic stimuli.
Materials and Methods
Isolation and cultivation of EPCs
Blood samples (40 mL) were obtained from healthy human volunteers aged from 22 to 28 years. The Institutional Review Board at “Istituto di Ricovero e Cura a Carattere Scientifico Policlinico San Matteo Foundation” in Pavia approved all protocols. Informed consent was obtained according to the Declaration of Helsinki. To isolate ECFCs, MNCs were separated from either PB or UCB by density gradient centrifugation on lymphocyte separation medium for 30 min at 400 g and washed twice in endothelial basal medium with 2% FCS (EBM-2). A median of 36 × 106 MNCs (range: 18–66) from PB and 16 × 106 MNCs (range: 7–23) from UCB were plated on collagen-coated culture dishes (BD Biosciences) in the presence of the EC growth medium EGM-2 MV Bullet Kit (Lonza) containing EBM-2, 5% fetal bovine serum, recombinant human (rh) EGF, rhVEGF, rhFGF-B, rhIGF-1, ascorbic acid, and heparin and maintained at 37°C in 5% CO2 and humidified atmosphere. Discard of nonadherent cells was performed after 2 days; thereafter, medium was changed 3 times a week. The outgrowth of ECs from adherent MNCs was characterized by the formation of a cluster of cobblestone-appearing cells, as previously described [3] (Supplementary Fig. S1a, b; available online at
Solutions
Physiological salt solution (PSS) had the following composition (in mM): 150 NaCl, 6 KCl, 1.5 CaCl2, 1 MgCl2, 10 glucose, 10 Hepes. In Ca2+-free solution (0Ca2+), Ca2+ was substituted with 2 mM NaCl, and 0.5 mM EGTA was added. Solutions were titrated to pH 7.4 with NaOH. The osmolality of the extracellular solution, as measured with an osmometer (Wescor 5500), was 300–310 mmol/kg.
[Ca2+]i measurements
EPCs were loaded with 4 μM fura-2 acetoxymethyl ester (fura-2/AM; 1 mM stock in dimethyl sulfoxide) in PSS for 30 min at room temperature. After washing in PSS, the coverslip was fixed to the bottom of a Petri dish and the cells were observed using an upright epifluorescence Axiolab microscope (Carl Zeiss), usually equipped with a Zeiss X63 Achroplan objective (water-immersion, 2.0 mm working distance, 0.9 numerical aperture). EPCs were excited alternately at 340 and 380 nm, and the emitted light was detected at 510 nm. A first neutral density filter (1 or 0.3 optical density) reduced the overall intensity of the excitation light and a second neutral density filter (optical density = 0.3) was coupled to the 380 nm filter to approach the intensity of the 340 nm light. A round diaphragm was used to increase the contrast. The excitation filters were mounted on a filter wheel (Lambda 10; Sutter Instrument). Custom software, working in the LINUX environment, was used to drive the camera (Extended-ISIS Camera; Photonic Science) and the filter wheel and to measure and plot on-line the fluorescence from 10 to 15 rectangular “regions of interest” (ROI) enclosing 10–15 single cells. Each ROI was identified by a number. As cell borders were not clearly identifiable, an ROI may not include the whole EPC or may include part of an adjacent EPC. Adjacent ROIs never superimposed. [Ca2+]i was monitored by measuring, for each ROI, the ratio of the mean fluorescence emitted at 510 nm when exciting alternatively at 340 and 380 nm (shortly termed “ratio”). An increase in [Ca2+]i causes an increase in the ratio [31]. Ratio measurements were performed and plotted on-line every 3–5 s. The experiments were performed at room temperature. The human umbilical cord rings were loaded with 4 μM fura-2/AM for 4 h at room temperature and analyzed using the same equipment described above.
RNA isolation and real-time reverse transcription–polymerase chain reaction
Total RNA was extracted from the EPCs using the QIAzol Lysis Reagent (Qiagen). Single cDNA was synthesized from RNA (1 μg) using random hexamers and M-MLV Reverse Transcriptase (Invitrogen S.R.L.). Reverse transcription was always performed in the presence or absence (negative control) of the reverse transcriptase enzyme. Quantitative-polymerase chain reaction (PCR) was performed in triplicate using 1 μg cDNA and specific primers (intron-spanning primers; Table 1), as previously described elsewhere [32]. MESA GREEN qPCR MasterMix Plus (Eurogentec) was used according to the manufacturer's instruction and qPCR was performed using Rotor Gene 6000 (Corbett). The conditions used were initial denaturation at 95°C for 5 min, 40 cycles of denaturation at 95°C for 30 s, annealing at 58°C for 30 s, and elongation at 72°C for 40 s. The q-reverse transcription (RT)-PCR reactions were normalized using β-actin as a housekeeping gene. Melting curves were generated to detect the melting temperatures of specific products immediately after the PCR run.
The triplicate threshold cycle (Ct) values for each sample were averaged, resulting in mean Ct values for both the gene of interest and the housekeeping gene β-actin. The gene Ct values were then normalized to the housekeeping gene by taking the following difference: ΔCt = Ct[gene] − Ct[β-actin], with high ΔCt values reflecting low mRNA expression levels. Relative gene expression quantitation was determined by the 2−ΔΔCt method [33]. Thus, to compare the relative levels of gene expression of UCB-EPC versus PB-EPC, ΔΔCt values were first calculated: ΔΔCt = ΔCt (UCB-EPC) − ΔCt (PB-EPC), and then fold changes were determined using transformation: fold increase = 2−ΔΔCt; fold decrease = −2ΔΔCt.
The sequences of the bands were checked by using the Big Dye Terminator Cycle Sequencing kit (Applied Biosystem). PCR products were also separated by agarose gel electrophoresis, stained with ethidium bromide, and acquired with the Image Master VDS (Amersham Biosciences Europe). The molecular weight of the PCR products was compared with the DNA molecular weight marker VIII (Roche Molecular Biochemicals).
Sample preparation and immunoblotting
PB-EPCs and UCB-EPCs were homogenized by using a Dounce homogenizer in a solution containing 250 mM sucrose, 1 mM ethylenediaminetetraacetic acid, 10 mM Tris-HCl (pH 7.6), 0.1 mg/mL phenylmethylsulfonyl fluoride (PMSF), 100 mM β-mercaptoethanol, and Protease Inhibitor Cocktail (P8340; Sigma). The homogenates were solubilized in Laemmli buffer [34] and 20–40 μg proteins were separated on 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to the Hybond-P PVDF Membrane (GE Healthcare) by electroelution. After 1 h blocking with Tris-buffered saline containing 3% bovine serum albumin and 0.1% Tween (blocking solution), the membranes were incubated for 3 h at room temperature with affinity-purified antibodies diluted 1:200 in the Tris-buffered saline and 0.1% Tween. Anti-Stim1 (sc-166840) and anti-TRPC4 (sc-15063) were obtained from Santa Cruz Biotechnology, anti-Orai1 was from Alomone Labs, and anti-TRPC1 (T8276) from Sigma-Aldrich.
The membranes were washed and incubated for 1 h with peroxidase-conjugated mouse, rabbit, and goat IgG (1:120,000 in blocking solution) (QED Bioscience). The bands were detected with the ECL™ Advance western blotting detection system (GE Healthcare Europe GmbH). Control experiments were performed by using the antibody preadsorbed with a 20-fold molar excess of the immunizing peptide and by incubating the blots with nonimmune serum. BenchMark prestained protein ladders (Invitrogen) were used to estimate the molecular weights.
Protein content
Protein contents of all the samples were determined by the Bradford's method, using bovine serum albumin as a standard [35].
Proliferation assays
A total of 1 × 105 ECFC-derived cells (first passage) were plated in 30-mm collagen-treated dishes in EGM-2 MV medium with or without 2–20 μM N-(4-[3,5-bis(trifluoromethyl)-1H-pyrazol-1-yl]phenyl)-4-methyl-1,2,3-thiadiazole-5-carboxamide (BTP-2). We chose this range after preliminary experiments, which showed no or unspecific toxic effect for lower or higher concentrations of BTP-2, respectively. Cultures were incubated at 37°C (in 5% CO2 and humidified atmosphere) and cell growth was assessed every day until confluence was reached in the control cultures (0 μM BTP-2). At this point, cells were recovered by trypsinization from all dishes and the cell number was assessed by counting in a hemocytometer. The percentage of growth inhibition by the drug was calculated by dividing the total number of cells obtained in presence of BTP-2 by the number of cells in control experiments and multiplying the ratio by 100.
Statistics
All the data have been collected from EPCs isolated from the PB of at least 3 healthy volunteers or 3 different samples of UCB. The amplitude of the peak Ca2+ response was measured as the difference between the ratio at the peak and the mean ratio of 1 min baseline before the peak. Plateau amplitude was measured by averaging 60 s of signal at 10 min after the peak. Pooled data are given as mean ± SE and statistical significance (P < 0.05) was evaluated by the Student's t-test for unpaired observations.
As to mRNA analysis, all data are expressed as means ± SEM. Statistical analysis of qRT-PCR experiments was primarily performed on ΔCt values. The significance of the differences of the means was evaluated with 1-way ANOVA followed by Newman–Keuls's Q test or Student's t-test. All statistical tests were carried out with GraphPad Prism 4.
Chemicals
EBM and EGM-2 were purchased from Clonetics (Cell System). Fura-2/AM was obtained from Molecular Probes Europe BV. N-(4-[3,5-Bis(trifluoromethyl)-1H-pyrazol-1-yl]phenyl)-4-methyl-1,2,3-thiadiazole-5-carboxamide (BTP-2) was purchased from Calbiochem. All other chemicals were obtained from Sigma Chemical.
Results
Stimulation of SOCE by CPA-induced store emptying in EPCs harvested from PB
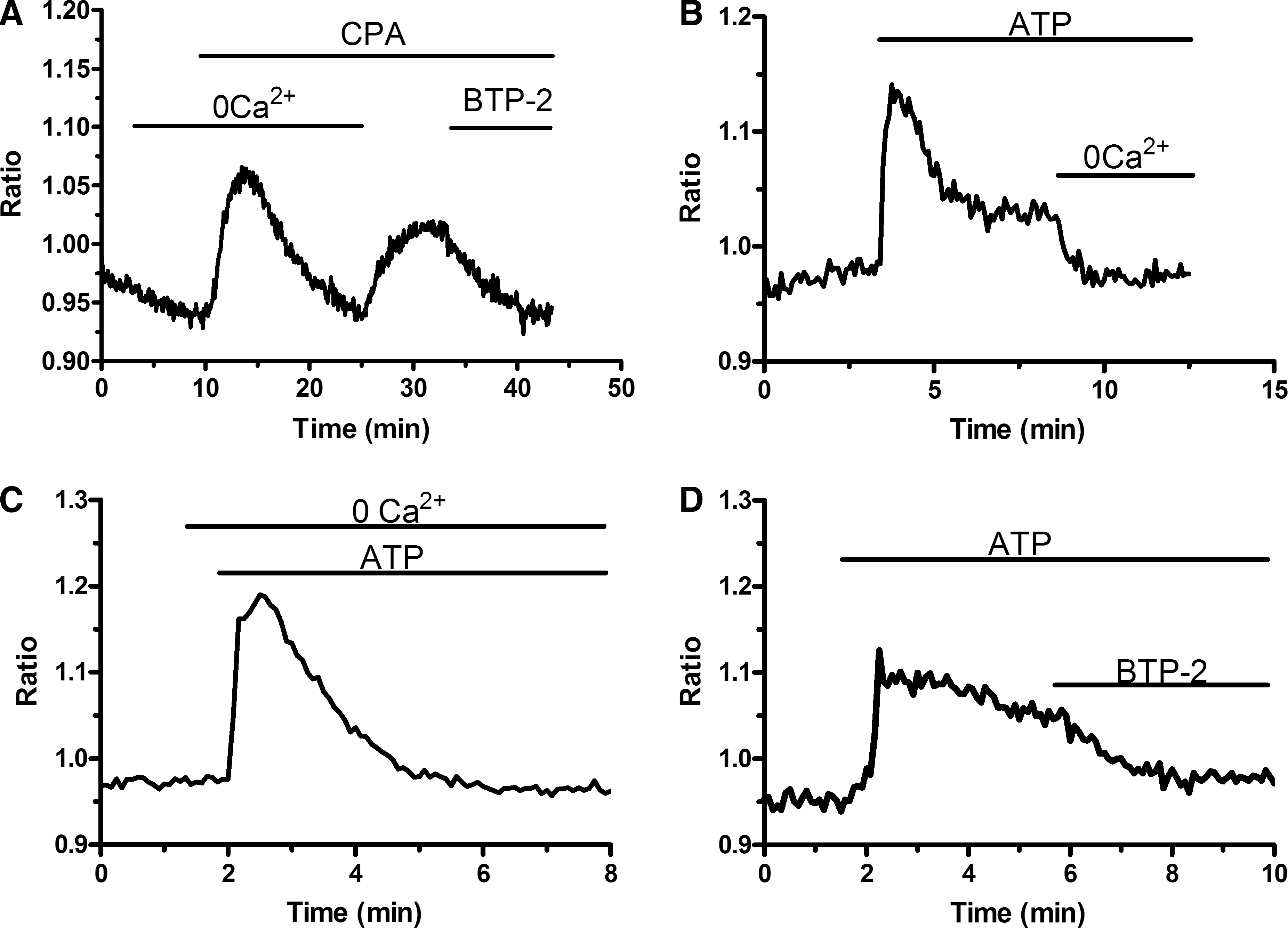
To assess the presence of a functional store-operated Ca2+ inflow, we carried out fluorescence measurements of EPCs loaded with the Ca2+-sensitive fluorochrome, Fura-2, and exposed to CPA. CPA, a widely employed SERCA blocker [21], prevents Ca2+ sequestration into the stores, thus leading to their depletion and to SOCE activation [21 –23]. As depicted in Fig. 1A, when PB-EPCs were bathed in absence of extracellular Ca2+ (0Ca2+), CPA (10 μM) induced a transient rise in [Ca2+]i because of passive emptying of Ca2+ stores through leakage channels in ER membrane. Thereafter, Ca2+ levels decayed to the baseline as the plasma membrane transporters (ie, plasma membrane Ca2+-ATPase and Na+/Ca2+ exchanger) extruded Ca2+ from the cytosol. Subsequent addition of Ca2+ (1.5 mM) to the extracellular medium (the classic “Ca2+ add-back” protocol) induced Ca2+ inflow through opened store-operated channels, which resulted in a robust increase in intracellular Ca2+ levels (Fig. 1A, C). In control experiments, replenishment of extracellular Ca2+ in absence of CPA did not elicit a detectable elevation in [Ca2+]i (data not shown). Removal of extracellular Ca2+ resulted in a drop in intracellular Ca2+ levels in a fraction of PB-EPCs (77 of 211 cells; 36.5%) (Fig. 1B). This result hinted at the presence of a resting Ca2+ permeability that was not investigated. The store-dependent nature of CPA-elicited Ca2+ entry into EPCs was further assessed by employing BTP-2, a specific inhibitor of SOCE [36]. Acute application of BTP-2 (20 μM) strongly inhibited CPA-dependent Ca2+ inflow in PB-EPCs (n = 156) (Fig. 1A). Accordingly, 30-min preincubation with BTP-2 (20 μM) prevented SOCE in PB-EPCs (Fig. 1B, C), whereas it did not affect the ER Ca2+ content (Fig. 1B, C). Altogether, these data indicate that a functional SOCE is expressed in PB-EPCs. Conversely, such cells lack functional voltage-gated Ca2+ channels (VGCCs), which promote Ca2+ inrush into excitable cells. Consistently, when PB-EPCs were exposed to high KCl extracellular solution, a protocol that may activate voltage-dependent Ca2+ transients in ECs obtained from bovine adrenal medulla [37], no Ca2+ signal could be detected (n = 59) (data not shown).
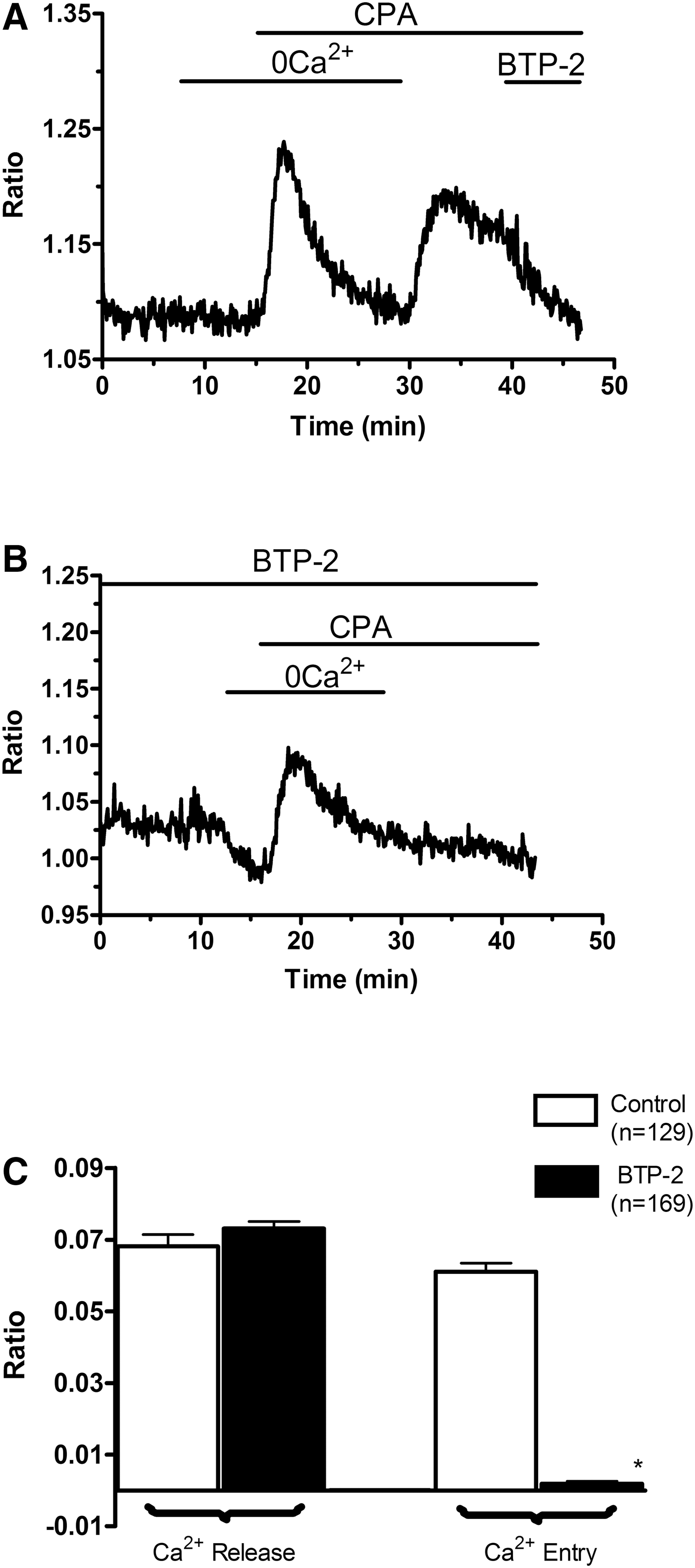
SOCE is expressed in EPCs harvested from PB.
SOCE may be engaged by physiological stimulation in PB-EPCs
SOCE engagement following activation of GPCRs was investigated by first testing the effect of a number of vasoactive agonists that mobilize Ca2+ from InsP3-sensitive intracellular stores [18]. Figure 2 shows that neither
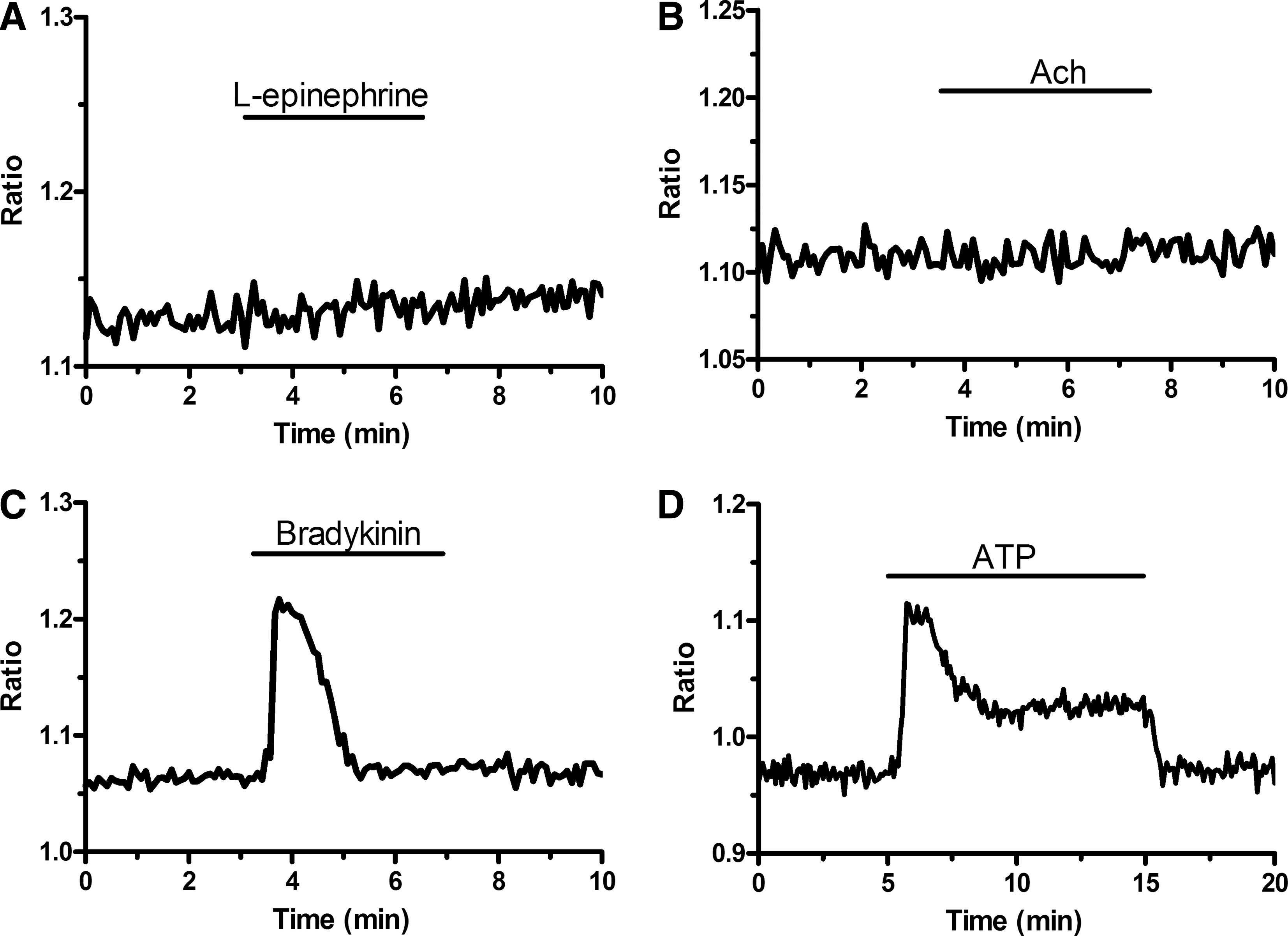
Different effects of endothelial agonists on [Ca2+]i in EPCs harvested from PB.
ATP-induced sustained elevation in [Ca2+]i requires Ca2+ entry.

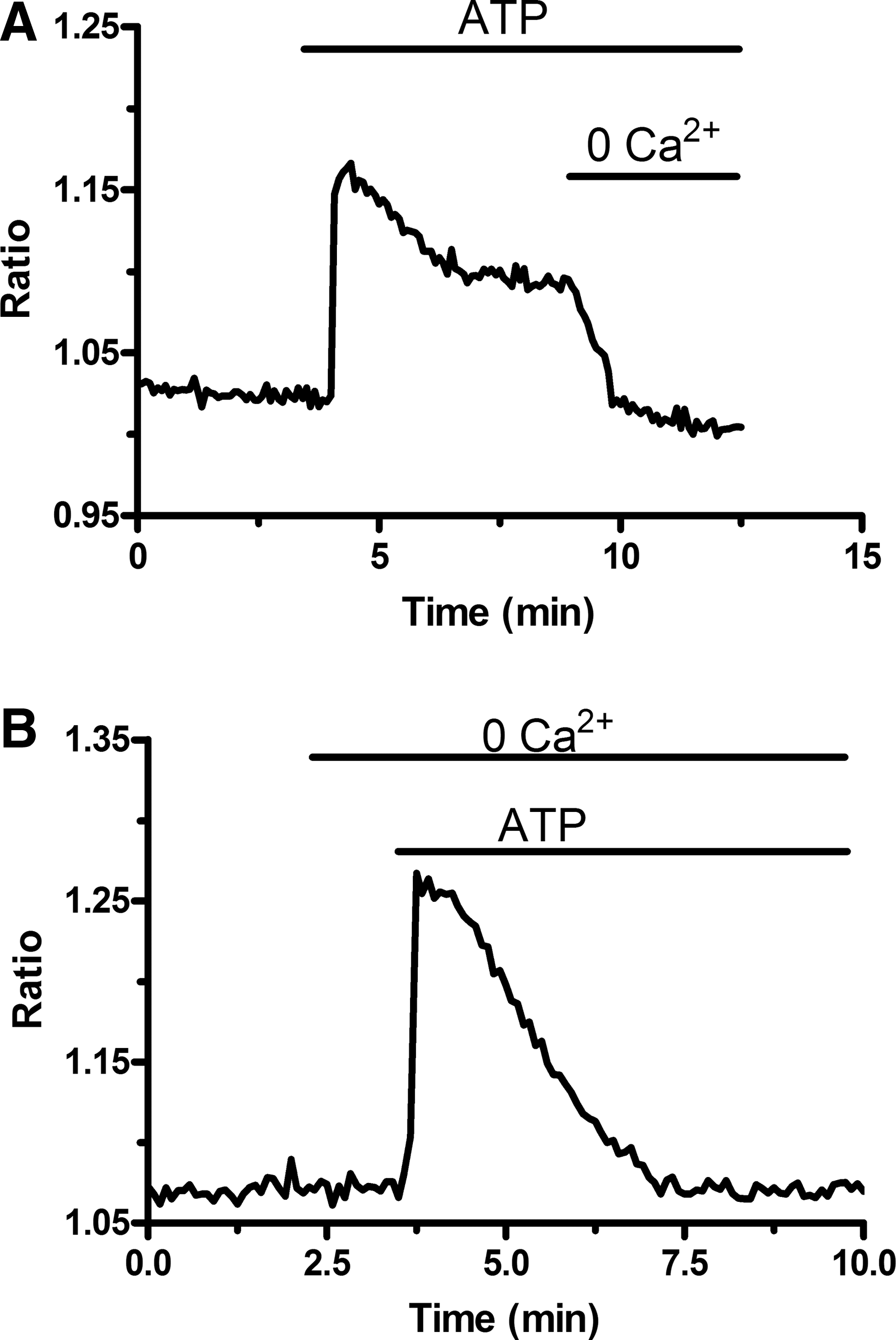
ATP engages SOCE in EPCs isolated from PB.

GPCRs-induced Ca2+ store depletion requires the PLC/InsP3 pathway
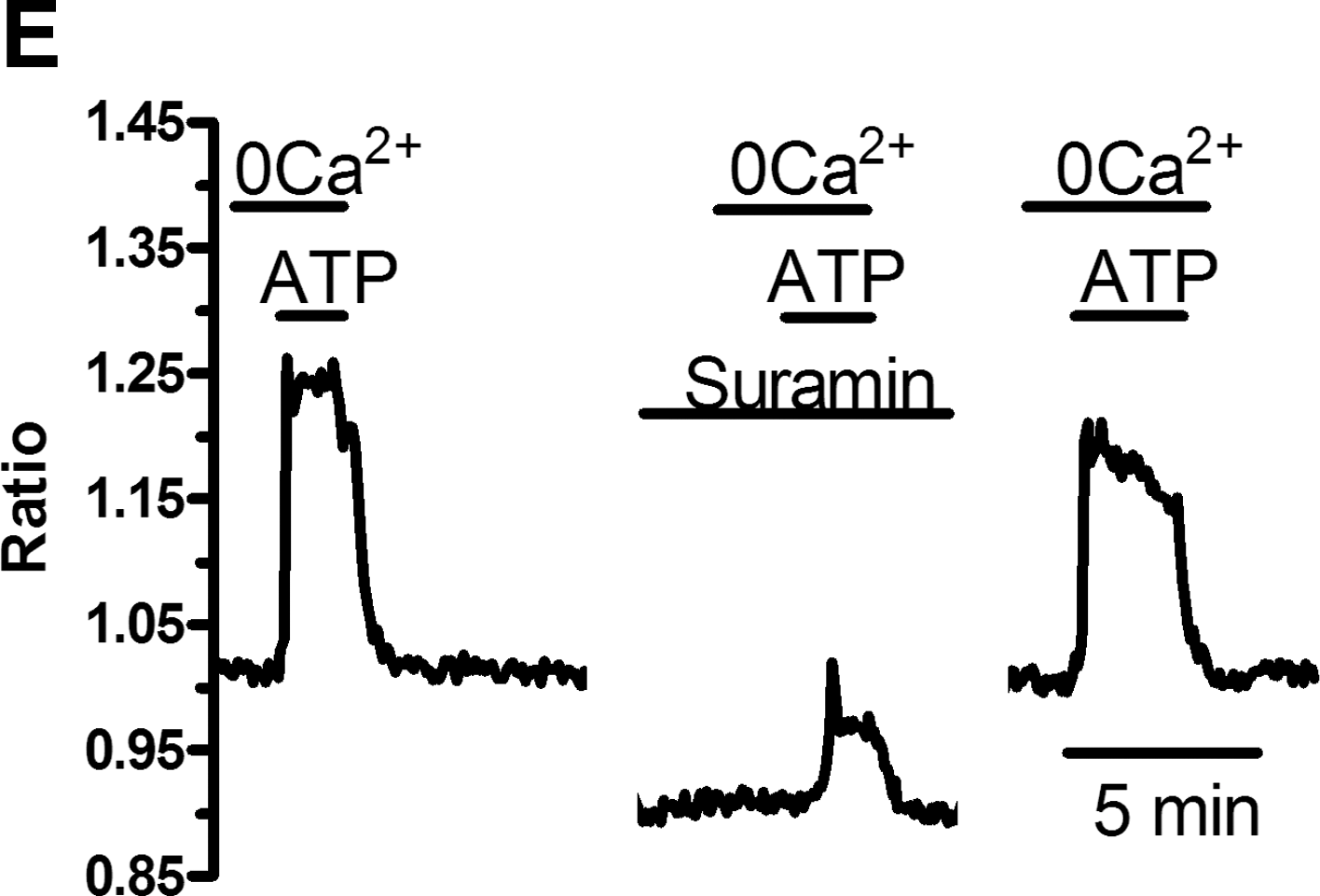
In mature ECs, SOCE recruitment by GPCRs is mediated by the PLC-β/InsP3 signaling pathway. The following experiments were carried out in 0Ca2+ to prevent any contaminating signal from Ca2+ entry and focus on the molecular mechanisms underlying ER Ca2+ store depletion. To assess the effects of the selective antagonists of the PLC-β/InsP3 pathway on the same cells, ATP was bath-applied 3 times (control, experiments, washout) for 100 s at 30-min (U73122) and 10-min (caffeine) intervals. U73122 (10 μM), a widely employed PLC-β blocker [31,45], prevented ATP-induced [Ca2+]i elevation in all 96 PB-EPCs (Fig. 5A). Washout of the drug led to the recovery of Ca2+ response to ATP (Fig. 5A). Subsequently, we assessed the effect of caffeine on intracellular Ca2+ homeostasis. Depending on the concentration, in vascular endothelium, caffeine may both inhibit InsP3Rs [31,45] and activate ryanodine receptors (RyRs) through the process of Ca2+-induced Ca2+ release (CICR) [46]. When applied at 20 mM, a dose able to stimulate RyRs in rabbit aortic ECs [46], caffeine did not elicit any detectable Ca2+ transient (n = 83) (Fig. 5B), a feature that suggested the absence of functional RyRs in EPCs. Notably, lower concentrations of the drug (100 μM), which selectively block InsP3Rs, reversibly hindered the Ca2+ response to ATP (Fig. 5C). The statistical analysis of these data has been summarized in Fig. 5D. Finally, depletion of InsP3Rs-sensitive store by previous addition of the SERCA inhibitor, CPA (10 μM), in absence of extracellular Ca2+ prevented ATP-induced elevation of [Ca2+]i (n = 50) (not shown). Collectively, these data indicate that, as in mature ECs, the PLC-β/InsP3 signaling pathway causes Ca2+ store depletion, which leads to SOCE. The latter route is engaged by metabotropic P2Y receptors when the incoming stimulus is provided by ATP. Consistently, the nonselective purinergic antagonist, suramin (300 μM) [47], reversibly inhibited the Ca2+ response to ATP in PB-EPCs (Fig. 5E). Indeed, the magnitudes of the Ca2+ peak were 0.314 ± 0.012, n = 119, and 0.059 ± 0.005, n = 119, in absence and presence, respectively, of the drug. Suramin is a fluorescent molecule and its application rapidly decreases the basal fluorescence ratio (Fig. 5E) [31]. However, the presence of the drug still allows the fluorimetric system to monitor intracellular Ca2+ signals, as indicated by exposing the cells to bradykinin (5 μM) after suramin addition (not shown).
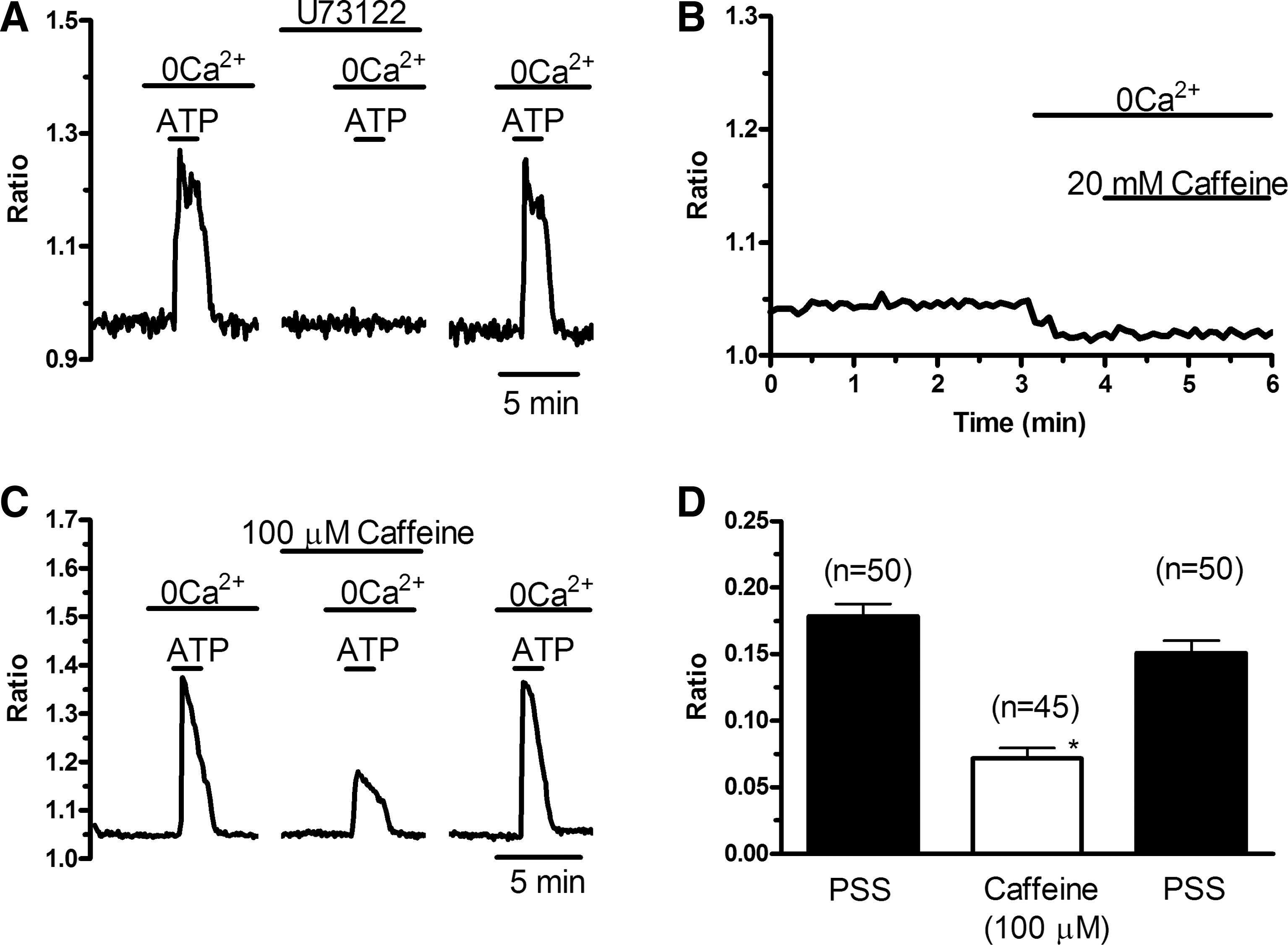
The phospholipase C/inositol-1,4,5-trisphosphate signaling pathway leads to endoplasmic reticulum Ca2+ store depletion in EPCs obtained from PB.
The mRNA encoding for putative mediators of SOCE is present in PB-EPCs
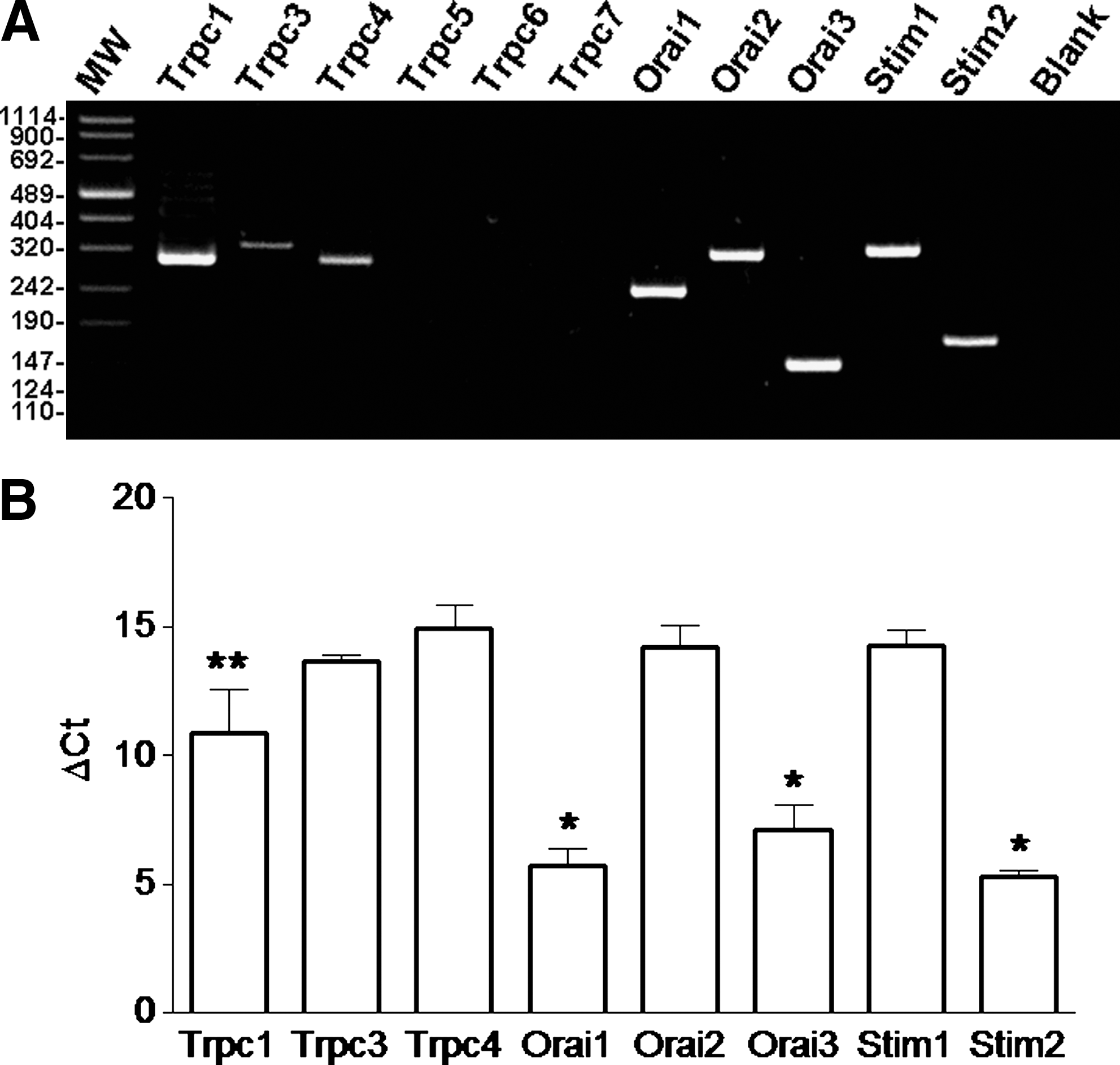
The expression of the molecular entities that have been proposed to underpin store-dependent Ca2+ entry in mature endothelium has been investigated in PB-EPCs by employing qRT-PCR. The specific primers depicted in Table 1 have been employed to evaluate the expression of mRNAs encoding for all TRPC (TRPC1 and TRPC3–7) channels expressed in humans [48] and for the recently cloned Orai (Orai1-3) and Stim (Stim1 and Stim2) genes [22,34]. TRPC1, TRPC4, Orai1, Orai2, Orai3, Stim1, and Stim2 transcripts were readily detectable in PB-EPCs (Fig. 6A), whereas there were no significant amount of mRNA for TRPC3, TRPC5, TRPC6, and TRPC7. Single bands of the expected size of cDNA fragments were amplified and the results of agarose gel electrophoresis of representative PCR products are shown in Fig. 6A. Negative controls were performed by omitting the reverse transcriptase (not shown). The comparison of ΔCt values of mRNA obtained by qRT-PCR in PB-EPCs showed that Orai1 transcript was significantly higher than Orai2, Orai3, Stim1, Stim2, and TRPC1 and that TRPC4 transcript was the lowest (P < 0.05, ANOVA followed by Newman–Keuls' Q test) (Fig. 6B).

Expression of classical transient receptor potential (TRPC) channels, Orai, and STIM proteins in EPCs harvested from PB.
SOCE is expressed in EPCs isolated from UCB
To assess SOCE expression in UCB-EPCs, the latter underwent the Ca2+ add-back protocol upon exposure to CPA (10 μM) in 0Ca2+. As illustrated in Fig. 7A, CPA induced a BTP-2–sensitive Ca2+ entry, which is the hallmark of SOCE, in 179 cells. Similar to PB-EPCs, removal of extracellular Ca2+ caused a decrease in intracellular Ca2+ levels in a minor percentage of UCB-EPCs (29 of 97 cells, 29.8%) (Fig. 7A). As observed in their peripheral counterparts, UCB-EPCs displayed a biphasic Ca2+ response to ATP (100 μM) (Fig. 7B). In these cells, the amplitudes of the transient Ca2+ rise and the sustained components were, respectively, 0.162 ± 0.004 (n = 224) and 0.064 ± 0.002 (n = 216). Both values are significantly (P < 0.05) smaller than those (see above) measured in PB-EPCs. As observed in the latter, when external Ca2+ was omitted during the plateau phase, [Ca2+]i returned to the resting levels in all 130 cells (Fig. 7B). Consistent with this finding, when ATP was administered in absence of extracellular Ca2+, the cells exhibited only the transient peak without the sustained component (Fig. 7C). As in PB-EPCs, the height of the Ca2+ response to ATP was significantly (P < 0.05) higher in 0Ca2+ (0.164 ± 0.006; n = 132) than in PSS (0.207 ± 0.007; n = 103). Notably, the plateau phase of ATP-elicited Ca2+ signal was strongly inhibited by BTP-2 (50 μM) in 131 cells (Fig. 7D). Collectively, these results indicate that also UCB-EPCs express a SOCE pathway that may be engaged following physiological stimulation of GPCRs. The qRT-PCR analysis revealed that, in addition to the transcripts found in PB-EPCs, UCB-EPCs express TRPC3 (Fig. 8A). Single bands of the expected size of cDNA fragments were amplified and the results of agarose gel electrophoresis of representative PCR reaction products are shown in Fig. 8A. Negative controls were performed by omitting the reverse transcriptase (not shown). The comparison of ΔCt values of the Ca2+ signal pathway mRNA obtained by qRT-PCR showed that TRPC1, TRPC3, TRPC4, Orai2, and Stim1 transcripts were significantly lower than Orai1, Orai3, and Stim2 (P < 0.01, ANOVA followed by Newman–Keuls's Q test) (Fig. 8B). Finally, when comparing the Ca2+ channel gene expression in the 2 cell populations, we found that Orai1, Orai3, and Stim2 levels were 6-, 22-, and 46-fold higher in UCB-EPCs, respectively (Fig. 9). Conversely, Orai2 and Stim1 levels were 18- and 12-fold higher in PB-EPCs, respectively, whereas TRPC1 and TRPC4 were similar (Fig. 9).
SOCE is expressed in EPCs isolated from UCB.
The mRNA encoding for canonical transient receptor potential (TRPC) channels, Orai, and Stim is expressed in UCB-EPCs.
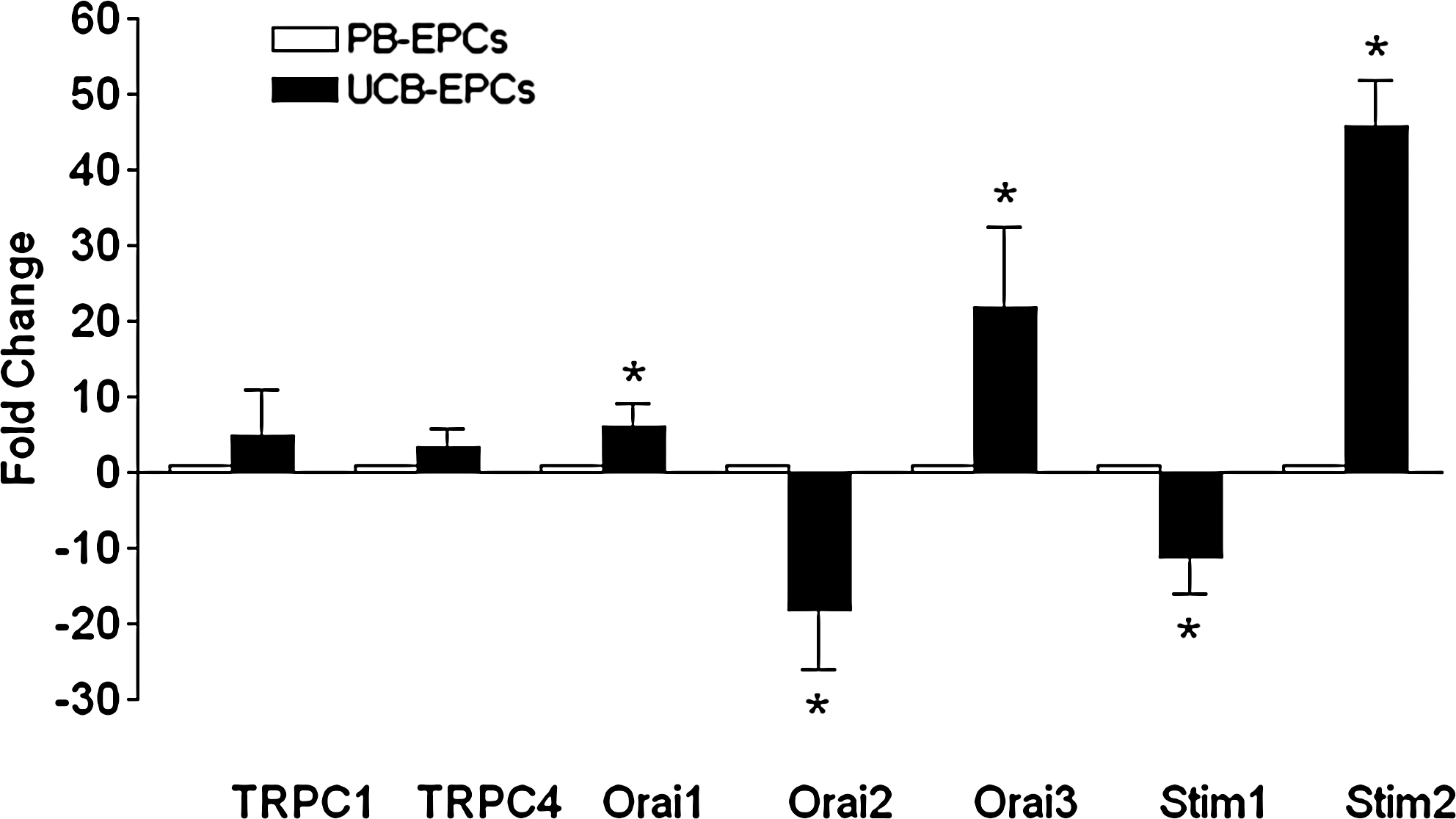
Relative expression of canonical transient receptor potential (TRPC) channels, Orai, and Stim genes in UCB-EPCs versus PB-EPCs. Fold change values for each gene were calculated versus PB-EPCs, as detailed in the Materials and Methods section. Statistical analysis was performed on ΔCt values and the significance of the differences of the means evaluated with Student's t-test. *P < 0.05 versus PB-EPCs.
SOCE-related proteins are expressed in both PB- and UCB-EPCs
Western blot analysis of Orai1, Stim1, TRPC1, and TRPC4 expression was conducted using affinity-purified antibodies. As observed for the RNA transcripts, all these proteins were detected in both PB-EPCs and UCB-EPCs (Fig. 10). Immunoblots showed major bands of about 33, 110, 110, and 100 kDa for Orai1, Stim1, TRPC1, and TRPC4, respectively. The molecular sizes were similar to those reported in the literature [19,49]. When the antibodies were preadsorbed with large amounts of the immunizing peptide or the antibodies substituted with nonimmune serum, the protein bands completely disappeared, thus indicating the specificity of the reaction. Along with the qRT-PCR analysis, these data demonstrate that both PB- and UCB-EPCs possess the molecular machinery that has been associated with SOCE in mature ECs.
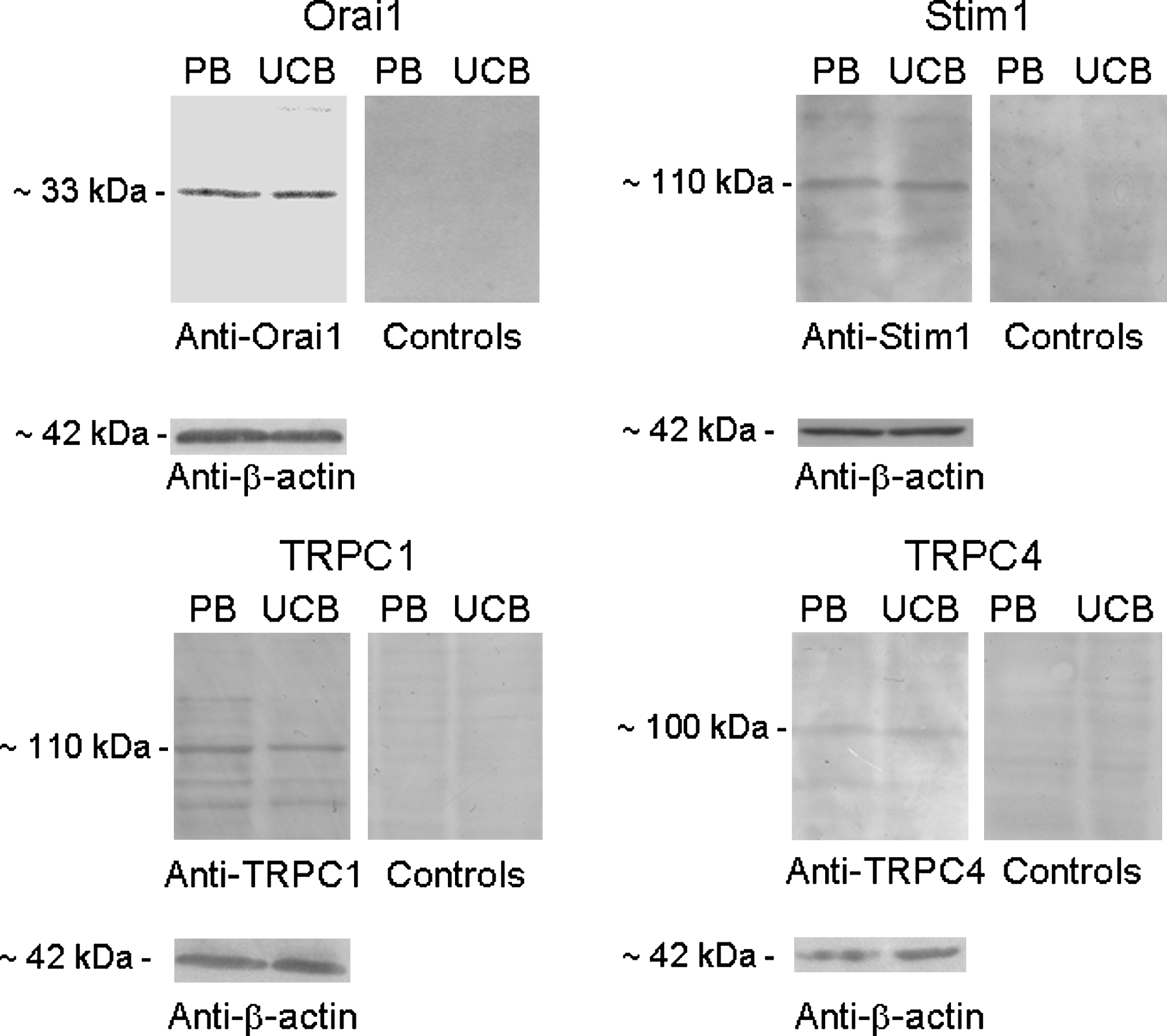
Expression of the proteins associated with store-operated Ca2+ influx in PB- and UCB-EPCs. Blots for Orai1, Stim1, TRPC1, and TRPC4 representative of 4 separate experiments are shown. Lanes were loaded with 20 μg of proteins for Orai1 detection and 40 μg of proteins for Stim1, TRPC1, and TRPC4 detection. The immunoblots were probed with affinity-purified antibodies diluted 1:200 and processed as described in the Materials and Methods section. Controls were performed as indicated in the Materials and Methods section. Major bands of the expected molecular weights were observed in both cell types. Conversely, no bands were observed in controls.
SOCE drives EPC proliferation
To assess the physiological outcome of SOCE, we focused on its involvement in EPC replication, as such a pathway may regulate the growth of mature ECs [19]. The proliferation assay was conducted on PB-EPCs. As shown in Table 2, BTP-2 inhibited cell growth in a dose-dependent manner (2–20 μM). None of the cultures carried on in the presence of BTP-2 reached confluence at the time (3 days) reached by control cultures. Confluence was never reached by BTP-2–exposed cells at longer incubation and cells usually began detaching and dying after 5–6 days of culture. Therefore, SOCE stands as a novel player in the signaling network underlying EPC proliferation and provides an additional target to enhance the outcome of cell-based therapy.
Results are expressed as percentage of growth compared with control (given as 100% growth).
Compared with control and after Bonferroni's correction (t-test for paired samples).
Discussion
EPCs are mobilized from BM to promote neovascularization and favor reendothelialization following vascular damage [1,4,11]. This observation prompted the injection of exogenous EPCs, harvested from both peripheral and UCB, as a novel therapeutic strategy to treat ischemic diseases [1,13]. Indeed, EPCs home to sites of tissue regeneration and contribute to neovascularization by acquiring an endothelial phenotype and being incorporated into neovessels [1,4]. In mature ECs lining the lumen of blood vessels, SOCE activation by GPCRs is central to the control of a wide number of functions, including proliferation, secretion, vascular permeability, and regulation of blood pressure [14 –19,24]. In the view of such a relevant role, it was worth investigating whether EPCs possess the SOCE pathway as well. Our results provide the first evidence that store-dependent Ca2+ inflow is present in both EPCs flowing in PB and EPCs harvested from UCB. Further, both PB-EPCs and UCB-EPCs express all the molecular candidates to mediate SOCE in mature ECs, that is, TRPC1, TRPC4, and Stim1/Orai1. We finally show that the signal transduction cascade involved in SOCE opening in differentiated endothelium, that is, ER Ca2+ store depletion by InsP3Rs activation, is also present in EPCs.
Several lines of evidence indicate that GPCRs recruit SOCE by emptying the ER reservoir via PLC-β/InsP3 signaling in EPCs. First, ligand (ie, ATP)-induced depletion of the intracellular Ca2+ pool in 0Ca2+ is a signal sufficient to activate Ca2+ inflow on Ca2+ readdition in the absence of agonist. As widely reported in the literature [21,23,41], this feature implies that Ca2+ entry occurs independently on receptor occupancy and does not rely on intracellular second messengers, such as InsP3 and DAG. When the agonist is withdrawn in the continuous presence of Ca2+ (Fig. 2D), [Ca2+]i recovers to the baseline as InsP3 synthesis terminates such that InsP3Rs close. As a consequence of the rapid interruption of Ca2+ efflux from ER, the intracellular Ca2+ pool refills and SOCE is inactivated [39,50,51]. On the other hand, exposure to ATP in the absence of extracellular Ca2+ leads to a transient Ca2+ signal (Fig. 4A), as most of Ca2+ released from InsP3Rs is extruded by plasma membrane transporters, such as Na+/Ca2+ exchanger and plasma membrane Ca2+-ATPase. The ensuing depletion of the intracellular Ca2+ reservoir results in SOCE activation and in the bump in [Ca2+]i observed on Ca2+ replenishment to the bath even in the absence of agonist [21,23,41,50,51]. Accordingly, passive depletion of the ER Ca2+ content with CPA led to the opening of Ca2+-permeable channels on the plasma membrane, which is the hallmark of SOCE [21,23], in both PB-EPCs and UCB-EPCs. Second, BTP-2, the hitherto most powerful and selective inhibitor of store-dependent Ca2+ inflow [36], abrogated CPA- and P2Y receptor-induced Ca2+ inflow in both cell types. Third, U73122, a widely employed pharmacological PLC blocker [31], strongly hindered ATP-elicited depletion of the ER Ca2+ pool. This finding is supported by a recent study that reported the expression of PLC-β2 in mice UCB-EPCs at both mRNA and protein levels [52]. Fourth, low doses of caffeine, which have long been known to antagonize InsP3-gated channels in several cell types, including rat cardiac microvascular ECs [31,45 and references therein], significantly reduced ATP-dependent Ca2+ mobilization from ER. Further, higher doses of caffeine, which stimulate RyRs to release Ca2+ via the so-called CICR, did not trigger any Ca2+ increase under control conditions. This result suggests either that functional RyRs are not expressed in EPCs or that resting Ca2+ levels are too low to enable the drug to sensitize RyRs [53]. Therefore, although our data indicate that RyRs are unlikely to participate in the signal transduction pathway leading to SOCE, further analyses are necessary to rule out their expression in EPCs. Notably, we found that bradykinin, which may promote Ca2+ signals via the G-protein–coupled B2 receptor [54], elicited a robust InsP3-dependent Ca2+ release, but not store-dependent Ca2+ entry (Fig. 3C). This feature, however, is not surprising when considering that partial dissociation between Ca2+ mobilization and SOCE activation may occur in several cells, including rat basophilic leukemia cells [55]. As reviewed in ref. [23], it has been suggested that 2 distinct InsP3-regulated Ca2+ stores exist within the ER: one mainly responsible for Ca2+ release and another tightly coupled to SOCE. According to Parekh and Penner [56], the InsP3-sensitive subcompartment devoted to the gating of store-operated channels is likely to lie in close proximity to the plasma membrane. It is, therefore, conceivable that, in human EPCs, ATP, but not bradykinin, engages such a pool and leads to store-dependent Ca2+ inflow. The same feature is likely to explain why SOCE amplitude is significantly higher in CPA- rather than ATP-treated cells (compare Fig. 1C with Fig. 4C). Indeed, CPA might deplete all the InsP3-dependent pool that is tightly coupled to plasmalemmal channels, whereas only a fraction of such a reservoir may be recruited by ATP [23,56].
The report of a functional SOCE pathway in both PB-EPCs and UCB-EPCs has 2 remarkable implications: (1) unlike other components of the endothelial signaling toolkit, such as muscarinic receptors or RyRs, this route is already expressed in immature cells, and consequently, (2) store-dependent Ca2+ inflow stands as a novel candidate among the mechanisms regulating EPC mobilization, proliferation, and homing.
The molecular underpinnings of SOCE in mature human endothelium, as well as in other cell types, include several candidates, such as TRPC1, TRPC4, and Stim1/Orai1 [19,20,27 –29]. Earlier studies provided the evidence that the molecular make-up of the Ca2+ channels gated by a drop in ER Ca2+ content in ECs comprised both TRPC1 and TRPC4, the latter being the subunit essential for coupling store depletion to channel activation [20,27 –29]. These data have been recently challenged by Trebak's group, who showed that small interference RNAs (siRNAs) targeted against either Stim1 or Orai1, but not against TRPC1 and TRPC4, inhibited SOCE in human umbilical vein ECs (HUVECs) [19]. This apparent discrepancy might arise from the well-known heterogeneity in the endothelial phenotype [57], so that ECs located in different vascular beds might mediate SOCE through distinct mechanisms [57,58]. Notably, qRT-PCR has revealed mRNA encoding for TRPC1/TRPC4 and Stim1/Orai1 in PB-EPCs and UCB-EPCs. The abundance of relative gene expression for these proteins was Orai1 > Stim1 = TRPC1 > TRPC4 in PB-EPCs and Stim1 = TRPC4 > TRPC1 > Orai1 in UCB-EPCs. When comparing the expression of such transcripts between the 2 cell types, we found that Orai1 and Stim1 are more abundant in PB-EPCs and UCB-EPCs, respectively, whereas TRPC1 and TRPC4 are expressed at the same level in both EPC types. Such a result cannot explain the fluorimetric measurements (ie, SOCE was higher in PB-EPCs) because of the tight stoichiometric coupling among the molecular candidates of SOCE [22,36]. Western blot experiments confirmed that all the transcripts encoding TRPC1/TRPC4 and Stim1/Orai1 are translated into proteins of the expected molecular mass in both PB-EPCs and UCB-EPCs. These data are consistent with the presence of all the putative mediators of store-operated Ca2+ channels in mature ECs. Therefore, siRNA experiments are required to unveil the molecular identity of store-dependent Ca2+ inflow in EPCs. The latter expressed also Stim2 and Orai2-3, which have been involved in SOCE in differentiated cells [22,36]. Stim2 has been reported to control the basal Ca2+ leak in several cell types [59], including HUVECs, and might regulate the resting Ca2+ inflow that we observed upon removal of external Ca2+ in a fraction of EPCs. As to Orai2 and Orai3, they have been also found in HUVECs [19] and might contribute to SOCE by forming heteromultimers with Orai1 in heterologous expression systems [22,36]. Nevertheless, their participation to GPCR-induced store-dependent Ca2+ entry in vascular endothelium is unlikely [19]. It is worth noting that we could not detect any transcript for other members of the TRPC family that are known to modulate endothelial function [17,60]. Indeed, unlike ECs from human arteries [60] and human microvessels [61,62], no transcripts of TRPC2, TRPC3, TRPC5, TRPC6, and TRPC7 were found by either quantitative or qualitative RT-PCR in PB-EPCs, whereas only TRPC3 was detected in UCB-EPCs. Preliminary experiments confirmed that the latter channel is also expressed at protein level. This feature is rather relevant when considering that peripheral EPCs may differentiate into mature ECs on mobilization from bone marrow. For instance, TRPC3, TRPC6, and TRPC7 mediate VEGF-triggered Ca2+ influx following an increase in the levels of the intracellular second messenger, DAG [17,60], in ECs isolated from several vascular beds [61,62]. It is, therefore, conceivable to assume that EPCs express the mRNA encoding for these channels when they are incorporated within the foci of neovascularization, where they acquire a mature phenotype and are exposed to site-specific environmental cues [57]. In agreement with this hypothesis, human cardiac progenitor cells do not express RyRs, although the latter drives Ca2+ cycling in differentiated cardiomyocytes [63]. Further, EPCs harvested from both peripheral and cord blood manifest a number of endothelial markers, such as CD31 and vascular cell adhesion molecule-1, and the ability to incorporate DiI-acetylated low-density lipoprotein, only following in vitro differentiation [2]. The same feature is likely to apply also for muscarinic and catecholamine receptors, which trigger intracellular Ca2+ elevations in mature ECs [18,39,40], but not in PB-EPCs. Similarly, voltage-dependent Ca2+ transients have been detected in bovine adrenal medulla capillary ECs [37], whereas they are absent in PB-EPCs. In this view, it is predictable that the reported effect of benidipine, a dihydropyridine Ca2+ channel blocker, on the differentiative outcome of mice PB-EPCs is not due to selective inhibition of Ca2+ entry [64]. This feature concurs with the notion that benidipine stimulates EPC differentiation by activating the phosphoinositide-3 kinase (PI3K)/Akt pathway [64].
It has long been known that Ca2+ entry is a key regulator of endothelial proliferation and migration [16 –19]. For instance, knocking-down either Stim1 or Orai1, which mediates SOCE in HUVECs, inhibited EC proliferation and caused cell cycle arrest at S and G2/M phases [19]. In addition, either reduction of extracellular Ca2+ or inhibition of plasmalemmal Ca2+ channels have been repeatedly shown to prevent cell growth and replication [16,65]. This feature also applies to immature cells, as blockage of SOCE dramatically impairs PB-EPC growth. Ca2+ entry may be translated into a mitogenic signal by direct binding of the ion to Ca2+-dependent targets or by Ca2+ binding to intracellular decoders such as calmodulin (CaM) and its downstream targets, including CaM kinase (CaMK) and calcineurin [16,17]. The recruitment of the CaM/CaMK pathway is crucial to cell cycle progression through G1 and mitosis by regulating the activation of several cyclin-dependent kinases (cdk), such as cdk2 and cdk4, whereas calcineurin controls the transcription factors that promote the G1/S transition, including cAMP-responsive element binding protein 1 and NFAT [16,65]. The finding that SOCE governs EPC proliferation highlights a novel alternative target to enhance the regenerative outcome of cell-based therapy. This latter approach mainly relies on 2 critical parameters: (1) EPC homing to ischemic organs and (2) EPC incorporation into newly growing vessels, proliferation, and differentiation into mature ECs [10 –12]. Therefore, once established the molecular machinery underlying SOCE in EPCs, transfecting autologous cells with genes encoding for the identified channels could be instrumental to augment EPC number and improve neovascularization. Consistent with this hypothesis, recruitment of PLC-β2 by the insulin-like growth factor 2 (IGF2) receptor is essential to ensure proper UCB-EPC homing to sites of hind-limb ischemia [52]. Further, CD133+ hematopoietic stem and progenitor cells isolated from PB and exposed to SDF-1α displayed a significantly reduced motility in a 3-dimensional collagen matrix migration assay upon pretreatment with BTP-2 [66]. In addition, SOCE-dependent Ca2+ signaling has been shown to drive the differentiation process of a number of both embryonic and adult stem cells [63,67 –69]. More specifically, SOCE inhibition prevented H19-7 hippocampal neuronal cells and osteoclast precursor cells from adopting their mature phenotype [68,69]. Indeed, store-dependent Ca2+ inflow may regulate the pattern of gene expression by selectively engaging a number of Ca2+-sensitive transcription factors, such as the nuclear factor of activated T-cells and nuclear factor-kappa B [16,17]. It will be interesting to assess whether downregulating SOCE will prevent EPC differentiation into mature ECs.
In conclusion, the present study provides the first evidence that both PB-EPCs and UCB-EPCs express store-dependent Ca2+ channels. In addition, PB-EPCs are endowed with both TRPC1/TRPC4 and STIM1/Orai1, which represent the most plausible molecular candidates to mediate SOCE in vascular endothelium. Finally, SOCE inhibition blocks PB-EPC proliferation, a finding that places such a pathway in the signaling network that controls EPC behavior [70]. Future work will be devoted to unveil the molecular underpinnings of SOCE to outline novel targets for EPC-based therapy.
Footnotes
Acknowledgments
The work was supported by FAR (Fondo Ateneo per la Ricerca) and CONACYT (Consejo Nacional de Ciencia y Tecnolog'ýa, Mexico) (grant no. 207991 to Y.S.-H.). The authors gratefully acknowledge Dr. Alessandra Fiorio Plà (University of Turin), Prof. Roberto Berra-Romani (University of Puebla), and Amparo Spezzia-Mazzocco for critical comments on western blot experiments, and Ms. Cinzia Bottino for performing some of the experiments described in ![]() .
.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.