
Abstract
The use of induced pluripotent stem cells (iPSCs) is an exciting frontier in the study and treatment of human diseases through the generation of specific cell types. Here we show the derivation of iPSCs from human nonmobilized peripheral blood (PB) and bone marrow (BM) mononuclear cells (MNCs) by retroviral transduction of OCT3/4, SOX2, KLF4, and c-MYC. The PB- and BM-derived iPSCs were quite similar to human embryonic stem cells with regard to morphology, expression of surface antigens and pluripotency-associated transcription factors, global gene expression profiles, and differentiation potential in vitro and in vivo. Infected PB and BM MNCs gave rise to iPSCs in the presence of several cytokines, although transduction efficiencies were not high. We found that 5 × 105 PB MNCs, which corresponds to less than 1 mL of PB, was enough for the generation of several iPSC colonies. Generation of iPSCs from MNCs of nonmobilized PB, with its relative efficiency and ease of harvesting, could enable the therapeutic use of patient-specific pluripotent stem cells.
Introduction
F
We previously reported that mouse bone marrow (BM) cells are highly susceptible to reprogramming [9]. It has been suggested that hematopoietic tissue cells are easily reprogrammed into iPSCs, but techniques for extracting human BM are highly invasive. It was recently reported that iPSCs can be generated from mobilized human CD34+ PB cells [10,11]. However, human CD34+ PB cells might not be an appropriate source of iPSCs in patients, because the pharmacological treatment used to mobilize the human hematopoietic stem cell compartment represents a health risk for the donor [12].
The growth of hematopoietic progenitor cells is known to be enhanced by activation of the gp130 signal-transducing molecule, a common signaling subunit of the interleukin (IL)-6 type cytokine family, though some hematopoietic cells express gp130 but not the IL-6 receptor α chain. The combination of soluble IL-6 receptor (sIL-6R) and IL-6 can stimulate gp130 in the absence of IL-6Rα and activate the Stat3 pathway [13 –15]. Further, fusion proteins of sIL-6R and IL-6 have much higher potency for activating the signal than does the combination [16,17]. Mouse embryonic stem cell (ESC) self-renewal depends on leukemia inhibitory factor (LIF)/signal transducer and activator of transcription (STAT3) signaling, whereas the signaling does not seem to be necessary for maintenance of human ESC (hESC) self-renewal. However, since the same transcription factors can reprogram both mouse and human somatic cells, it is still possible that a common transcriptional network governs stemness, but extrinsic factors in terms of pluripotency are different between mouse and human cells. Thus, we have tried to use cytokine combinations including fusion proteins of sIL-6R and IL-6 in the early phase of the iPS induction.
In this study, we show that nonmobilized PB mononuclear cells (MNCs) as well as BM MNCs are competent as donor cells and can be directly reprogrammed into iPSCs. We verified that these iPSCs corresponded nearly perfectly to human ES cells in molecular-expression profiles and in the capacity to form teratomas in immune-deficient mice. Generation of PB or BM MNC-derived iPSCs did not require long preculture and mobilization before harvesting PB. Here we established methods to generate iPSCs using less than 1 mL of PB. Hence, we suggest that PB MNCs could be an ideal adult cell source to generate patient-specific pluripotent cells for cell therapy applications and iPSC banks.
Materials and Methods
Cells and cell culture
BM and PB MNCs were obtained from Allcells (ABM007F and PB003F each). Frozen BM MNCs purchased from Allcells were collected from a 25-year-old male donor (lot#BM1705), a 25-year-old female donor (lot#BM1703), and a 27-year-old female donor (lot#BM1715). Frozen PB MNCs were collected from a 28-year-old female donor (lot#A1609), a 26-year-old male donor (#A1611), and a 27-year-old male donor (#A1628). Human embryonic kidney (HEK) 293 (ATCC: CRL-1537), HEK293T (CRL-11268), Jurkat (TIB-152), and Daudi (CCL-213) cells were acquired from ATCC; and a human multiple myeloma cell line, OPM-2 (ACC50) cells, were purchased from DSMZ. G3T-hi cells (Takara-Bio) were cultured and maintained with Dulbecco's modified Eagle's medium (DMEM; Sigma-Aldrich) containing 10% fetal bovine serum (FBS). Established iPSCs were cultured on irradiated mouse embryonic fibroblasts (MEFs) (purchased from Dainippon Sumitomo Pharma Co. or Kitayama Labes Co.) in human ES medium (DMEM/F12; Sigma-Aldrich) containing 20% Knock-out Serum Replacement (Invitrogen), human basic fibroblast growth factor (bFGF), penicillin/streptomycin, 2 mM L-glutamine, 0.1 mM beta-mercaptoethanol, and nonessential amino acid. For feeder-free culture of iPSCs, the plate was coated with 0.3 mg/mL Matrigel (BD-Bioscience); and mTeSR (StemCell Technologies) was used according to the manufacturer's instructions. All iPSCs could be maintained in feeder-free culture and continuously passaged by harvesting with 1 mg/mL dispase according to the manufacturer's instructions.
Viral constructs and production
The cDNA fragments for human OCT3/4, SOX2, KLF4, and c-MYC were amplified by reverse transcription-polymerase chain reaction (RT-PCR) and cloned into the retroviral pMXs vector [18,19]. The retroviral pMXs vector, pVSV-G vector, and pGP vector were transfected into G3T-hi cells (Takara-Bio) by FuGene6 (Roche Diagnostics) according to the manufacturer's instructions. Two days later, viral supernatants were harvested, combined, filtered through a 0.45-μm filter, and concentrated by centrifugation. Resuspended viruses with DMEM containing 10% FBS were added to the PB or BM MNCs culture for infection. pMXs/IRES–green fluorescent protein (GFP) vector was used as control and also transfected into G3T-hi cells for control GFP viruses.
Retroviral infection and iPSC induction
Two days before infection, PB or BM MNCs were plated at 5 × 105 cells per well of a 6-well nontissue culture treated dish and incubated in DMEM containing 10% FBS supplemented with 100 ng/mL human steel factor (SCF; Kyowa Hakko Kirin), 100 ng/mL human thrombopoietin (TPO; Kyowa Hakko Kirin), 100 ng/mL human IL-3 (Kyowa Hakko Kirin), and 20 ng/mL IL-6/IL-6 receptor fusion protein (FP6; Kyowa Hakko Kirin); or with 100 ng/mL human SCF, 100 ng/mL human TPO, 100 ng/mL human IL-3, 20 ng/mL FP6, 100 ng/mL human granulocyte-macrophage colony-stimulating factor (GM-CSF; Kyowa Hakko Kirin), 50 ng/mL human macrophage colony-stimulating factor (M-CSF; Invitrogen), and 100 ng/mL human flt3 ligand (Flt3/L; R&D Systems); or with 100 ng/mL human SCF, 100 ng/mL human TPO, 100 ng/mL human IL-3, 20 ng/mL FP6, 100 ng/mL Lipopolysaccharide (LPS; Sigma-Aldrich), 1 μg/mL anti-CD3 antibody (Janssen Pharmaceutical), 1 μg/mL anti-CD28 antibody (eBioscience), and 6 μg/mL anti-CD40 antibody (Kyowa Hakko Kirin). For infection, each well of a 12-well dish coated with a fragment of RetroNectin (Takara-Bio) was covered with virus-containing supernatants and centrifuged for 40 min. After the adhesion of viruses onto the surface of plates, 5 × 105 cells of PB and BM MNCs were inoculated into each well filled with DMEM containing FBS supplemented with SCF, TPO, and IL-3; or with SCF, TPO, IL-3, and FP6; or with SCF, TPO, IL-3, FP6, GM-CSF, M-CSF, and Flt3/L; or with SCF, TPO, IL-3, FP6, LPS, anti-CD3 antibody, anti-CD28 antibody, and anti-CD40 antibody. Two days after infection, cells were harvested with 0.05% trypsin 0.5 mM ethylenediaminetetraacetic acid in phosphate-buffered saline (PBS) and seeded on irradiated MEF in DMEM containing FBS supplemented with a combination of cytokines as described, or collected and analyzed for control GFP infection efficiency by fluorescence-activated cell sorting (FACS). Six days after the infection, the medium was replaced with hES medium as described above. Subsequently, the cells were incubated in ES medium; and the medium was changed every day. After 12–23 days, some granulated cell clusters were picked up and seeded on feeder MEFs in ES medium or feeder-free culture. Cloned ES-like colonies were collected for FACS analysis and used for various experiments.
Antibodies, FACS analysis, and immunocytochemistry
The following fluorescently conjugated antibodies were used for FACS analysis and immunocytochemistry: anti-human tumor-related antigen TRA1-60 (BD-Bioscience) conjugated with Alexa Fluor 647 (Invitrogen) according to manufacturer's instructions, anti-human TRA1-60 PE conjugated (BD-Bioscience), antistage-specific embryonic antigen SSEA-4 (BD-Bioscience) conjugated with Alexa Fluor 488 (Invitrogen) according to manufacturer's instructions, anti-SSEA-4 PE conjugated, anti-CD38 PE conjugated, anti-CD117 PE conjugated, anti-CD11b APC conjugated, anti-CD13 PE conjugated (BD-Bioscience), anti-CD45 APC conjugated, anti-CD45 PE conjugated, anti-CD3 PE conjugated, anti-CD19 PE conjugated (Immunotech), anti-CD34 APC conjugated, anti-CD34 PE conjugated (Beckman Coulter), anti-Oct3/4 (Santa Cruz Biotechnology), anti-Nanog (Cosmo Bio), anti-human alpha smooth muscle actin (α-SMA), anti-human alpha 1-fetoprotein (AFP), anti-human glial fibrillary acidic protein (GFAP), and antineurofilament 68 (Dako).
Anti-human TRA1-60 and anti-SSEA-4 were used for FACS analysis performed by using FACSCalibur or FACSAria (BD-Bioscience).
For immunocytochemistry, cells were fixed with PBS containing 4% paraformaldehyde for 15 min at room temperature. After washing with PBS, the cells were treated with PBS containing 0.1% Triton X for 5 min and 1% bovine serum albumin (Sigma-Aldrich) for 30 min at room temperature. Anti-Oct3/4, anti-Nanog, anti-human α-SMA, anti-human AFP, anti-human GFAP, and anti-neurofilament 68 antibodies were used for immunocytochemistry. For immunohistochemistry of paraffin-embedded and sliced teratoma, we used a Dako Envision kit (Dako) following the manufacturer's instructions. Nuclei staining with 4,6-diamidino-2-phenylindole (Sigma-Aldrich) was performed according to manufacturer's instructions.
In vitro differentiation
For embryoid body formation, human iPSCs were harvested using collagenase IV, transferred to a low-attachment dish, and cultured in hES medium without bFGF. Three days later, embryoid bodies were transferred to a gelatin-coated plate and cultured in the same medium with 100 μM ascorbic acid (Wako Chemical) or KNOCKOUT DMEM (Invitrogen) supplemented with 10% FBS, penicillin/streptomycin, 2 mM L-glutamine, 0.1 mM beta-mercaptoethanol, and nonessential amino acid. For mesoderm differentiation, iPSCs were maintained on a Matrigel-coated plate in mTeSR medium for 6 days; and the medium was replaced with RPMI1640 (Sigma-Aldrich) and B27 supplement (Invitrogen) medium (RPMI-B27) supplemented with the following cytokines: 100 ng/mL human recombinant activin A (R&D Systems) for 24 h, followed by 10 ng/mL human recombinant bone morphologenic protein 4 (R&D Systems) for 4 days. The medium was then exchanged for RPMI-B27 without any cytokines. The medium for each condition was changed every other day.
Teratoma formation
One quarter of iPSC suspension from a confluent 100 mm dish was injected into the testis of 10-to-14 week-old SCID mice (CB17; CLEA Japan). Eight and 9 weeks after injection, tumors were surgically removed from the mice. Samples were fixed with PBS containing 4% paraformaldehyde and embedded in paraffin. Paraffin-embedded tissues were sliced and stained with hematoxylin and eosin or subjected to histological analysis. Animal experiments were reviewed and approved by The Ethics Committee for Animal Experiments, Kyowa Hakko Kirin Co. Ltd., in full compliance with Japanese laws and regulations.
Determination of karyotypes
Karyotypes were determined with quinacrine-Hoechst staining at Chromocenter, Inc. iPSCs were pretreated with 0.25 mg/mL colcemid (Sigma-Aldrich) for 4 h, incubated with 0.075M KCl for 15 min, and then fixed with Carnoy's fixative.
Rearrangement analysis
Immunoglobulin heavy chain (IgH), T cell receptor β, (TCRβ), and T cell receptor γ (TCRγ) rearrangement analysis by PCR was performed by Mitsubishi Chemical Medience Co.
Gene expression analysis
Total RNA was reverse-transcribed using SuperScriptIII (Invitrogen) and the oligo dT primer. PCR was done with LATaq (Takara-Bio). Oligonucleotide primer sequences were previously described [6,20] and as follows: glyceraldehyde 3-phophate dehydrogenase forward, 5′-ACCACAGTCCATGCCATCAC -3′, and glyceraldehyde 3-phophate dehydrogenase reverse, 5′-TCCACCACCCTGTTGCTGTA -3′.
Total RNA for microarray analysis was labeled with Cy3. Samples were hybridized to a Whole Human Genome Microarray 4 × 44 K (Agilent Technologies) according to the manufacturer's protocol. Each sample was hybridized once with the 1-color protocol. Arrays were scanned with a Microarray Scanner System (Agilent Technologies), and processed signals were called by Feature Extraction ver. 10.5 (Agilent Technologies). We have deposited the microarray data to GEO DataSets with the accession number GSE20127. They were combined with data from the NCBI GEO database for the human ES cell lines (GSM194390, GSM194391, GSM194392 [21], and GSM336012 [22]), human iPSC lines 201B7 (GSM241846) [6], clone 1-8 (GSM257570), clone 2-4 (GSM257572) and clone 3-2 (GSM257574) [20], and human adult fibroblasts (GSM336057 [22] and GSM242095 [6]); and they were normalized by the quantile method followed by the baseline correction to median of all samples using GeneSpring GX11 (Agilent Technologies). For hierarchical clustering, we selected 660 probes that were greater than or equal to 256-fold changes to median of all the samples. Two-dimensional hierarchical clustering on entities and conditions were performed with similarity measure by Euclidean distance and average linkage.
Southern blotting
Genomic DNA was isolated with a Puregene DNA purification Kit (Gentra Systems). Each DNA sample was digested with PstI or HindIII restriction enzymes (Takara-Bio), electrophoresed on a 0.7% agarose gel, transferred to nylon membranes (Hybond-N+; GE Healthcare), and hybridized with 32P-labeled probes. Probes were detected using a Typhoon imaging system (GE Healthcare). The probes were generated as previously described [6].
Bisulfite genomic sequencing
Bisulfite treatment of gDNA was carried out using a EpiTect Bisulfite Kit (Qiagen) according to the manufacturer's protocol. Converted gDNA was amplified by PCR using primers within the OCT3/4 and NANOG promoters as previously described [23]. PCR products were cloned using TOPO TA cloning (Invitrogen).
Results
Generation of iPSCs from BM MNCs
We first examined whether the combinations including sIL-6R/IL-6 fusion protein (FP6) could enhance human hematopoietic cell reprogramming. We introduced retroviral constructs driving the expression of human Oct3/4, Sox2, Klf4, and c-Myc (4 defined factors) into BM MNCs (5 × 105 cells). BM MNCs were cultured in DMEM containing FBS supplemented with SCF, TPO, and IL-3 (designated as ST3) or SCF, TPO, IL-3, and FP6 (designated as ST3FP6) for 2 days before viral infection and were infected with the retroviruses of the 4 defined factors for 2 days. Two days after transduction, we reseeded cells onto MEFs. At this time, we analyzed cells infected with control GFP virus using FACS (Fig. 1A; Supplementary Fig. S1, available online at

Generation of iPSCs from BM MNCs.
Generation of iPSCs from PB MNCs
We next attempted to generate iPSCs from PB MNCs using a similar process as was used for BM MNCs. To generate iPSCs from PB MNCs, we introduced retroviruses containing 4 defined factors into 5 × 105 PB MNCs in the presence of ST3FP6 or ST3 stimulation as well as the transduction to BM MNCs. Control GFP transduction efficiency was quite low (Fig. 2A). Nine to 14 days after the transduction, 3 small adherent colonies containing round-shaped cells appeared in the culture with ST3FP6, but the number of colonies was smaller than BM MNCs. We picked and transferred them onto MEFs or Matrigel-coated dishes without feeder cells. Twenty days after the transduction, we obtained colonies with hESC-like morphology from the ST3FP6 culture but not ST3 (Fig. 2C, Table 1). Isolated hESC-like colonies from PB MNCs were designated as PB iPSCs. Further, we could generate only 1 colony from a culture in the absence of any cytokines in all tests (11 times tried for generation of iPSC from a culture in the absence of any cytokine). We then characterized clones as either FP#1 iPS or Nega#8 iPS, indicating that they were generated from PB MNCs cultured with ST3FP6 or without cytokines, respectively (Fig. 2C). Both clones positively express Oct3/4, Nanog, SSEA4, and TRA1-60 (Fig. 2D, E).
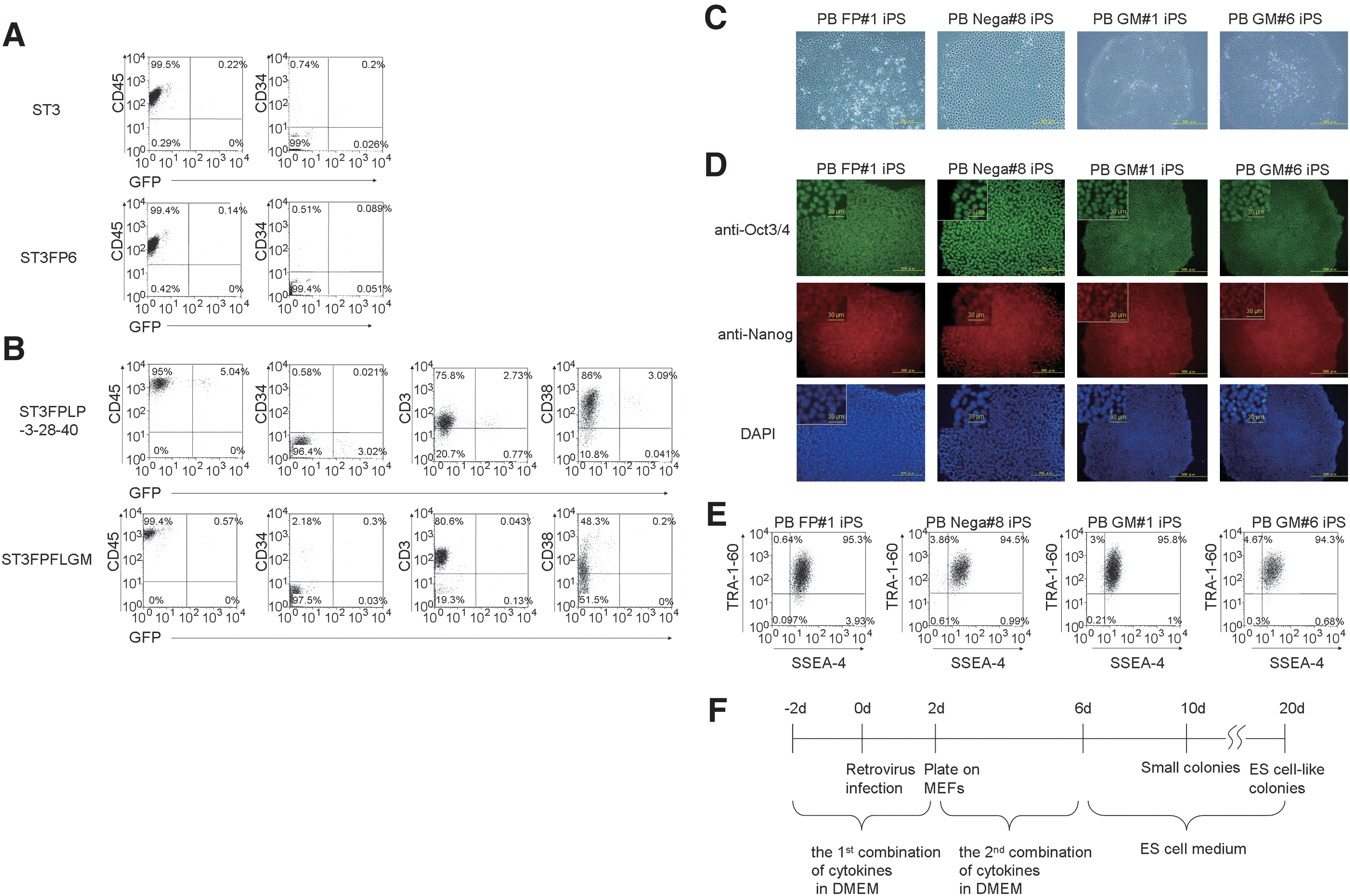
Generation of iPSCs from PB MNCs. FACS analysis of PB MNCs after infection of control GFP retrovirus.
Since the frequency of the cytokine combination–responsive hematopoietic cells might be quite low in the PB, we examined whether combinations of additional cytokines could improve reprogramming efficiency of PB MNCs. In addition to ST3FP6, LPS, anti-CD3 antibody, anti-CD28 antibody, and anti-CD40 antibody (designated as ST3FPLP-3-28-40), or human Flt3/L, human GM-CSF, and human M-CSF (designated as ST3FPFLGM), slightly increased the control GFP transduction to PB MNCs (Fig. 2B). Since the former combination of cytokines is linked with proliferation and expansion of lymphoid cells, most of the cells expressing control GFP were CD45+CD3+ T lymphocytes (CD34−) when PB MNCs were cultured and infected with control GFP retroviruses in vitro with ST3FPLP-3-28-40. On the other hand, when PB MNCs were cultured and infected with control GFP retroviruses in vitro with ST3FPFLGM as well as ST3 or ST3FP6, most of the control GFP-expressing cells were nonlymphoid CD45+ leukocytes that partially expressed CD34 (Fig. 2B; Supplementary Fig. S2, available online at
We then performed viral transduction of 4 defined factors for PB MNCs in the presence of ST3FPFLGM or ST3FPLP-3-28-40 cytokines. PB MNCs were cultured with the cytokine combinations for 2 days before viral transduction and then infected with viruses of the 4 defined factors in DMEM containing FBS supplemented with ST3FPFLGM or ST3FPLP-3-28-40 for an additional 2 days (the first combination of cytokines). Two days after transduction, we reseeded cells onto MEFs in the same medium with ST3FP6 (the second combination of cytokines) until day 6 after the transduction, and the medium was replaced with hES medium (Fig. 2F). We obtained some small attached colonies and hESC-like colonies from PB MNCs stimulated with ST3FPFLGM but not ST3FPLP-3-28-40 (Table 1). After picking and cloning these colonies, we characterized 2 lines: PB GM#1 iPS and PB GM#6 iPS (iPSC clones generated from PB MNCs cultured with ST3FPFLGM at the first combination of cytokines) (Fig. 2C). They have hESC-like morphology, as do the other PB iPSCs. They were positive for Oct3/4 and Nanog in immunofluorescence staining and SSEA4 and TRA1-60 in FACS (Fig. 2D, E).
To confirm which combination of cytokines was superior for generating hESC-like colonies from PB MNCs, we examined several combinations at a time (Table 1). We found that the most colonies were generated from PB MNCs cultured with ST3FPFLGM cytokines first and subsequently ST3FP6 (Supplementary Fig. S3, available online at
Marker expression and promoter methylation analysis in human iPSCs from BM and PB MNCs
We further confirmed the clonal origin and the characters of 5 iPSC clones: BM FP#17 iPS, PB FP#1 iPS, PB Nega#8 iPS, PB GM#1 iPS, and PB GM#6 iPS. Southern blot analyses using probes recognizing OCT3/4, SOX2, KLF4, and c-MYC confirmed the independent origin of all assayed iPSC clones, because each iPSC clone has a unique transgene integration pattern (Fig. 3A). Individual clones contained zero to 5 copies of each transgene. Notably, the absence of KLF4 retroviral integrations in BM FP#17 iPS suggests that Klf4 was not necessarily required for reprogramming of hematopoietic cells.
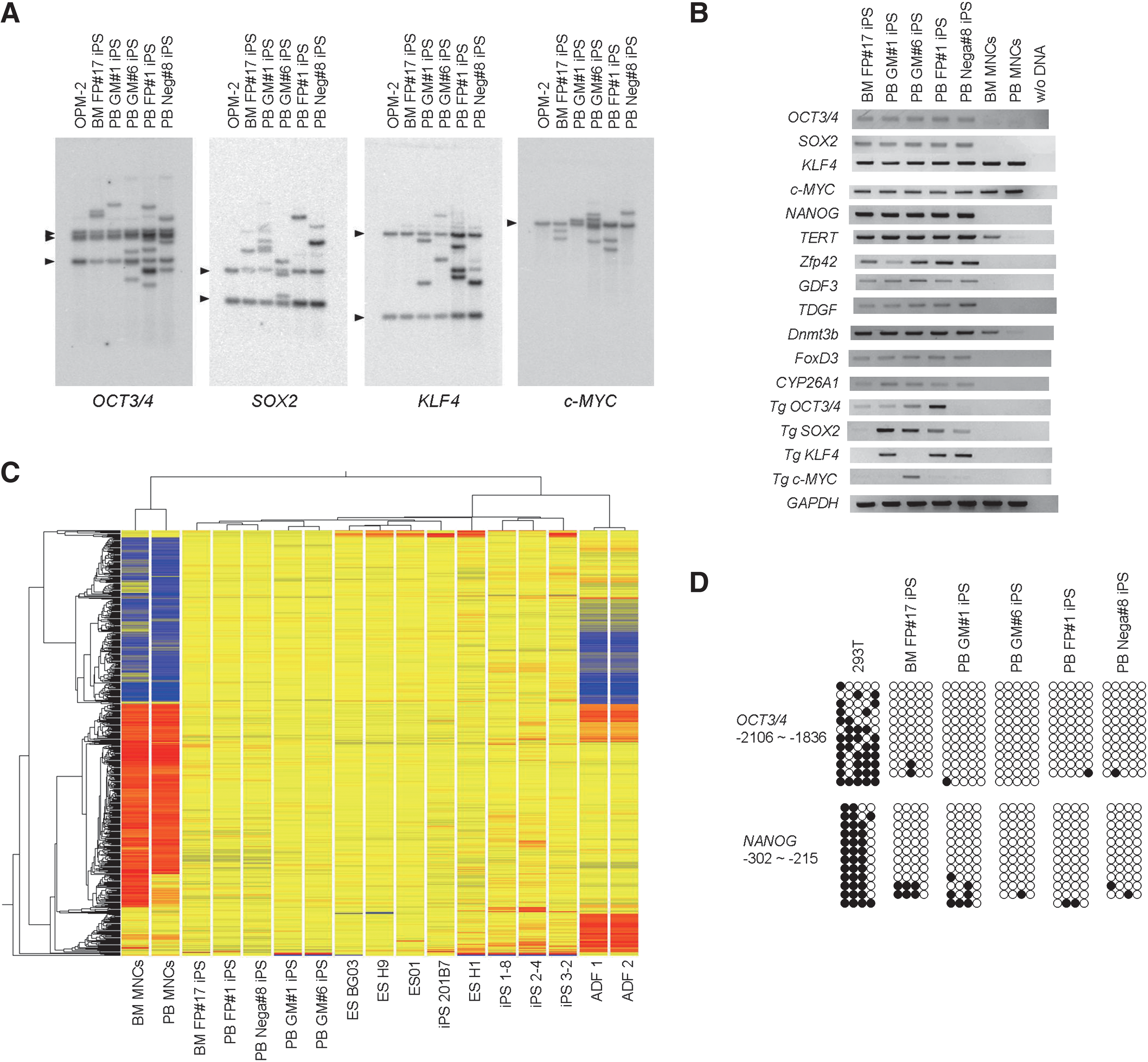
Characterization of BM and PB iPSC clones.
RT-PCR showed that all BM and PB iPSC lines highly expressed endogenous OCT3/4, SOX2, KLF4, and c-MYC as well as the ES cell marker genes NANOG, GDF3, TDGF1, FOXD3, DNMT3b, ZFP42, CYP26A1, and hTERT, as compared with those in BM and PB MNCs (Fig. 3B).
We next carried out genome-wide transcriptional profiling of BM MNCs, PB MNCs, human fibroblasts, hESCs, and BM and PB iPSC clones using microarray technology. Hierarchical analysis revealed that both BM and PB iPSC clones were clustered with human ES cell lines and fibroblast-derived iPSCs but with neither BM nor PB MNCs (Fig. 3C). These data support similarities among our 5 human iPSC clones and the other human pluripotent stem cell lines.
Bisulfite sequencing was performed to determine the status of DNA methylation at OCT3/4 and NANOG promoter regions in BM and PB iPSC clones. The results show that DNA methylation at both promoter regions was present in HEK293T cells whereas BM and PB iPSC clones displayed widespread demethylation at both promoter regions (Fig. 3D).
Cytogenetic analysis showed that each iPSC clone had a normal karyotype (Fig 1H; Supplementary Fig. S4, available online at
Pluripotency of BM and PB iPSCs
To test the pluripotency of iPSCs, we tested BM and PB iPSC clones in assays of differentiation in vitro and teratoma induction in vivo. All iPSC lines differentiated in vitro into all 3 germ layers including ectoderm, mesoderm, and endoderm derivatives that stained positive for neurofilament, α-SMA, and AFP immunoreactivity, respectively (Supplementary Fig. S5, available online at
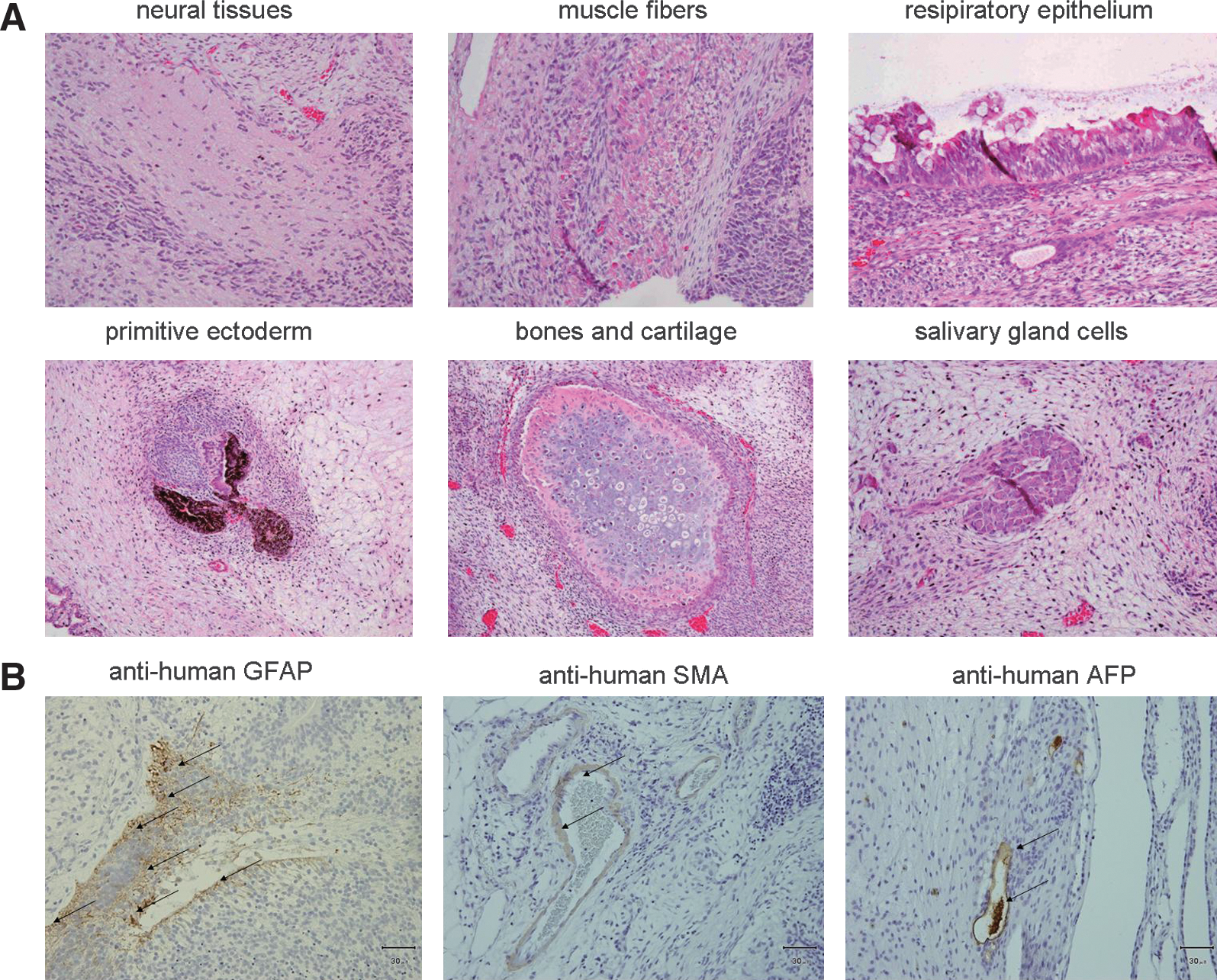
Differentiation potential of BM and PB iPSC.
Germline IgH, TCRβ, and γ loci in BM and PB iPSCs
Hematopoietic tissues include many types of cells such as leukocytes, vascular progenitors, and mesenchymal cells. To confirm whether or not our iPSC clones were derived from mature lymphocytes, we characterized the genomic rearrangements of the TCRβ, TCRγ, and IgH loci in BM and PB iPSC clones by PCR (Table 2) [24]. The V-J and D-J rearrangements of TCRβ alleles and the V-J rearrangements of TCRγ alleles were observed in genomic DNA of PB Nega#8 iPS as well as in that of Jurkat cells (TCR alleles rearrangements control samples); whereas any rearrangements of TCRβ, TCRγ, and IgH loci were not observed in genomic DNA of BM FP#17, PB FP#1, and PB GM#6 iPS as well as that of HEK293 cells (embryonic type genome control samples). These results suggest that BM and PB iPSCs, established under the stimulation of ST3FP6 and ST3FPFLGM, tended to be derived from nonlymphoid cells of hematopoietic tissues and harbored an embryonic type of genome in IgH and TCR loci.
BM, bone marrow; PB, peripheral blood; HEK, human embryonic kidney.
Discussion
The present study demonstrates that normal human BM and PB MNCs can be reprogrammed into human iPSCs, without the mobilization or the enrichment of any kind of cells, and it took less time than iPSC derivation from dermal fibroblasts. We estimate that as little as 500 μL of collected PB is enough for the generation of several iPSC colonies, an amount that seems easily accessible and minimally invasive, suggesting that our methods could be applied to various patients with less cost and difficulty. Several groups have reported that inactivation of the Ink4/Arf locus or suppression of the p53-p21 pathway using p53 short interfering RNA elevated the reprogramming efficiency. Some small molecules were also reported to improve the efficiency. A combination of those methods and ours might increase the number of iPSC colonies derived from PB MNCs [22,25 –30].
We have previously shown that mouse BM MNCs could be reprogrammed into mouse iPSCs, which gave rise to adult chimeras with germline contribution, in a shorter time and with higher efficiency compared with MEFs [9]. In this study, we succeeded in the generation of iPSCs from hematopoietic tissues in spite of low retroviral infection efficiency. In a recent report, it was shown that myeloid and multiple progenitors might be more susceptible to reprogramming than lymphocytes and mature myeloid cells [31,32]. Considering these results, it has been suggested that some hematopoietic cells included in both BM and PB MNCs might be efficiently reprogrammed into iPSCs. The cytokine combinations, including FP6, might be able to stimulate those susceptible hematopoietic cells. We did not figure out which type of cells in BM or PB was the most susceptible to reprogramming, although we obtained more BM iPSC colonies with the stimulation of ST3FP6 than with ST3, and more PB iPSC colonies were obtained with ST3FPFLGM than with ST3. The control infection revealed that most of the infected BM and PB MNCs expressed CD45, CD34, and CD38 antigen under the stimulation of those cytokines. These results imply that the susceptible cells might be highly concentrated in CD34+-enriched cells in mobilized PB [10,11]. In addition, our BM or PB iPSC clones, except for PB Nega#8, did not show intrinsic genetic DNA rearrangements of IgH, TCRβ, and TCRγ loci. BM or PB iPSC generation using ST3FP6 or ST3FPFLGM cytokines is likely to harbor embryonic-type immunoglobulin and TCRs.
Readily available frozen tissues or less-invasive adult tissues enable us to set up the field of disease research and clinical medicine. Although the success of the iPSC generation from various tissues such as keratinocytes [7], ASCs [33], cord blood [30,34,35], and PB [10,11] increase the opportunities for research and development, the identity among iPSCs derived from various tissues is still not clear. Although the efforts to establish standardization of iPSCs are neccesary, iPSCs derived from much less invasive tissues could hasten the development of comprehensive clinical applications of iPSCs.
Footnotes
Acknowledgments
We are indebted to all members of Frontier Laboratory of Kyowa Hakko Kirin for helpful discussions of the results. We are especially grateful to Dr. Ikegami for valuable discussions on the histological data. We thank Dr. Toshio Kitamura for the pMXs retrovirus vectors.
Author Disclosure Statement
A.K., M.W., H.S., T.O, I.I., and K.N. are (or formerly were) employees of Kyowa Hakko Kirin Co. Ltd. in the area of stem cell research.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.