
Abstract
Many studies have shown that it is possible to use culture conditions to direct the differentiation of murine embryonic stem (ES) cells into a variety of cell types, including cardiomyocytes and neurons. However, the molecular mechanisms that control lineage commitment decisions by ES cells remain poorly understood. In this study, we investigated the role of the 3 major mitogen-activated protein kinases (MAPKs: extracellular signal-regulated kinase, c-Jun N-terminal kinase, and p38) in ES cell lineage commitment and showed that the p38 MAPK-specific inhibitor SB203580 blocks the spontaneous differentiation of ES cells into cardiomyocytes and instead induces the differentiation of these ES cells into neurons. Robust p38 MAPK activity between embryoid body culture days 3 and 4 is crucial for cardiomyogenesis of ES cells, and specific inhibition of p38 MAPK activity at this time results in ES cell differentiation into neurons rather than cardiomyocytes. At the molecular level, inhibition of p38 MAPK activity suppresses the expression of bmp-2 mRNA, whereas treatment of ES cells with bone morphogenetic protein 2 (BMP-2) inhibits the neurogenesis induced by SB203580. Further, luciferase reporter assays and chromatin immunoprecipitation experiments showed that BMP-2 expression in ES cells is regulated directly by the transcription factor myocyte enhancer factor 2C, a well-known substrate of p38 MAPK. Our findings reveal the molecular mechanism by which p38 MAPK activity in ES cells drives their commitment to differentiate preferentially into cardiomyocytes, and the conditions under which these same cells might develop into neurons.
Introduction
M
The extracellular signal-regulated kinases (ERKs), c-Jun N-terminal kinases (JNKs), and p38 kinases are the 3 major groups of mitogen-activated protein kinases (MAPKs) found in mammals [6,7]. ERK1 and ERK2 are widely expressed and involved in the regulation of meiosis, mitosis, and postmitotic functions in differentiated cells. In contrast, the JNK enzymes are important for controlling cell survival and apoptosis. The p38 kinases, which regulate the expression of many cytokines, were first identified in lipopolysaccharide-stimulated murine macrophages and in a screen for drugs able to inhibit tumor necrosis factor-α-mediated inflammatory responses in human monocytes [8,9]. Immune system cells that encounter inflammatory cytokines respond by activating p38, and this MAPK then supports the activation of immune responses. Four different p38 isoforms, p38α, p38β, p38γ, and p38δ, have been identified in mammalian cells [10]. The p38α and p38β isoforms are expressed in murine heart, whereas p38γ and p38δ are expressed at low levels in this organ. Deletion of the p38α gene in mice leads to early embryonic lethality between E11.5 and E12.5 [11,12]. On the other hand, p38β gene-targeted mice are viable and exhibit no apparent health problems [13]. It has been demonstrated that p38α associates with the transcription factor myocyte enhancer factor 2C (MEF2C), which is a member of the MADS-box family [14]. Phosphorylation of MEF2C by p38 stimulates MEF2C's ability to activate transcription of its target genes. In mammals, there are 4 MEF2 family genes, namely MEF2A, MEF2B, MEF2C, and MEF2D, which form homo- and heterodimers and bind to the DNA consensus sequence CTA(A/T)4TA(G/A) [15,16]. This sequence is found in the promoter regions of numerous muscle-specific genes, as well as in genes induced by growth factors or stress. A major role of the MEF2C protein is to regulate muscle-specific gene expressions. For example, loss-of-function mutations in myocyte enhancer factor 2C (mef2c) severely disrupt early cardiogenesis [17] and vascular development [18], suggesting that MEF2C may be critical for cardiomyogenesis by ES cells. However, the role of MEF2C in neural commitment is unknown.
Several extracellular signaling pathways are important for both embryonic and tissue stem cell determination, including pathways involving the Wnt proteins and the bone morphogenetic proteins (BMPs) [19,20]. However, a detailed understanding of the molecular mechanisms underlying the regulation of stem cell fate by these extracellular factors is lacking. BMPs are members of the transforming growth factor-β (TGF-β) superfamily and are known to function in the development and regulation of a wide range of biological systems. These extracellular ligands were originally isolated as components of bone extracts that induced ectopic cartilage and bone formation when implanted in muscle [21]. However, BMPs have since been demonstrated to function in multiple developmental processes, including dorsoventral patterning within the neural tube, the induction of mesoderm during gastrulation, and hematopoiesis [22]. As might be expected from these complex in vivo functions, BMPs also play key roles in regulating fate choices during tissue stem cell differentiation. For example, BMPs direct mesenchymal stem cells to differentiate into chondrogenic and osteogenic cell lineages [20]. BMPs have also been shown to regulate fate choices in neural crest stem cells [23].
In this study, we demonstrate that p38 MAPK controls an ES cell fate choice between cardiomyocytes and neurons. Further, our results show that this choice is mediated by the action of BMP-2, whose transcription is directly regulated by the p38 MAPK substrate MEF2C.
Materials and Methods
Cell culture and ES cell differentiation
Feeder cell-independent E14K ES cells were maintained on a gelatin-coated dish with Dulbecco's modified Eagle's medium (Gibco) containing 15% fetal bovine serum (FBS) (Hy-Clone, Lot No. AMC15813), 0.1% 2-mercaptoethanol (Sigma), and 1000 U/mL LIF (propagation medium), as described previously [24 –27]. To induce cardiomyocyte differentiation, LIF was removed from the propagation medium and 3×103 ES cells suspended in a 25 μL hanging drop. The drop was placed on the lid of an inverted bacterial Petri dish so that the cells would eventually attach and form embryoid bodies (EBs). After 2 days (on day 3), the EBs were collected and transferred into a bacterial Petri dish. After 4 days of suspension culture (on day 7), the EBs were plated on a gelatin-coated tissue culture dish. Areas of tissue showing a spontaneous “heartbeat” were readily detected on day 12.
For MAPK inhibition experiments, SB203580 (10 μM; Calbiochem), U0126 (10 μM; Promega), SP600125 (5 μM; Biomol), or wortmannin (1 μM; Wako) was added to EB cultures on day 1. For BMP-2 experiments, recombinant human BMP-2 (3 ng/mL; R&D Systems) or BMP-2 antagonist Noggin (100 ng/mL; R&D Systems) was added to EB culture on days 4–6.
Microscopic analysis of cardiomyocytes and neurons
Individual EBs, prepared as described earlier, were plated onto gelatin-coated 96-well tissue culture plates on day 7. The numbers of spontaneously beating EBs and EBs with neurite outgrowths were counted on day 12 under a phase-contrast microscope. Data were expressed as the percentage of the total number of EBs plated.
Immunofluorescence and immunohistochemistry
Immunofluorescence staining was performed as described previously [26]. For immunohistochemistry (IHC), EBs were fixed in 4% paraformaldehyde (PFA)–phosphate-buffered saline (PBS) for 2 h at 4°C, washed sequentially with PBS, 10% sucrose/PBS/0.02% NaN3, 15% sucrose/PBS/0.02% NaN3, and 20% sucrose/PBS/0.02% NaN3, and embedded in OCT compound (Tissue Tek) with liquid nitrogen. Frozen sections were cut at 10 μm and placed on 3-aminopropyltriethoxy-silane (APES)-coated slides. After air drying, sections were fixed in acetone at room temperature for 10 min, rinsed in PBS, and incubated in 0.3% H2O2/PBS for 30 min to block endogenous peroxidases. After preincubation with blocking solution (5% bovine serum albumin/PBS/0.1% Tween 20) for 1 h, slides were incubated overnight at 4°C with a 1:800 dilution of anti-α-actinin antibody (cardiac specific) or a 1:500 dilution of anti-TuJ-1 (neuron-specific class III β-tublin) antibody. After 3×5 min washes in PBS/0.05% Tween 20 (PBST), sections were incubated with biotinylated secondary antibodies (Vectastain Elite ABC Kit) for 2 h. Slides were washed again in PBST and incubated for 1 h with Vectastain Elite ABC Reagent. Following a last wash in PBST, sections were incubated in 3,3′-Diamino-benzidine (DAB) solution (200 μg/mL DAB, 0.015% H2O2/PBS) until a color change was observed (2–10 min), and slides were rinsed in PBS. Finally, sections were counterstained with hematoxylin at room temperature for 1 min, washed, dehydrated, mounted, and inspected using a phase-contrast microscope.
Reverse transcriptase–polymerase chain reaction analysis
ES cells, EBs, or mouse organs (brain, heart, liver) from E12.5 mouse embryos were lysed with Trizol reagent (Invitrogen), and first-strand cDNA was synthesized using SuperScript III RNase H-reverse transcriptase (Invitrogen). The primers used in polymerase chain reactions (PCRs) are shown in Supplementary Table S1 (available online at
Western blotting analysis
EBs were lysed and fractionated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and immunoblotted with antibody against total p38 (C-20; Santa Cruz Biotechnology) or phospho-p38 MAPK (Thr180/Tyr182) (Cell Signaling Technologies, No. 9211). Horseradish peroxidase-conjugated goat anti-rabbit IgG was used as the secondary antibody. Bands were observed using SuperSignal West Pico Chemiluminescent Substrate (Pierce) for total p38, or SuperSignal West Femto maximum sensitivity substrate (Pierce) for phospho-p38, as described previously [28].
Luciferase reporter assays
A proximal promoter region (−1703/−1) of mouse bmp-2 containing the MEF2 binding site was amplified by PCR using the upstream primer 5′-CGACGCGTCTGTCCAGAGGCATCCATTT-3′ and the downstream primer 5′-CGCTCGAGAACACCTCCCCCTCGGA-3′. The sequence was confirmed and cloned into the pTAL-Luc reporter vector (pTAL-BMP-2-Luc). HeLa cells were cotransfected with pTAL-BMP-2-Luc and pcDNA3-Mef2c expressing MEF2C using FuGENE 6 transfection reagent (Roche Molecular Biochemicals). Luciferase activity was assayed at 24 h after transfection using the dual-luciferase reporter assay system (Promega) following the manufacturer's protocols.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) assays were performed according to published protocols from Cosmo Bio (
Results
The p38-specific inhibitor SB203580 and the ERK-specific inhibitor U0126 block spontaneous ES cell cardiomyogenesis
To identify the kinases most important for ES cell lineage commitment, we treated EBs with the ERK-specific inhibitor U0126, the JNK-specific inhibitor SP600125, the p38 MAPK-specific inhibitor SB203580, or the PI3K-specific inhibitor wortmannin. The inhibitors were applied to the EB cultures for the interval spanning days 1–6 during the EB differentiation process. We found that only the ERK-specific inhibitor U0126 and the p38 MAPK-specific inhibitor SB203580 blocked spontaneous cardiomyocyte differentiation, and that this block was so profound that fewer than 5% of EBs contained beating foci at day 12 (Fig. 1A). In contrast, more than 90% of untreated control EBs contained beating areas, which were confirmed as cardiac in commitment by phenotypic testing. Intriguingly, more than 90% of SB203580-treated EBs (but not U0126-treated EBs) continued to exhibit prominent outgrowths even though cardiomyogenesis was inhibited (Fig. 1B). These outgrowths stained positively with an anti-βIII-tubulin antibody specific for neurons, in contrast to the negative staining displayed by untreated EBs and EBs treated with U0126 (Fig. 1C, D). These results demonstrate that SB203580 blocks cardiomyocyte differentiation and induces neural differentiation, but that neural differentiation does not depend solely on the inhibition of cardiomyogenesis.

Effects of specific mitogen-activated protein kinase inhibitors on embryonic stem (ES) cell differentiation. (
SB203580-mediated inhibition of p38 MAPK blocks cardiomyogenesis and commits ES cell differentiation to the neuronal lineage
To confirm that SB203580 had a switch effect on cardiac versus neural ES cell differentiation, frozen sections from EBs that had been untreated or treated with SB203580 between days 1 and 6 were subjected to IHC at day 12. SB203580-treated EBs did not stain positively with an antibody recognizing the cardiac-specific marker α-actinin, but did stain with an anti-TuJ-1 antibody specific for neurons (Fig. 2A). RT-PCR analysis of a set of embryonic genes revealed that SB203580 treatment completely inhibited the mRNA expression of the cardiac-associated mef2c, α-cardiac myosin heavy chain (mhc), and myosin light chain 2v (mlc2v) genes (Fig. 2B), but induced significant increases in the mRNA levels of the neuronal lineage genes nestin, hairy and enhancer of split 5 (hes5), mammalian achate schute homolog 1 (mash1), mouse atherosclerosis 3 (math3), and microtubule-associated protein 2 (map2) (Fig. 2C).
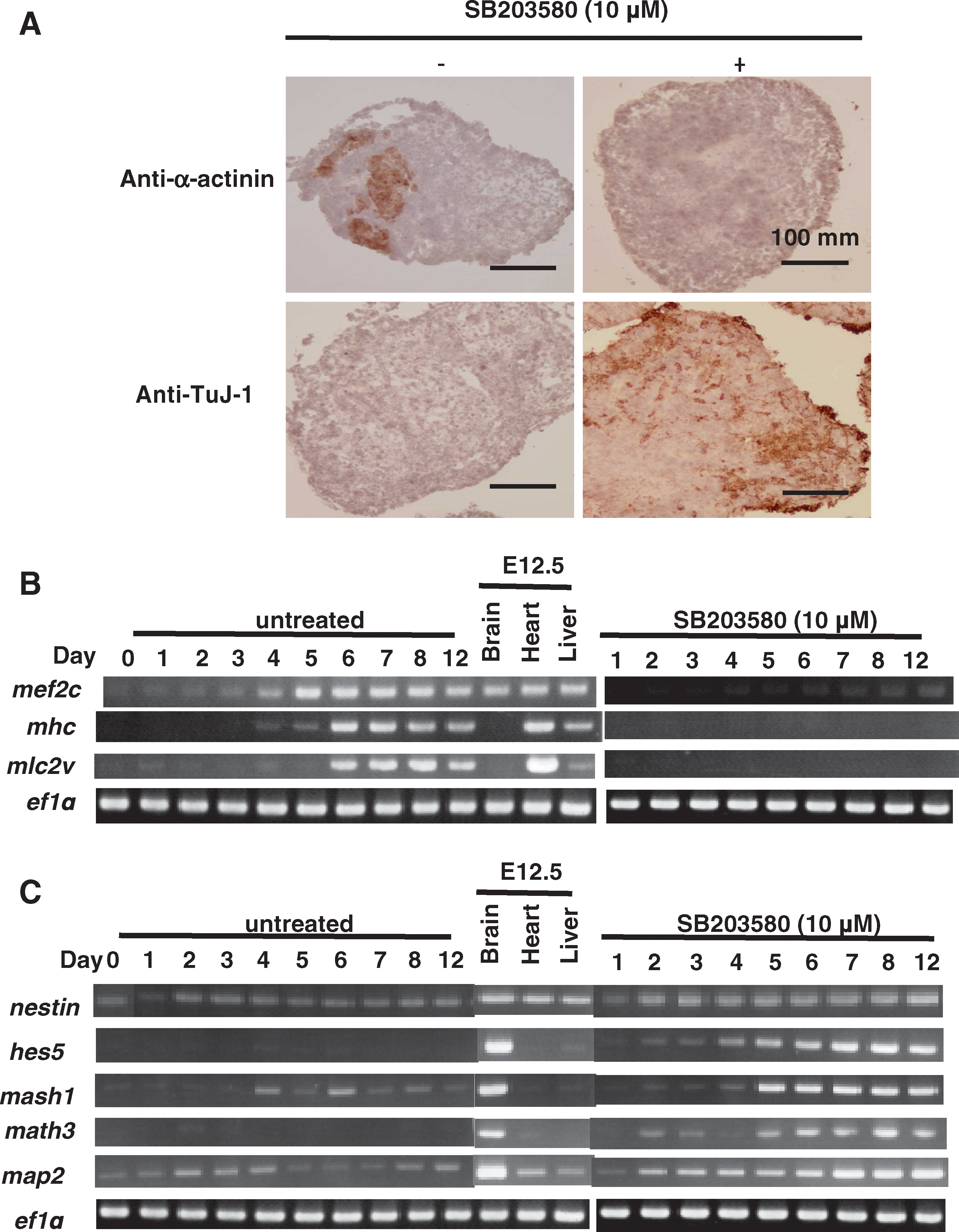
SB203580-mediated inhibition of p38 mitogen-activated protein kinase blocks cardiomyogenesis and commits ES cells to the neuronal lineage. (
p38 MAPK activity between days 3 and 4 serves as a switch determining cardiac or neural commitment of ES cells
To define the role of p38 MAPK in ES cell commitment, we used immunoblotting to measure p38 MAPK activation during the earliest stages of ES cell differentiation. At day 0, when ES cells were cultured as a monolayer, no detectable phospho-p38 MAPK (activated enzyme) could be detected in whole cell lysates. However, after EB formation at day 2, high levels of phospho-p38 MAPK spontaneously appeared and were maintained until day 6; total p38 MAPK protein levels were not affected (Fig. 3A). To determine at what time point p38 MAPK acts during ES cell differentiation, we treated EBs with SB203580 for specific time intervals. As shown in Fig. 3B, when EBs were exposed to SB203580 between days 3 and 4, neuron differentiation was promoted at the expense of cardiomyocyte differentiation, an effect replicated by SB203580 treatment from day 0 to 6. In contrast, exposure to SB203580 for other intervals did not interfere with spontaneous cardiomyocyte generation and did not induce neurogenesis. To examine dose-dependent effects of SB203580 on ES cell differentiation, we treated EBs with various concentrations of SB203580. As shown in Fig. 3C, when EBs were exposed to SB203580 between days 3 and 6, neuron differentiation was promoted at the expense of cardiomyocyte differentiation in a dose-dependent manner. RT-PCR analysis confirmed that the expression of the neuronal markers hes5 and map2 was induced only when SB203580 was applied to EBs between days 3 and 4, or between days 1 and 6, whereas expression of the cardiomyocyte-specific genes mhc and mlc2v was strongly decreased at these times (Fig. 3C). These results clearly demonstrate that p38 MAPK activity spanning days 3 and 4 is critical for the cardiac commitment of ES cells, and that interference with this p38 MAPK activity drives neurogenesis instead.
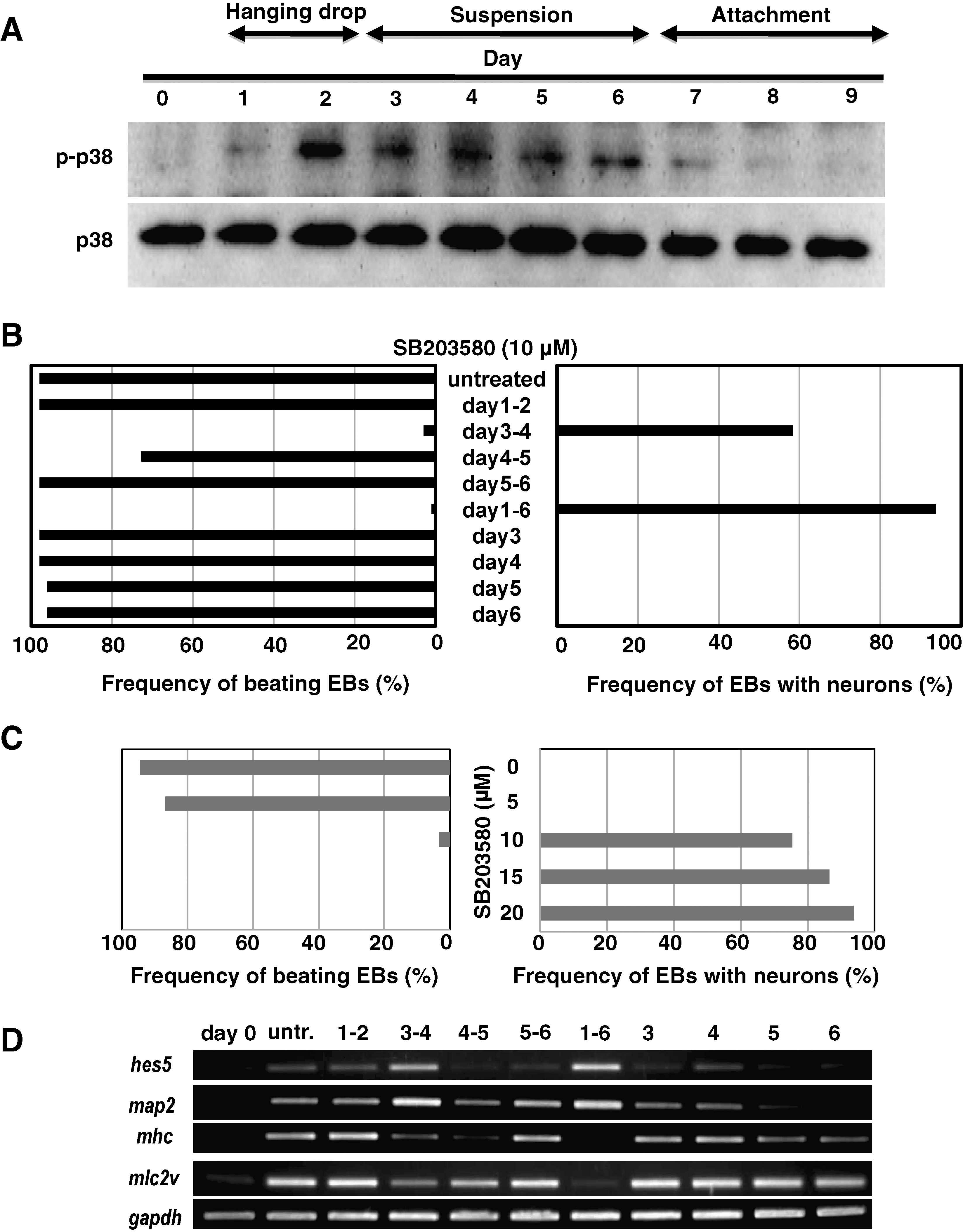
p38 mitogen-activated protein kinase (MAPK) activity spanning days 3 and 4 serves as a switch determining cardiac or neuronal commitment of ES cells. (
p38 MAPK controls the expression of BMP-2 during ES cell differentiation
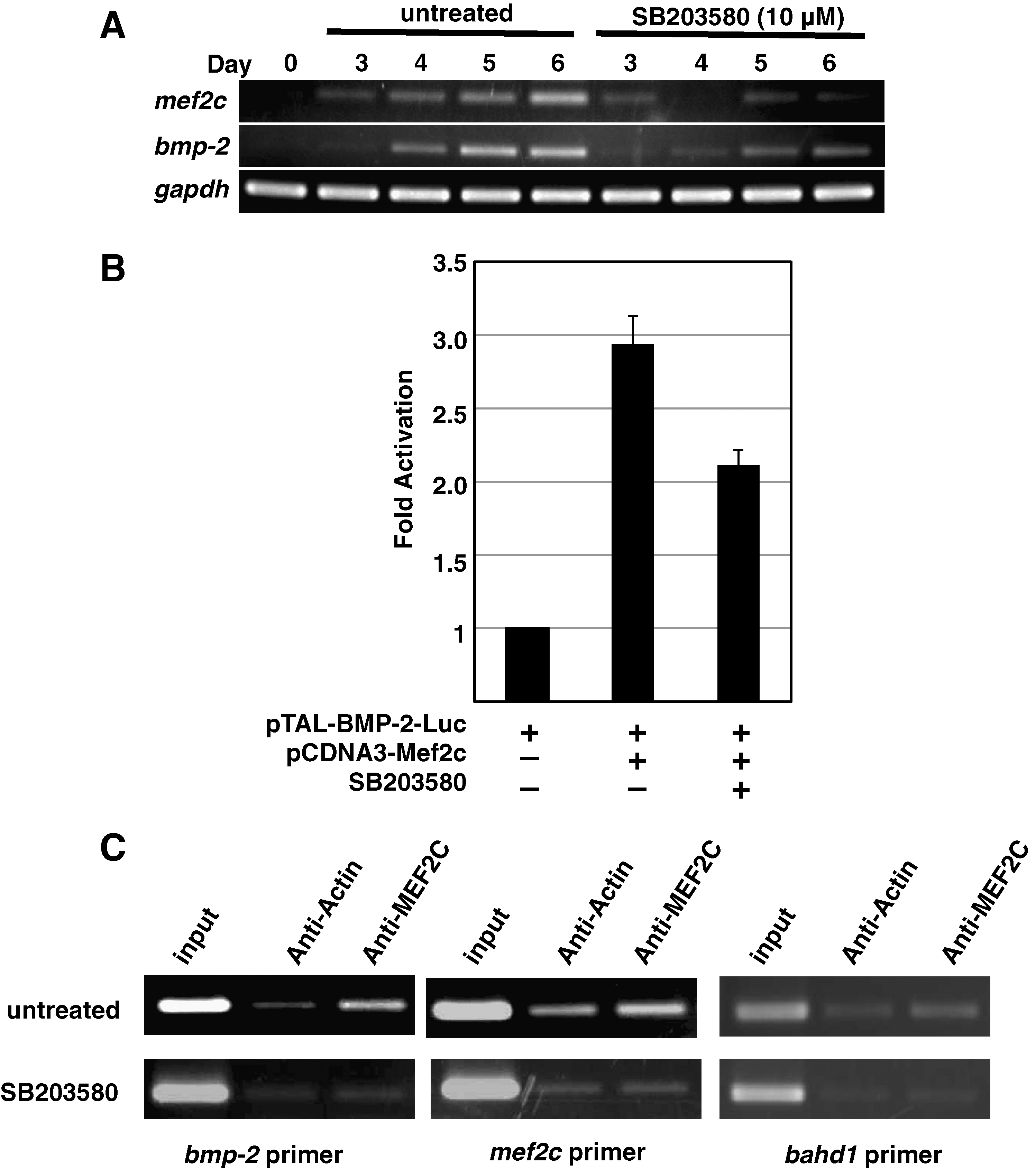
To elucidate the molecular mechanism by which p38 MAPK governs cell fate choices in ES cells, we sought to identify relevant downstream targets of this kinase. Because p38 MAPK activity spanning days 3 and 4 was found to be crucial for lineage commitment, we investigated whether the mRNA expression of genes known to be involved in ES cell fate decisions was affected by treating the EBs with SB203580 for this interval. Notably, RT-PCR analysis showed that the expression of the bmp-2 mRNA, which normally commences on day 4, was decreased by SB203580 exposure compared with the untreated control (Fig. 4A). This effect was specific to bmp-2 as none of the other genes examined showed any SB203580-induced difference in mRNA expression. Further analysis confirmed that bmp-2 mRNA was first induced on day 4, and that SB203580 strongly repressed (but did not extinguish) this induction (Fig. 4B). On the other hand, U0126 did not repress the induction of bmp-2 mRNA (Fig. 4C). These results indicate that p38 MAPK but not ERK regulates the induction of bmp-2 mRNA in this ES cell differentiation.

p38 mitogen-activated protein kinase (MAPK) controls the expression of bone morphogenetic protein 2 (BMP-2) during ES cell differentiation. (
BMP-2 inhibits SB203580-induced neuronal differentiation
To determine whether it was the downregulation of BMP-2 in SB203580-treated EBs that committed them to neurogenesis, we treated EBs that had been exposed to SB203580 on days 3–6 with recombinant human BMP-2 (rhBMP-2) on days 4–6. As predicted, the neuronal differentiation induced by SB203580 treatment was dramatically repressed by rhBMP-2 treatment (Fig. 5A). A quantitative analysis showed that SB203580 treatment on days 3–6 induced nearly 80% of EBs to generate neuronal lineage cells, whereas the addition of rhBMP-2 on days 4–6 reduced this rate to fewer than 5% of EBs (Fig. 5B). Consistent with the microscopic analysis, RT-PCR confirmed that rhBMP-2 strongly inhibited SB203580-induced expression of the neuron-specific gene map2 (Fig. 5C, top row). In contrast, expression levels of the cardiomyocyte-specific genes mhc and mlc2v in SB203580-treated EBs were not improved by the addition of rhBMP-2 (Fig. 5C, middle row).

Bone morphogenetic protein 2 (BMP-2) inhibits SB203580-induced neuronal differentiation. Embryoid bodies (EBs) were left untreated or treated with 3 ng/mL recombinant human BMP-2 on days 4–6 in the presence or absence of 10 μM SB203580 on days 3–6. (
The BMP-2 antagonist Noggin blocks cardiomyogenesis and induces neural differentiation
On the basis of the above results, we postulated that vigorous interference with endogenous BMP-2 function might prevent the differentiation of ES cells into cardiomyocytes and induce neurogenesis. To test this hypothesis, we treated EBs with the BMP-2 antagonist Noggin on days 4–6. Like SB203580 treatment, Noggin treatment of EBs at this time dramatically blocked cardiomyogenesis and promoted neuronal differentiation (Fig. 6A). A quantitative analysis showed that more than 60% of EBs treated with 100 ng/mL Noggin on days 4–6 differentiated into neurons, a rate similar to the 75% of EBs induced to undergo neurogenesis by SB203580 treatment on days 3–6 (Fig. 6B). Moreover, RT-PCR analysis confirmed that Noggin treatment strongly induced the expression of the neuronal gene map2 and repressed expression of the cardiac gene mhc (Fig. 6C). Taken together, these results indicate that p38 MAPK controls ES cell lineage commitment (at least with respect to cardiomyocyte vs. neuron differentiation) by regulating the expression of BMP-2.
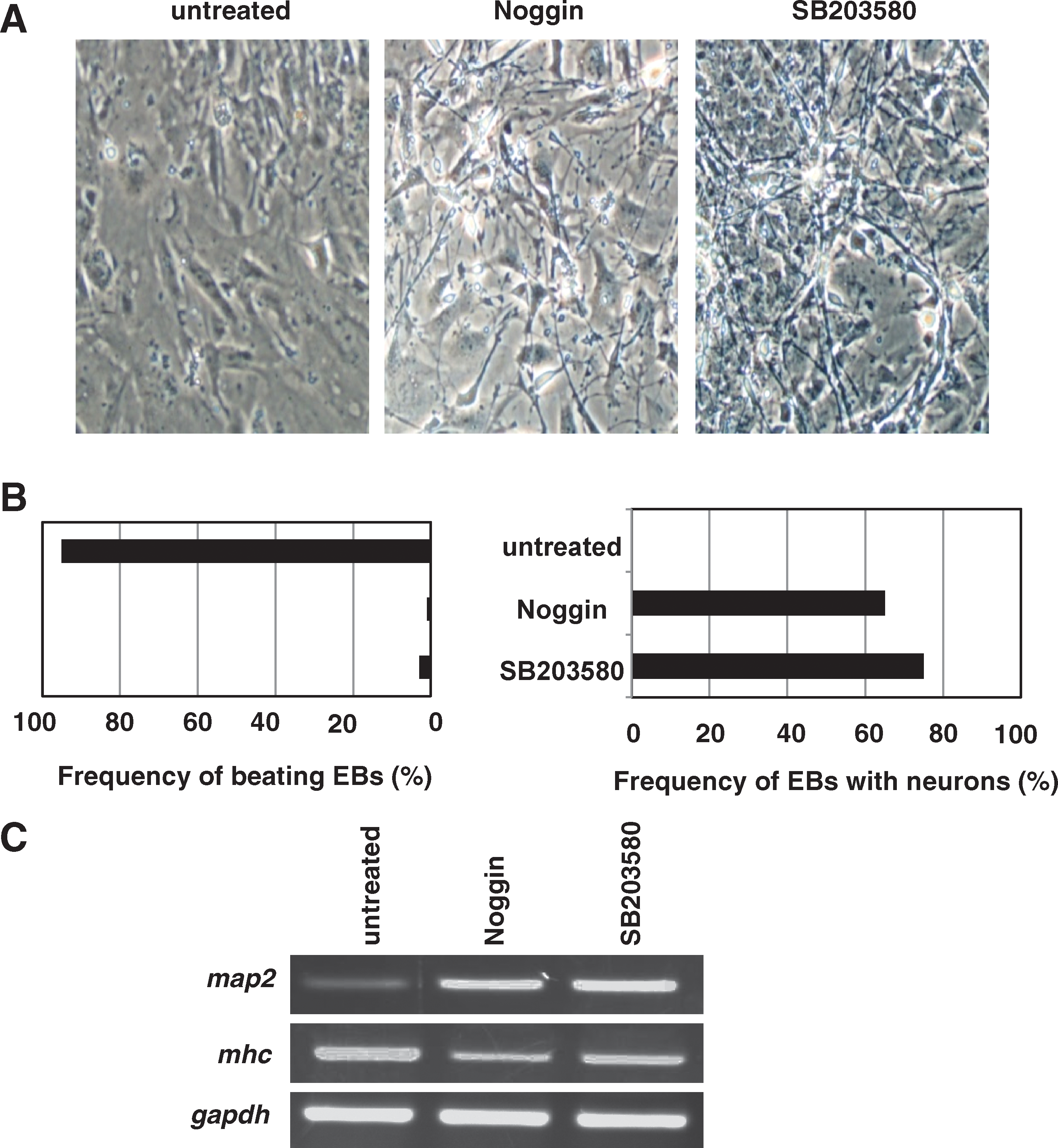
The bone morphogenetic protein 2 antagonist Noggin blocks cardiomyogenesis and induces neuronal differentiation. Embryoid bodies (EBs) were left untreated or treated with 100 ng/mL Noggin on days 4–6, or with 10 μM SB203580 on days 3–6. (
BMP-2 is a direct transcriptional target of MEF2C
The above experiments revealed that treatment of EBs with SB203580 resulted in a dramatic decrease in the mRNA expression of the transcription factor MEF2C, a well-known substrate of p38 MAPK (refer to Fig. 2B). We therefore compared the expression patterns of mef2c and bmp-2 during SB203580-induced neuronal differentiation and found that they were strikingly similar (Fig. 7A). Once activated by p38 MAPK-mediated phosphorylation, MEF2C activates the transcription of many cardiac-specific genes. Our observations suggested that p38 MAPK might induce BMP-2-regulated cardiomyogenesis by EBs via direct regulation of MEF2C. Importantly, a highly conserved consensus binding site for MEF2 has been identified in both the mouse and human proximal BMP-2 promoters. To test whether MEF2C could directly regulate BMP-2 transcription, we first carried out reporter assays in HeLa cells in which luciferase was placed under the control of a proximal region (−1703/−1 bp) of the mouse bmp-2 promoter; this region contains the MEF2-binding site. HeLa cells engineered to overexpress MEF2C showed a 3-fold increase in luciferase activity, whereas SB203580 treatment repressed this transactivation (Fig. 7B).
Bone morphogenetic protein 2 (BMP-2) is a direct transcriptional target of myocyte enhancer factor 2C (MEF2C). (
To determine whether MEF2C could physically bind to the BMP2 promoter region, we carried out ChIP analyses of day 6 EBs that had been allowed to spontaneously differentiate. The region of the mouse bmp-2 promoter encompassing the −656/ −635 bp MEF2-binding site was successfully immunoprecipitated using anti-MEF2C antibody, indicating that MEF2C can indeed bind to the endogenous bmp-2 promoter (Fig. 7C, top left panel). MEF2C did not bind to the promoter region of the control bahd1 gene present on the same chromosome (Fig. 7C, top right). Further, SB203580 treatment inhibited the binding of MEF2C to the MEF2-binding site (Fig. 7C, bottom left). As a positive control, we examined the binding of MEF2C to the promoter of the mouse mef2c gene, which itself is a target of MEF2 transactivation. The resulting ChIP pattern was similar to that derived for bmp-2 (Fig. 7C, middle). Collectively, these data indicate that BMP-2 is a direct transcriptional target of MEF2C.
Discussion
In this study, we analyzed the influence of the 3 major MAP kinases, ERK, JNK, and p38, on ES cell lineage commitment. Our results show that ERK and p38 MAPK play an essential role in the cardiomyogenesis of mES cells. An interesting cellular response of our work is that, at the same time as it promotes the induction of cardiomyocyte differentiation, p38 MAPK activity specifically inhibits neuronal differentiation. We demonstrate that p38 MAPK achieves these effects by activating the transcription factor MEF2C, which in turn directly regulates BMP-2 expression. Several previous studies also reported that p38 MAPK regulates both murine and human ES cell survival and lineage commitment, including cardiomyocyte differentiation [29 –32]. Our work revealed the molecular mechanism of a switch between cardiomyocyte and neuronal commitment of mES cells.
The pyridinylimidazole compound SB203580 inhibits the catalytic activity of p38α and p38β MAPKs via competition for ATP, but SB203580 does not inhibit the closely related ERK or JNK enzymes or any other serine–threonine protein kinases [33]. Graichen et al. reported that SB203580 at concentrations lower than 10 μM induced cardiomyogenesis of human ES cells, whereas at concentrations more than 15 μM, it strongly inhibited cardiomyogenesis [30]. These results indicate the dose-dependent differences in lineage determination in human ES cells. However, we could not observe the phenomena using mES cells in the presence of SB203580 at concentrations of 5–20 μM (Fig. 3C). In our mES cell differentiation system, more than 90% EBs differentiated into cardiomyocytes in the absence of SB203580, and so it may be difficult to evaluate the enhanced induction of cardiomyogenesis by low concentrations of SB203580.
Consistent with our findings, Aouadi et al. have reported that loss of p38 MAPK activity due either to treatment with the chemical inhibitor PD169316 or to genetic p38α deficiency is sufficient to block cardiomyogenesis and induce a high level of neurogenesis [34]. These results clearly show that it is p38α that is mainly responsible for p38 MAPK functions during ES cell lineage commitment: the control of p38α activity constitutes an early switch, committing ES cells into either cardiomyogensesis (p38 on) or neurogenesis (p38 off). However, the molecular mechanism of p38 off-dependent neurogenesis was unclear.
P38 MAPK induces cell cycle exit and differentiation in many cell types, and activated p38 has been shown to phosphorylate several downstream signaling molecules important for cardiomyocyte differentiation and hypertrophy in murine P19 cells and mice [35 –37]. In our study, we found that p38 MAPK is spontaneously activated between days 2 and 6 after the formation of EBs. Further, our data indicate that this spontaneous p38 MAPK activity is critical between days 3 and 4 for the cardiac commitment of ES cells. Inhibition of p38 MAPK activity at this early juncture drives ES cells toward the neuronal lineage. These findings stand in sharp contrast to those of other groups investigating the role of p38 MAPK in later stages of neuronal differentiation [38]. P38 MAPK activation is required for neurite formation and neuron survival in PC12 and P19 cells during the late stages of differentiation. Okamoto et al. reported that the p38α/MEF2 pathway prevents cell death during neuronal differentiation in P19 cells [39]. Thus, the role of p38 MAPK during the complex process of neuronal differentiation appears to be stage dependent.
BMPs are part of the larger superfamily of TGF-β ligands, which signal through a well-defined molecular pathway [21]. BMPs were found to be required for maintaining cultured mES cells in an undifferentiated state [40]. In our study, we demonstrate both that p38 MAPK regulates the expression of BMP-2, and thereby controls mES cell lineage commitment, and that BMP-2 treatment inhibits SB203580-induced neuronal differentiation. Further, like SB203580, the exogenous BMP antagonist Noggin prevents the spontaneous differentiation of mES cells into cardiomyocytes and promotes neuronal differentiation. These data suggest a dynamic role for BMP in specifying cell fate and emphasize that defining the molecular context of BMP signaling is critical for understanding how ES cells are regulated at a physiological level.
MEF2C is an important transcription factor that transactivates many genes encoding cardiac structural proteins, and p38 MAPK is a well-known regulator of MEF2C [14,41 –43]. Gene-targeted mouse embryos lacking MEF2C have cardiogenic defects [17]. BMP-2 is also required for cardiogenesis, and BMP2-deficient embryos exhibit an early defect in cardiac development [44]. In our study, we found that BMP-2 is a direct transcriptional target of MEF2C, and that p38 MAPK may regulate BMP-2 by controlling MEF2C activation. However, we found that simple stimulation of ES cells with BMP-2 did not augment cardiomyocyte generation (data not shown), suggesting that BMP-2 is essential but not sufficient for cardiac induction. It is likely that other MEF2C-dependent genes encoding cardiac structural proteins are also required for normal cardiac development. It will be interesting to investigate whether MEF2C−/− ES cells can differentiate spontaneously into neurons. Additionally, unknown factors in FBS contribute to the frequency of beating EBs and play important roles in cell lineage commitment.
In conclusion, our study has revealed an intriguing role for p38 MAPK as a cell fate switch during ES cell differentiation. The choice between cardiac and neuronal cell development depends on the early stage function of BMP-2, whose expression in turn depends on transactivation by the p38 MAPK target MEF2C.
Footnotes
Acknowledgments
This work was supported by research grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan and the Japanese Society for the Promotion of Science. The authors are grateful to numerous members of the Nishina and Katada Laboratories for critical reading of this manuscript and helpful discussions.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.