
Abstract
Ochratoxin A (OTA) is a ubiquitous fungal metabolite with nephrotoxic, carcinogenic, and apoptotic potential. Although the toxic effects of OTA in various cell types are well characterized, it is not known whether OTA has an effect on stem cell differentiation. In this study, we demonstrate that OTA inhibits adipogenesis in human adipose tissue-derived mesenchymal stem cells, as indicated by decreased accumulation of intracellular lipid droplets. Further, OTA significantly reduces expression of adipocyte-specific markers, including peroxisome proliferator-activated receptor-γ (PPAR-γ), CCAAT enhancer binding protein-α (C/EBP-α), lipoprotein lipase (LPL), and adipocyte fatty acid-binding protein (aP2). At the molecular level, OTA phosphorylates PPAR-γ2 through extracellular signal-related kinase activation and inhibits PPAR-γ activity. We also found that treatment with the mitogen-activated protein kinase kinase inhibitor, PD98059, significantly blocked the OTA-induced inhibition of adipogenesis. These results indicate that OTA suppresses adipogenesis in an extracellular signal-related kinase-dependent manner. Taken together, our results suggest a novel effect of OTA on adipocyte differentiation in human adipose tissue-derived mesenchymal stem cells and the possibility that OTA might affect the differentiation of other types of stem cells.
Introduction
T
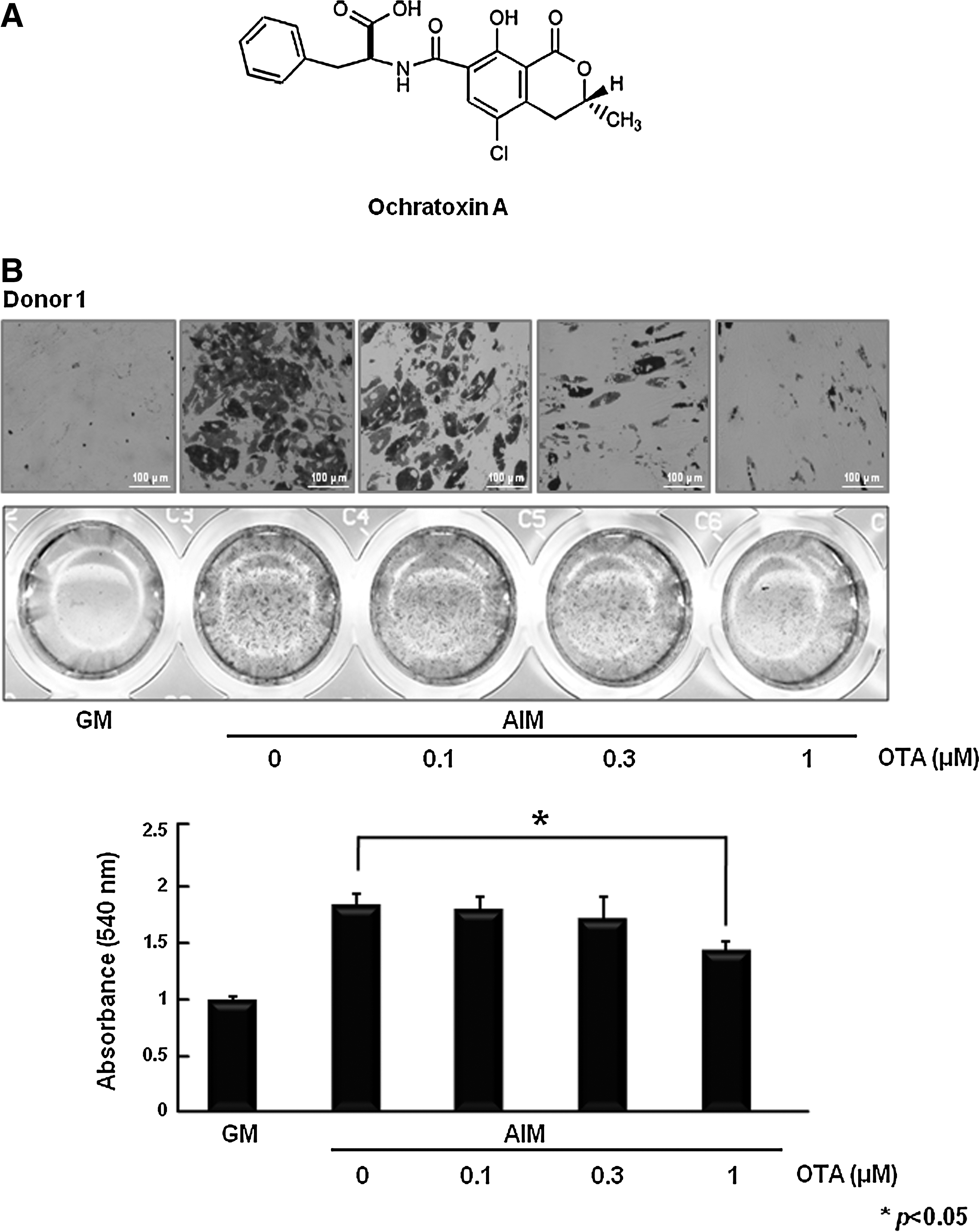
OTA inhibits fat accumulation in hAMSCs.

Mesenchymal stem cells (MSCs) are multipotent cells that can be isolated from several tissues, including bone marrow, adipose tissue, and fetal tissue. MSCs have the potential to differentiate into myoblasts, chondroblasts, osteoblasts, and adipocytes under defined conditions [8]. Because of these properties, MSCs might be very interesting target for potential therapeutic use in regenerative medicine and tissue engineering. Therefore, identification of the factors to regulate MSC differentiation is essential for the use these cells in regenerative medicine.
Adipogenesis, the process of fat cell development, has been extensively studied using various cell and animal models. The first hallmark of the adipogenesis process is alteration in cell shape in parallel with changes in the type and expression levels of extracellular matrix components and cytoskeletal components [9,10]. These events are required for expression of adipogenic transcription factors, including CCAAT enhancer binding protein-α (C/EBP-α) and peroxisome proliferator-activated receptor-γ (PPAR-γ). C/EBP-α is involved in PPAR-γ expression and together with C/EBP-α, PPAR-γ promotes terminal differentiation by transactivating expression of downstream adipocyte-specific genes, including adipocyte fatty acid-binding protein (aP2) and lipoprotein lipase (LPL) [11].
Mitogen-activated protein kinases (MAPKs) are serine/threonine kinases activated by dual phosphorylation on both a tyrosine and a threonine residue [12,13]. These enzymes have important roles in membrane-to-nucleus signaling pathways in mammalian cells. There are at least 3 distinctly regulated groups of MAPKs: extracellular signal-related kinases (ERK), Jun N-terminal kinases (JNK), and p38 MAPKs (p38). Among them, ERK is related to adipogenesis. ERK activation inhibits adipocyte differentiation via ERK-mediated PPAR-γ phosphorylation on serine 112 [14,15]. PPAR-γ activity is attenuated serine 112 phosphorylation by ERK. To date, there are several reports suggesting that ERK regulates adipocyte differentiation under different condition. ERK is involved in adipogenesis of MSCs in hyperglycemia or hypoxia condition [16,17]. Moreover, activation of the MAPK kinase (MEK)/ERK signaling pathway with the increase of serum concentration in the medium promoted PPAR-γ expression and phosphorylation, and subsequently enhanced adipogenic differentiation [18]. These results indicate that ERK plays a crucial role in the regulation of adipogenesis.
Several reports indicate that OTA is involved in cellular differentiation and dedifferentiation. OTA triggers cell dedifferentiation in MDCK-C7 cells and inhibits neuronal cell differentiation [4,19,20]. Moreover, OTA also induces an epithelial to mesenchymal transition in proximal tubular cells [21]. These articles provide a relationship between OTA and cell differentiation. On the basis of these reports, we examined the effect of OTA on stem cell differentiation, especially on adipogenesis in human adipose tissue-derived MSCs (hAMSCs). We found that OTA induced phosphorylation of PPAR-γ2 and decreased PPAR-γ activity through long-term activation of ERK. In addition, treatment with the MEK inhibitor PD98059 restored OTA-mediated suppression of adipogenesis in a dose-dependent manner. These results not only give more insight into the influence of OTA on adipogenesis, but also have important implications for understanding the ability of OTA to modulate stem cell differentiation.
Materials and Methods
Materials
OTA, 3-isobutylmethylxanthine, dexamethasone, rosiglitazone, indometacin, insulin, methyl thiazolyl tetrazolium (MTT), and oil red O were purchased form Sigma-Aldrich. Anti-C/EBP-α, PPAR-γ, ERK2, poly-ADP ribose polymerase (PARP)-1, and aP2 antibodies were from Santa Cruz Biotechnology, Inc., and anti-p-ERK1/2 and p-JNK antibodies were from Cell Signaling Technologies. The anti-p-PPAR-γ antibody MAB3632 was purchased from Chemicon, and anti-β-actin antibody was purchased from MP Biomedicals, Inc. PD98059, SP600125, and SB203580 were purchased from Calbiochem.
Cell culture
Subcutaneous adipose tissues were obtained from individuals undergoing elective surgery after obtaining informed consent from each individual. The patients were women aged 50, 39, and 28 years, respectively. The hAMSCs were isolated from adipose tissues according to the methods as described by Lee et al. [22] with modifications. Briefly, adipose tissues were digested at 37°C for 30 min with 0.075% type I collagenase. The enzyme activity was neutralized with α-modified Eagle's medium (α-MEM), containing 10% fetal bovine serum (FBS). The floating adipocytes were separated from the stromal-vascular fraction by centrifugation (1,200 g for 10 min). The pellet was resuspended in the growth medium (GM) consisting of α-MEM supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL of streptomycin, and plated in tissue culture dishes at 3,500 cells/cm2. After incubation, the tissue culture plates were washed to remove any residual nonadherent cells and maintained at 37°C/5% CO2 in GM. The primary hAMSCs were cultured for 5–6 days until confluence and were defined as passage 0. The passage number of hAMSCs used in the experiments was 3–10. When the monolayer of adherent cells reached 80% confluency, the cells were trypsinized (0.25% trypsin; Sigma), resuspended in GM, and subcultured at a concentration of 2,000 cells/cm2. Since hAMSCs derived from different donors were shown to exhibit varying degrees of differentiation potentials either to adipocytes or osteoblasts, we used hAMSCs derived from at least 2 different donors in this study.
Adipogenic induction
Before inducing differentiation of cells, hAMSCs were grown to confluence. Adipogenic differentiation was induced by culturing hAMSCs for 14 days in the adipogenic induction medium (AIM, 10% FBS, 1 μM dexamethasone, 0.5 mM 3-isobutyl-1-methylxanthine, 1 μM insulin, and 100 μM indomethacin in α-MEM). After 6 days the cells were cultured in AIM, but containing only 10% FBS and 1 μM insulin in α-MEM, and AIM was changed every 3 days. Differentiated adipocytes were stained with oil red O as an indicator of intracellular lipid accumulation.
Oil red O staining
Cells were cultured in AIM as described above for 14 days. Lipid droplets within differentiated adipocytes from hAMSCs were observed using a modified oil red O staining method. In brief, cells were washed twice with PBS and fixed with 4% paraformaldehyde for 2 h at 4°C. After 2 washes in PBS, cells were stained for 2 h in freshly diluted oil red O solution (6 parts oil red O stock solution and 4 parts H2O; oil red O stock solution is 0.5% oil red O in isopropanol) at 4°C. The stain was then removed, and the cells were washed twice with PBS. Images of cells stained with oil red O were obtained with a Zeiss Axiovert 135 microscope plus Olympus DP71 CCD camera (Olympus Corporation). For quantitative analysis of oil red O staining, cells were exposed to 1 mL isopropanol for 30 min, and absorbance of the supernatant was measured at 540 nm using a Biotrak II plate reader (Amershan Biosciences, Biochrom Ltd., Cambridge England).
RNA extraction and real-time quantitative polymerase chain reaction
Total RNA was extracted from hAMSCs using easy-BLUE Total RNA extraction kit (iNtRON Biotechnology). cDNA was reverse-transcribed from 1.5 μg total cellular RNA using oligo(dT) primers and moloney murine leukemia virus reverse transcriptase (Promega Corporation). cDNA was amplified for 45 cycles using the following primers: PPAR-γ1 and γ2 5′-atggagtccacgagatcatt-3′ (sense), 5′-cgcaggctctttagaaactc-3′ (antisense); C/EBP-α 5′-aaccttgtgccttggaaatg-3′ (sense), 5′-cctgctcccctccttctct-3′ (antisense); adiponectin 5′-accactatgatggctccact-3′ (sense), 5′-ggtgaagagcatagccttgt-3′ (antisense); aP2 5′-aaccttagatgggggtgtcctg-3′ (sense), 5′-tcgtggaagtgacgcctttc-3′ (antisense); LPL 5′-ctggacggtaacaggaatgtatgag-3′ (sense), 5′-catcaggagaaagacgactcgg-3′ (antisense) and reference gene, ribosomal protein large P0 (RPLP0) 5′- ggaatgtgggctttgtgttc -3′ (sense), 5′- tgcccctggagattttagtg -3′ (antisense). For real-time polymerase chain reaction (PCR), total RNA (100 ng) was amplified with a One Step SYBR RT-PCR kit using a Light Cycler 2.0 PCR system (Roche Diagnostics). PCR conditions consist of a 10-min hot start at 95°C, followed by 45 cycles of 15 s at 95°C, 10 s at 60°C, and 30 s at 72°C. Expression levels of each mRNA were compared after normalization against expression of RPLP0. This experiment was repeated twice.
Western blot analysis
Whole cell lysates were prepared in lysis buffer (1% Triton X-100, 10% glycerol, 150 mM NaCl, 50 mM HEPES, pH 7.3, 1 mM EGTA, 1 mM sodium orthovanadate, 1 mM sodium fluoride, 1 mM phenylmethysulfonyl fluoride, 10 mg/mL leupeptin, and 10 mg/mL aprotinin). Lysates were then centrifuged at 14,000 g for 10 min at 4°C. Electrophoresis was performed with 8% polyacrylamide gels and the proteins were then electrotransferred to nitrocellulose membranes. Membranes were immunoblotted with mouse anti-PARP-1(Santa Cruz; sc-8007), PPAR-γ (Santa Cruz; sc-7273), aP2 (Santa Cruz; sc-18661), or rabbit anti-C/EBP-α (Santa Cruz; sc-61) in Tween 20/Tris-buffered saline containing 5% skim milk. After incubation with the appropriate peroxidase-conjugated secondary antibody, proteins were detected with the enhanced chemiluminescence system (ECL system; Amersham).
Plasmids and luciferase activity assay
The 3xPPRE-tk-LUC plasmid, which contains 3 copies of the PPAR-γ-response element and pSV- PPAR-γ2, was received from Dr. Jae Bum Kim [23]. The pRL-SV40 construct, which was the expression vector of renilla luciferase, was purchased from Promega Corporation. Human embryonic kidney 293 (HEK293) cells were seeded into 24-well plates and cultured for 24 h before transfection. A DNA mixture containing the PPRE-luciferase reporter plasmid (0.1 μg), pSV-PPAR-γ2 (0.1 μg) and an internal control plasmid pRL-SV-40 (25 ng) was transfected using Lipofectamine transfection reagent according to the manufacturer's recommendations. After 24 h of transfection, the cells were incubated for an additional 36 h following treatment with positive control or the test materials. The luciferase activity of the cell lysates was measured using the Dual-Luciferase® Reporter Assay System according to the manufacturer's instruction (Promega Corporation). Relative luciferase activity was normalized for transfection efficiency using the corresponding Renilla luciferase activity.
MTT assay
hAMSCs were seeded at a density of 1 × 104 cells/well onto a 96-well plate and cultured for 24 h and cells were cultured for the appropriate length of time, as indicated. MTT was dissolved in phosphate-buffered saline (PBS) at a concentration of 5 mg/mL. The stock solution was filtered and added to the culture medium to a final concentration of 0.5 mg/mL. The plates were incubated at 37°C for 2 h. Dark brown formazan crystals formed after reduction of the tetrazolium ring by the mitochondria of living cells. The crystals were dissolved in dimethylsulfoxide, and the absorbance values of the samples were measured at 540 nm.
Statistical analysis
Data were analyzed using Student's t-test. P < 0.05 was considered significant.
Results
OTA reduces lipid droplet formation
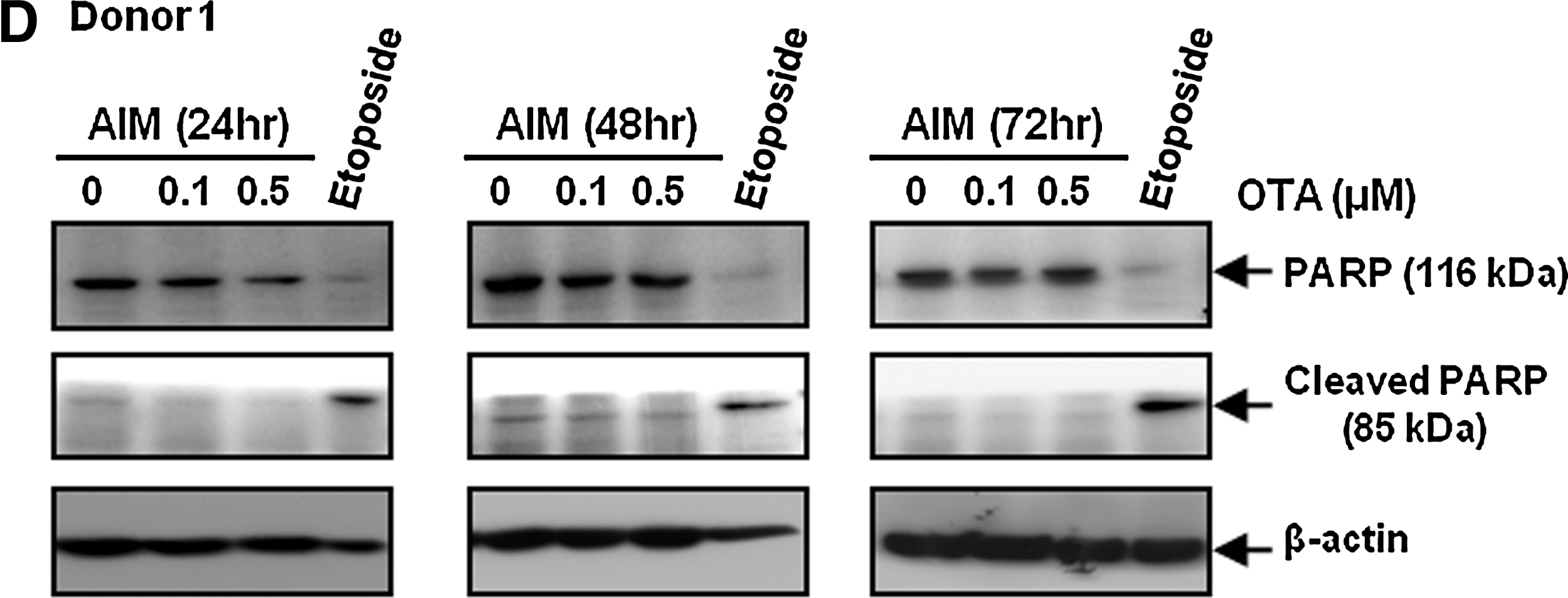
To investigate the effect of OTA on adipocyte differentiation, hAMSCs were treated with AIM in the presence of OTA during adipogenic differentiation. At 10 days after the initiation of differentiation, accumulated lipid droplets were detected by staining with oil red O. Treatment of OTA significantly reduced adipocyte differentiation in a dose-dependent manner (Fig. 1B). To further validate these visual results, we measured triglyceride content in these cells; triglyceride content was reduced by OTA treatment (Fig. 1B). We also confirmed that OTA reduced lipid droplet formation and triglyceride content of hAMSCs derived from different donors (Fig. 1B). These results indicate that OTA suppresses adipogenesis in hAMSCs. In addition, because OTA is known to have potent toxic effects in many cell types, we performed an MTT assay to check the effect of OTA on cellular toxicity under GM and AIM. The MTT assay showed that OTA does not affect cell viability in cells cultured in AIM at 0.1 or 0.5 μM OTA (Fig. 1C). Moreover, we also confirmed that OTA had no significant inhibitory effects on cell viability in hAMSCs derived from other different donors (Fig. 1C). To further confirm the effect of OTA on apoptosis, we examined PARP cleavage analysis. Because PARP is known to be cleaved by caspase during apoptosis, cleaved PARP was shown to be a marker of apoptosis [24]. We used etoposide, the apoptosis inducer, as a positive control [25]. Etoposide is a topoisomerase II inhibitor that induces double-strand breaks in DNA, thus leading to the apoptosis [26]. After 12 h, etoposide (100 μM) markedly induced PARP cleavage (Fig. 1D). However, OTA had no effect on PARP cleavage under AIM condition in the presence of 0.1 or 0.5 μM OTA (Fig. 1D). These results suggest that OTA inhibits adipocyte differentiation without affecting cellular toxicity.
OTA decreases mRNA levels and protein expression of adipocyte-specific genes
Adipocyte differentiation is accompanied by altered expression of various transcription factors and adipocyte-specific genes [27]. To further confirm the effect of OTA on adipocyte differentiation, we performed quantitative real-time PCR to determine expression of PPAR-γ and C/EBP-α, 2 key transcription factors known to be involved in adipogenesis. We also checked the mRNA levels of fatty acid binding protein (aP2), adiponectin and LPL. Figure 2A shows that the mRNA levels of C/EBP-α, PPAR-γ, LPL, aP2, and adiponectin were significantly decreased by treatment with 500 nM OTA. Moreover, after 6 days of adipogenic induction, we also examined the protein levels of C/EBP-α, PPAR-γ, and aP2. The expression level of adipocyte-specific proteins was reduced by OTA in a dose-dependent manner (Fig. 2B). These results suggest that OTA inhibits adipocyte differentiation.
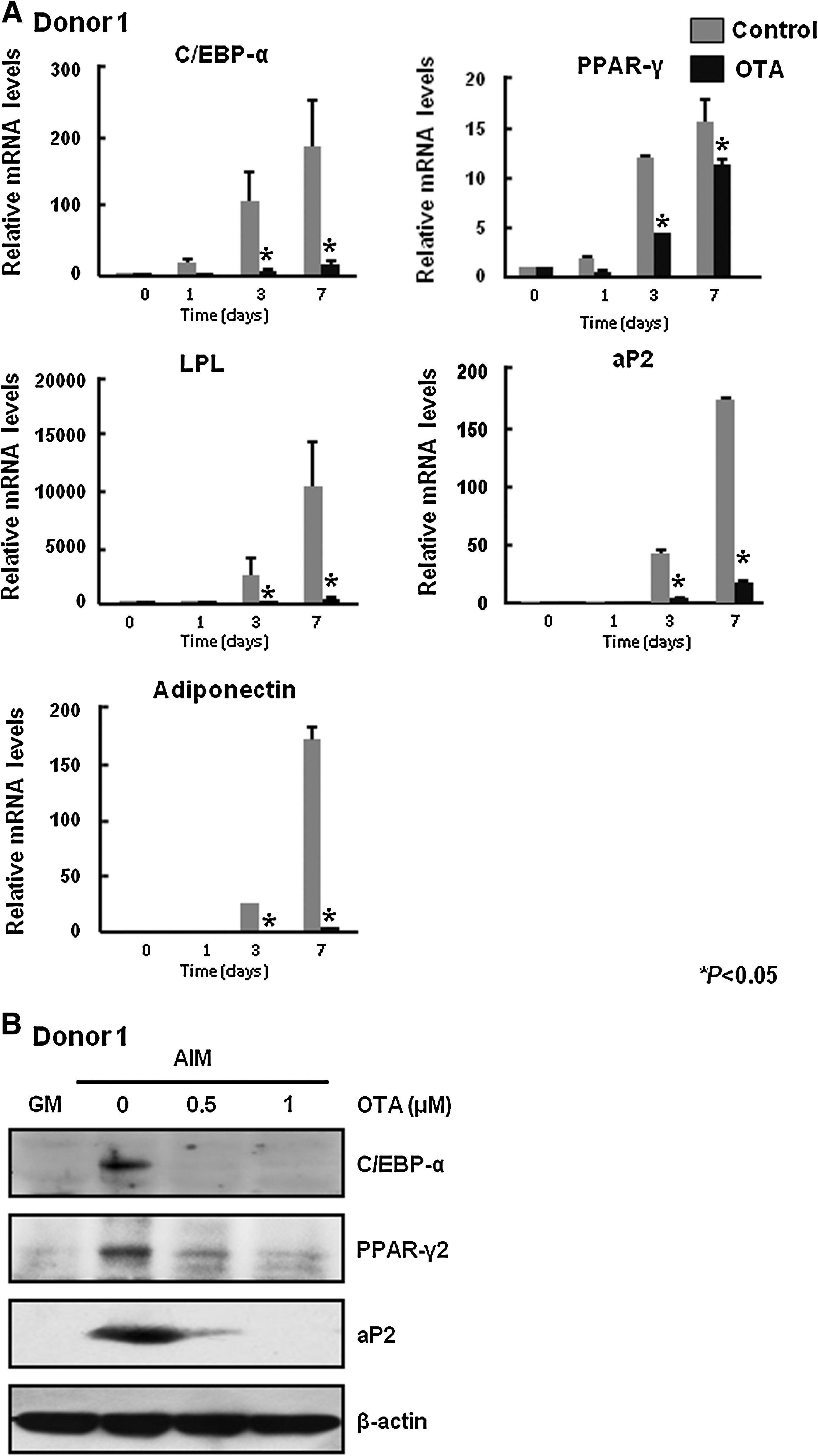
OTA decreases mRNA levels and protein expression of adipocyte-specific genes.
OTA inhibits PPAR-γ activity
PPAR-γ is a critical transcription factor for adipogenesis [27,28]. In addition, some natural products regulate adipogenesis by modulating PPAR-γ activity [29 –31]. On the basis of these reports, we tested whether OTA inhibits adipocyte differentiation through regulation of PPAR-γ activity. Because the gene transfection efficiency of hAMSCs is very low, we used HEK293 cells to examine the effect of OTA on PPAR-γ activity. HEK293 cells were cotransfected with a PPREx3-tk-luciferase reporter plasmid and mouse PPAR-γ2 expression vector. We used rosiglitazone, a synthetic PPAR-γ ligand, as a positive control. After 36 h, rosiglitazone (100 nM) markedly increased PPAR-γ activity (Fig. 3A). However, OTA reduced transcriptional activity of PPAR-γ in a dose-dependent manner (Fig. 3A). To further confirm the effect of OTA on PPAR-γ activity, we next examined mRNA expression of aP2, which is a known PPAR-γ target gene [32]. HEK293 cells were transfected with a mouse PPAR-γ2 expression vector and treated with rosiglitazone (100 nM), a synthetic PPAR-γ ligand, in the presence or absence of OTA for 36 h. Generally, HEK293 cells do not express aP2 mRNA, but transfection of PPAR-γ2 expression vector and treatment of rosiglitazone, PPAR-γ ligand, significantly increases aP2 mRNA levels (Fig. 3B). As shown in Fig. 3B, OTA inhibited rosiglitazone-induced aP2 mRNA expression in a dose-dependent manner. These data suggest that OTA decreases adipogenic differentiation through inhibition of PPAR-γ activity.
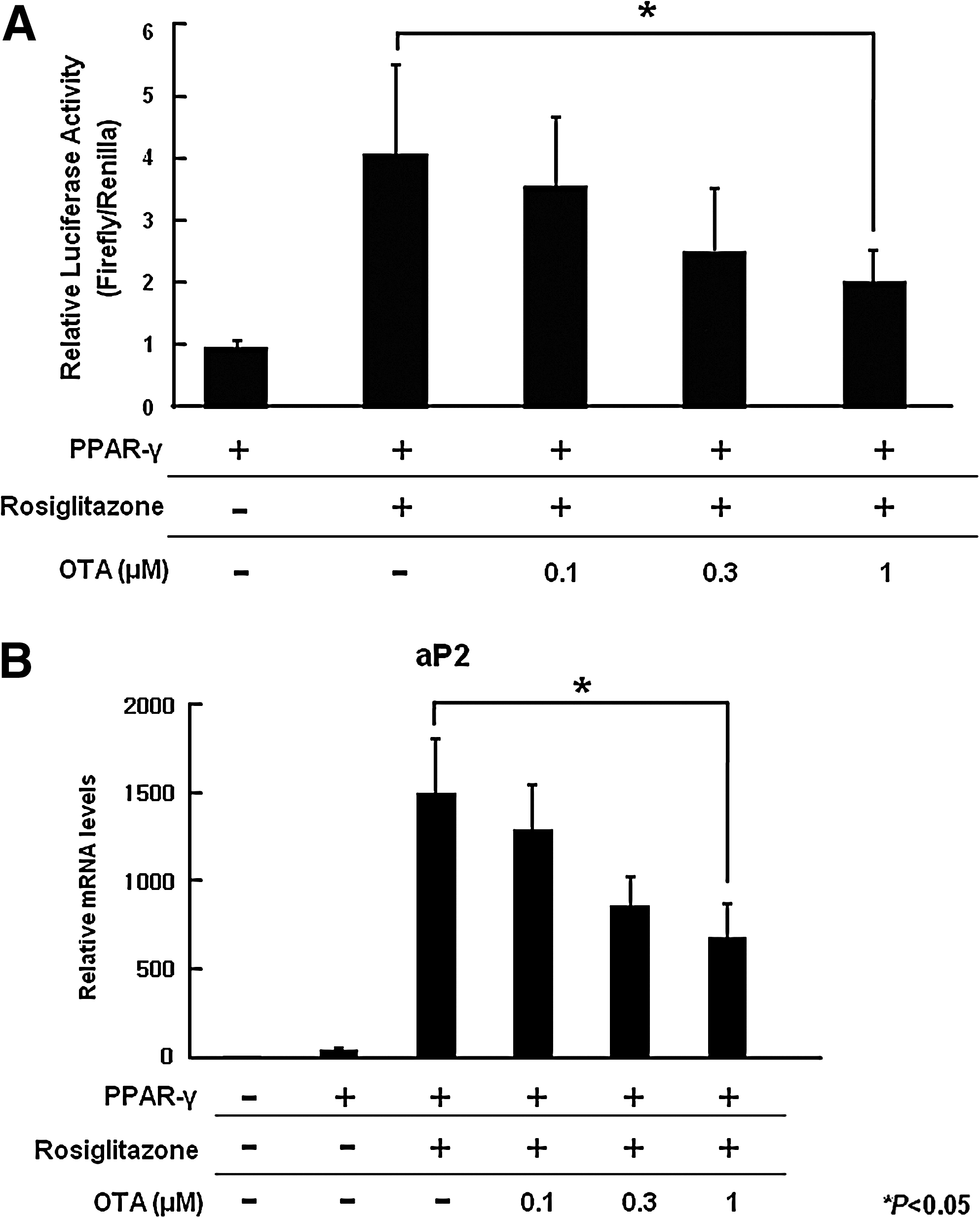
OTA inhibits PPAR-γ activity.
OTA causes phosphorylation of PPAR-γ through ERK activation
It has been reported that OTA stimulates ERK phosphorylation in a time- and dose-dependent manner [33]. Interestingly, OTA induces long-term activation of ERK, which leads to epithelial dedifferentiation and transformation in MDCK-C7 cells [19]. On the basis of these reports, we examined whether OTA activates ERK in hAMSCs. Figure 4A shows that OTA (500 nM) induces ERK and JNK phosphorylation, but not p38 phosphorylation, compared with a control, in the serum-free medium. Interestingly, stimulation of hAMSCs with OTA showed a biphasic activation of ERK. OTA transiently activated ERK at 15 min and decreased to near baseline levels by ∼6 h after stimulation. A second wave of increase in p-ERK levels appeared around 12 h and was sustained for up to 36 h (Fig. 4A). Although the molecular mechanism of biphasic activation of ERK is still unclear, we observed that OTA induced biphasic ERK phosphorylation in repeated experiments. Similar results were observed in another donor-derived hAMSCs (data not shown). In addition, OTA (500 nM) stimulates long-term activation of ERK, compared with AIM only condition (Fig. 4B). Because active ERK phosphorylates serine 112 of PPAR-γ2, which inhibits PPAR-γ's transcriptional activity, we investigated whether OTA treatment causes serine 112 phosphorylation of PPAR-γ2. At 6 days after the initiation of adipogenic differentiation, treatment with 500 nM OTA induces serine 112 phosphorylation of PPAR-γ2 (Fig. 4C). Taken together, these results suggest that OTA induces long-term activation of ERK, which leads to phosphorylation of PPAR-γ2 and inhibits PPAR-γ2 activity in hAMSCs.
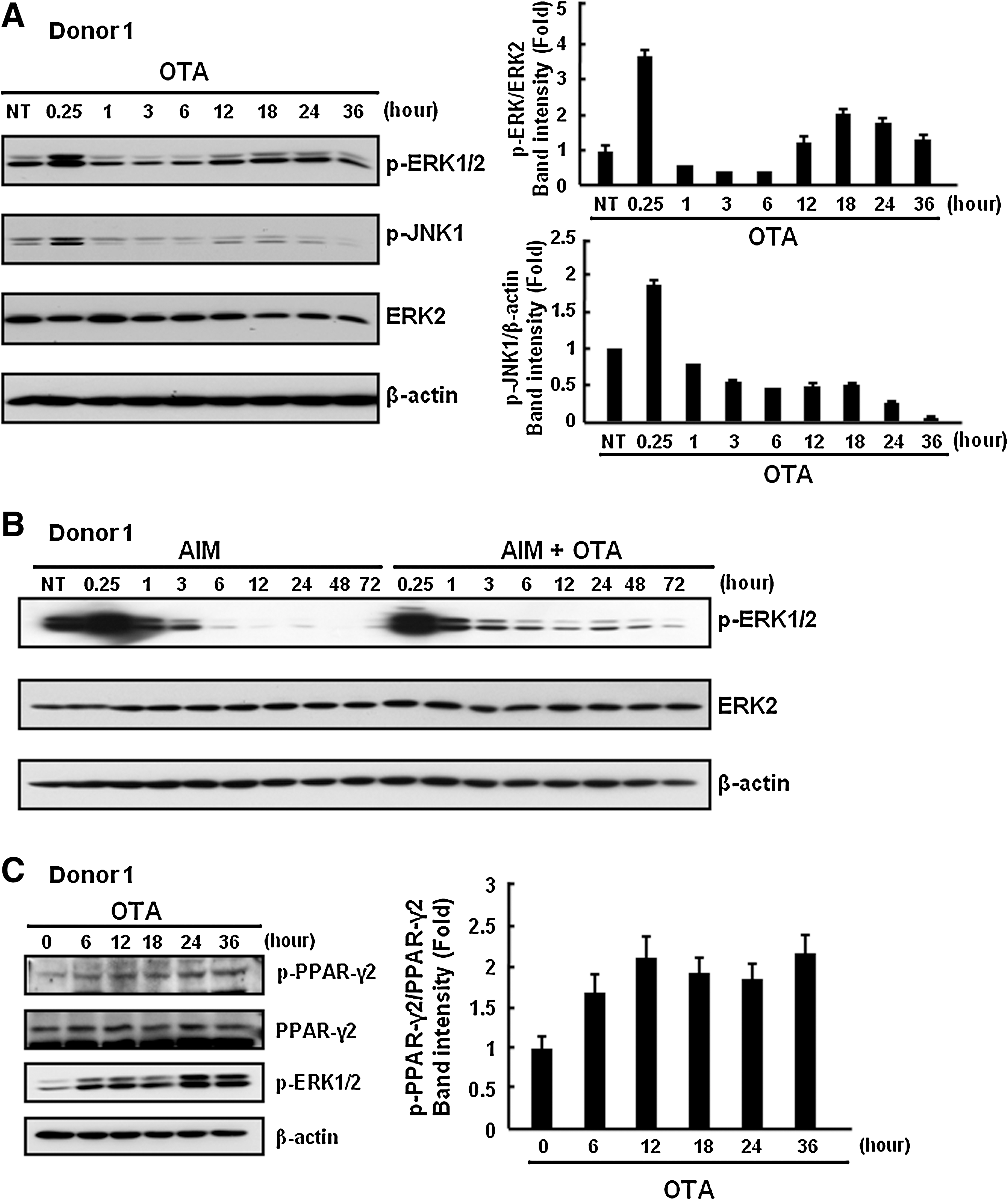
OTA causes phosphorylation of PPAR-γ via ERK activation.
OTA reduces adipogenesis in an ERK-dependent manner
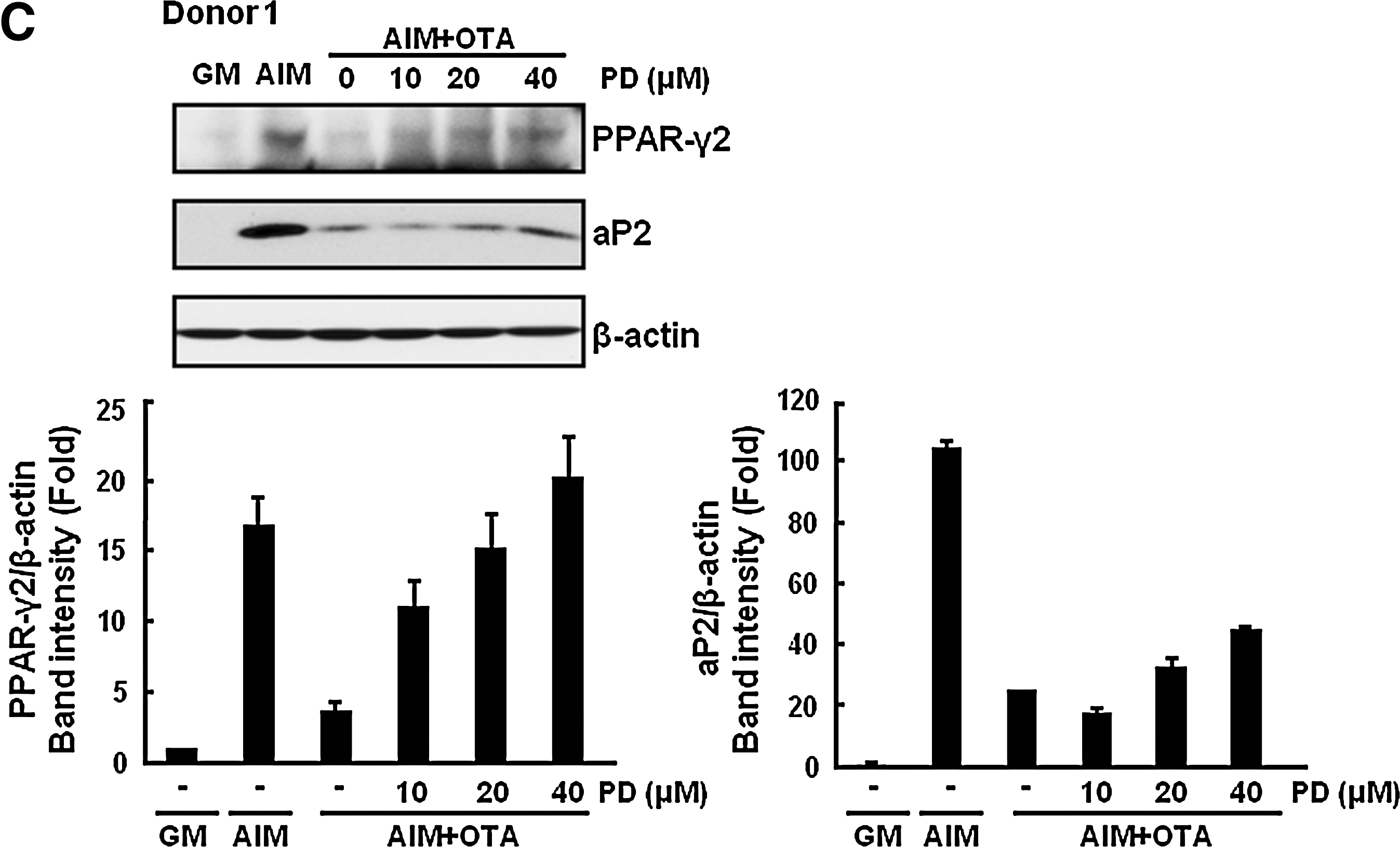
Because activation of ERK is known to suppress adipogenesis, we examined whether OTA mediates the inhibition of adipogenesis in an ERK-dependent manner. To test this possibility, we used PD98059, which is a specific MEK inhibitor. Because MEK is an upstream kinase of ERK, blocking of MEK activity prevents ERK activation [34]. Treatment with PD98059 restored OTA-mediated inhibition of lipid droplet formation in a dose-dependant manner (Fig. 5A). From these results, ERK is involved in regulation of adipogenesis by OTA. However, blocking the JNK or p38 pathway by specific inhibitors SP600125 or SB203580, respectively, did not reverse the suppression of adipogenesis by OTA (data not shown). To further confirm the involvement of ERK on OTA-mediated inhibition of adipogenesis, we performed real-time PCR analysis for adipogenic transcription factors and marker gene expression. As shown in Fig. 5B, treatment with 40 μM PD98059 rescued the cells from OTA (500 nM)-induced reduction of adipocyte-specific markers including ap2 and PPAR-γ. We also confirmed that treatment with PD 98059 recovered OTA-mediated decrease of aP2 mRNA levels of hAMSCs derived from different donors (Fig. 5B). In addition, expression of adipocyte-specific proteins, such as PPAR-γ2 and aP2 was also rescued by ERK inhibition in a dose-dependent manner (Fig. 5C). Therefore, our data indicate that OTA suppresses adipogenesis in an ERK-dependent manner.
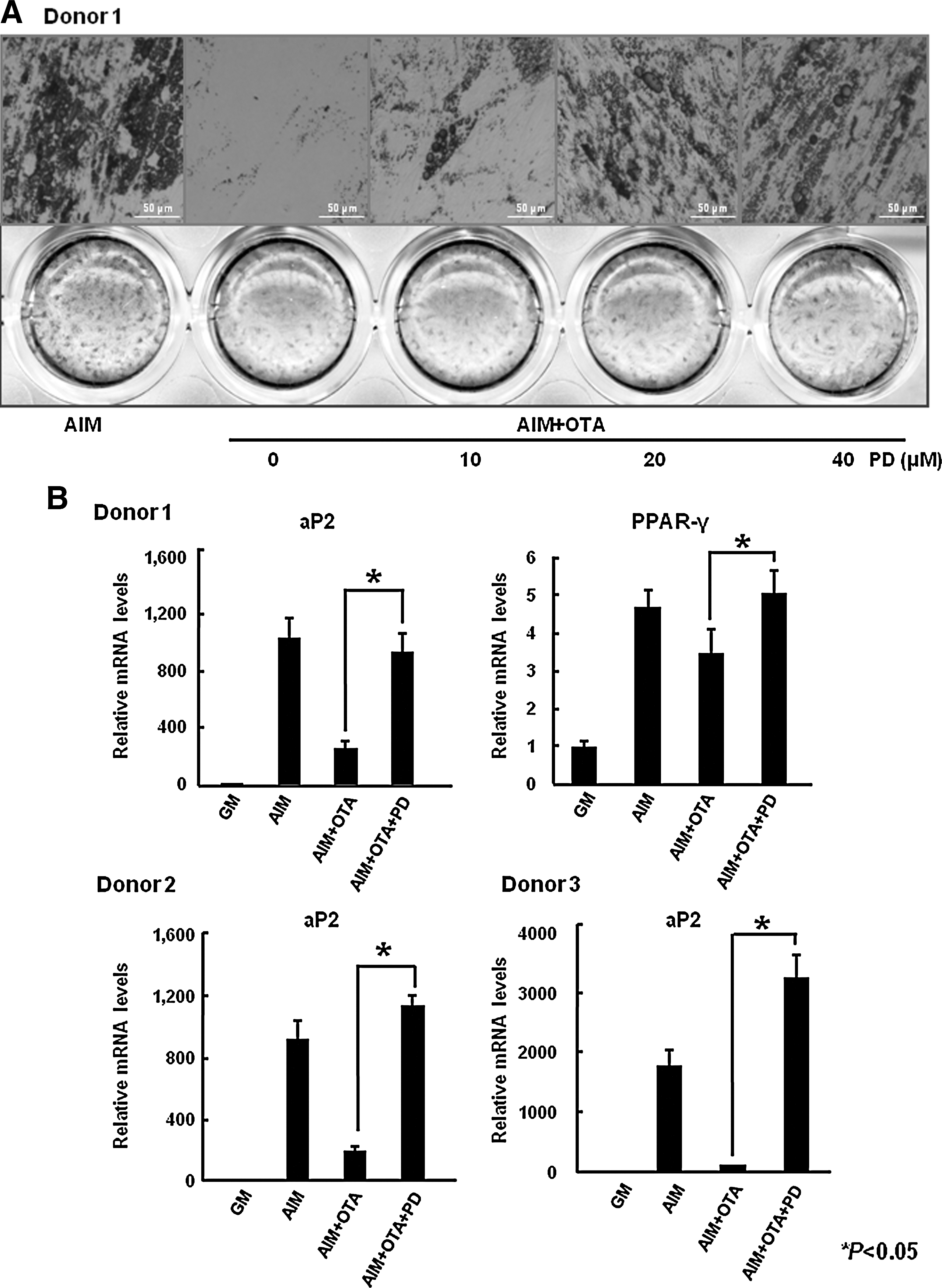
OTA reduces adipogenesis in an ERK-dependent manner.
Discussion
It has been reported that OTA induces various cellular events, such as inhibition of protein synthesis, oxidative stress, and alteration of specific cell signaling pathways [3]. However, the potential effect of OTA on stem cell differentiation has not been studied. In this study, we investigated the effect of OTA on adipogenic differentiation in hAMSCs. Our results show that OTA can inhibit adipogenic differentiation, as measured by oil red O staining and RT-PCR of adipocyte-specific markers. OTA causes phosphorylation of PPAR-γ and decreases PPAR-γ activity via long-term activation of ERK. Moreover, inhibition of ERK activation restores OTA-mediated suppression of adipogenesis in a dose-dependent manner. To our knowledge, this is the first report to demonstrate an effect of OTA on stem cell differentiation.
Previous studies on OTA have focused on the toxicological aspects in various cultured cell system and animal models. However, several studies were performed using high concentrations of OTA, at the micromolar concentrations range, and these high concentrations do not represent naturally occurring conditions [6,7]. Recently, some reports showed that OTA has an ability to alter physiological signals, at nanomolar concentrations, thereby influencing cell functions, including proliferation and dedifferentiation [4]. For instance, OTA changed cellular pH- and energy-homeostasis in immortalized human kidney epithelial cells [35]. Further, OTA potentiated the effect of angiotensin II and epidermal growth factor on cytosolic calcium homeostasis [36]. These reports imply a function of OTA as a signal modulator. With regard differentiation, OTA can lead to cell dedifferentiation of renal epithelial MDCK-C7 cells through ERK activation [33]. In addition, OTA also has effect on human hematopoietic progenitors' proliferation and differentiation [37]. These reports provide further evidence that OTA could relate to cell differentiation. On the basis of previous reports, we hypothesized that OTA might affect stem cell differentiation and demonstrated a novel effect of OTA on adipogenesis at nanomolar concentrations.
During adipocyte differentiation, expression and modification of PPAR-γ, a master transcription factor in adipogenesis, play crucial roles. Previous studies found that ERK phosphorylates PPAR-γ and reduces its transcriptional activity [14,15]. Recently, several reports demonstrated that activation of ERK is involved in suppression of adipogenesis. For example, preadipocyte factor 1 activates MEK/ERK signaling, which is required for preadipocyte factor 1 inhibition of adipogenesis [38]. In the IL-6 family, oncostatin M inhibits adipogenesis through the RAS/ERK and STAT5 signaling pathways [39]. These reports also showed that inhibition of adipogenesis is reversed by treatment with the MEK inhibitor PD98059. Because we also observed prolonged activation of ERK and phosphorylation of PPAR-γ2 after OTA treatment in hAMSCs (Fig. 4), we speculated that ERK was involved in the reduction of adipogenesis by OTA. In the presence of OTA, blocking of ERK activation induces adipocyte differentiation (Fig. 5). Beside ERK, another MAPK, JNK, is involved in the negative regulation of adipogenesis. JNK also phosphorylates PPAR-γ and inhibits its transcriptional activity [40]. In addition, treatment with a JNK inhibitor, SP600125, promotes adipogenesis in human MSCs [41]. We observed that OTA activates JNK under serum-free conditions (Fig. 4A); however, a JNK-specific inhibitor did not restore adipogenesis (data not shown). Therefore, our data indicate that OTA suppresses adipogenesis in a specific ERK-dependent manner.
ERK plays critical roles in the regulation of stem cell differentiation. For example, inhibition of ERK pathway promotes self-renewal of mouse embryonic stem cells and leads to suppression of primitive endoderm and trophoectoderm formations [42 –44]. Moreover, ERK is also important for osteogenesis via upregulation of expression and DNA binding activity of Runx2, the master regulator of osteogenic differentiation [45 –47]. On the basis of these results, OTA might be related to control the lineage commitment in stem cell differentiation through prolonged ERK activation.
To date, a number of studies have demonstrated that several phytochemicals from natural products inhibit adipogenesis through regulating the key molecules for adipogenesis. [9]. For example, resveratrol decreases adipocyte differentiation through increasing expression of sirt1, which promotes fat mobilization by repressing PPAR-γ [48]. In addition, genistein, epigallocatechin gallate, and capsaicin were shown to inhibit adipogenesis by activating AMPK [49]. In this study, our data show a novel role of OTA for regulating adipocyte differentiation of hAMSCs through ERK-PPAR-γ pathway. These results provide the better understanding about the novel effect of OTA derived from fungal metabolite on cellular differentiation.
In conclusion, this study demonstrates that OTA inhibits adipogenesis via the ERK-PPAR-γ pathway in hAMSCs. These results provide a novel effect of OTA on differentiation, specifically adipogenesis, and also suggest that OTA might affect the differentiation of other types of stem cells. Further studies will be needed to reveal the effects of OTA on stem cell differentiation in vitro and in vivo.
Footnotes
Acknowledgments
This work was supported by a grant from the Korean Ministry of Education, Science and Technology (The Regional Core Research Program/Anti-aging and Well-being Research Center) and by the National Research Foundation of Korea Grant funded by the Korean Government (KRF-2007-341-C00027).
Author Disclosure statement
No competing financial interests exist.