
Abstract
Human adipose-derived stromal cells (hASCs) represent a multipotent stromal cell type with a proven capacity to undergo osteogenic differentiation. Many hurdles exist, however, between current knowledge of hASC osteogenesis and their potential future use in skeletal tissue regeneration. The impact of frozen storage on hASC osteogenic differentiation, for example, has not been studied in detail. To examine the effects of frozen storage, hASCs were harvested from lipoaspirate and either maintained in standard culture conditions or frozen for 2 weeks under standard conditions (90% fetal bovine serum, 10% dimethyl sulfoxide). Next, in vitro parameters of cell morphology (surface electron microscopy [EM]), cell viability and growth (trypan blue; bromodeoxyuridine incorporation), osteogenic differentiation (alkaline phosphatase, alizarin red, and quantitative real-time (RT)–polymerase chain reaction), and adipogenic differentiation (Oil red O staining and quantitative RT–polymerase chain reaction) were performed. Finally, in vivo bone formation was assessed using a critical-sized cranial defect in athymic mice, utilizing a hydroxyapatite (HA)-poly(lactic-co-glycolic acid) scaffold for ASC delivery. Healing was assessed by serial microcomputed tomography scans and histology. Freshly derived ASCs differed significantly from freeze–thaw ASCs in all markers examined. Surface EM showed distinct differences in cellular morphology. Proliferation, and osteogenic and adipogenic differentiation were all significantly hampered by the freeze–thaw process in vitro (*P < 0.01). In vivo, near complete healing was observed among calvarial defects engrafted with fresh hASCs. This was in comparison to groups engrafted with freeze–thaw hASCs that showed little healing (*P < 0.01). Finally, recombinant insulin-like growth factor 1 or recombinant bone morphogenetic protein 4 was observed to increase or rescue in vitro osteogenic differentiation among frozen hASCs (*P < 0.01). The freezing of ASCs for storage significantly impacts their biology, both in vitro and in vivo. The ability of ASCs to successfully undergo osteogenic differentiation after freeze–thaw is substantively muted, both in vitro and in vivo. The use of recombinant proteins, however, may be used to mitigate the deleterious effects of the freeze–thaw process.
Introduction
I
Various cytokines, mechanical signals, and stimuli have been observed to enhance the osteogenic differentiation of ASCs. The most well studied include the bone morphogenetic protein (BMP) signaling pathway. BMP-2 and BMP-4 have been shown to promote the osteogenic differentiation of ASCs [8,14 –16], and in fact ASCs require intact BMP/BMPR1B signaling to remain an osteo-competent cell type [6]. The retinoid signaling pathway has likewise been thoroughly documented to enhance ASC osteogenesis [7,17,18]. Numerous studies have explored other signaling pathways in a candidate fashion to enhance ASC osteodifferentiation, including, but not limited to, connective tissue growth factor, insulin-like growth factor (IGF), and Hedgehog signaling [19 –21]. The successful utilization of ASCs for skeletal tissue engineering hinges on a continued expansion of our understanding of those factors that both enhance and inhibit ASC osteogenic differentiation.
The potential breakthroughs in the use of human stem and stromal cells have prompted some individuals to store their own tissues in a fee-for-service fashion. The most obvious example is the cryostorage of umbilical cord-derived blood [22]. The process of cell freezing and the long-term storage under such conditions inevitably alters cellular processes [23]. Parameters such as cell cycle progression and proliferation, viability, and differentiation have all been observed to significantly differ based on freezing conditions [23 –25]. A small number of articles have examined the effects of freezing and cold storage on ASC biology. Thirumala et al. showed that the post-thaw viability of ASCs differs significantly based on method of freezing and rate of thaw [26]. Other studies have examined the effects of various cryoprotective agents on cell viability [27 –31]. In vitro cellular parameters, including proliferation and osteo- and adipogenic differentiation, have also been assessed after freeze–thaw [28 –30]. Most studies suggest that the osteo- and adipogenic differentiation of ASCs can be maintained in vitro post-thaw. These conclusions based on in vitro analysis, however, are not quantitative and are often based on high-powered fields of representative alkaline phosphatase (ALP) or Oil red O staining, and did not include in vivo correlations. Observations from our laboratory suggested that the freezing process of hASCs has deleterious effects on the ability of these cells to undergo osteogenic differentiation. In this study, we therefore examined the potentially deleterious effects of freeze–thaw on hASC proliferation and osteo- and adipogenic differentiation, both in vitro and in vivo. Finally, we sought to rescue the osteogenic potential of hASCs subjected to freeze–thaw with the use of recombinant human IGF-1 (rhIGF-1) and recombinant human BMP-4 (rhBMP-4).
Methods
Cell harvest
ASCs were harvested from human lipoaspirate from 4 separate women aged <50 years as previously described [19]. All specimens were derived from the flank, abdomen, and thigh regions only. Briefly, specimens were digested with a Type II collagenase solution at 37°C. The stromal vascular fraction was then pelleted and filtered at 100 μm pore size, and primary cultures were established at 37°C, 21% O2, 5% CO2. After 72 h in culture, hASCs were either passaged and placed in experiments or frozen in 90% fetal bovine serum (FBS) and 10% dimethyl sulfoxide at a density of 1 × 107 cells/mL. Two separate methods of cell freezing were used. First, ASCs were placed overnight in a Styrofoam container at −80°C, and subsequently transferred to liquid nitrogen for storage (data presented in Figs. 1 –7). Second and as a method of verification, a Mr. Frosty freezing container was used (Nalgene Labware), to approximate a −1°C/min cooling rate (data presented in Fig. 4, Method 2). Cryopreserved ASCs by either method were stored in liquid nitrogen for 2 weeks. Thereafter, cells were quickly thawed and placed in a standard growth medium for 24–48 h expansion before seeding of experiments. Thus, passage 1 hASCs were used for all experiments unless otherwise stated.
Osteogenic differentiation among hASCs subjected to a 2-week freeze–thaw (frozen) as compared with control (fresh). (
Comparison of freezing methods on in vitro cellular parameters. ASCs were either used freshly after isolation, or frozen for 2 weeks under 2 conditions: either (1) frozen at −80°C overnight using a Styrofoam container followed by storage in liquid nitrogen, or (2) using a Mr. Frosty freezing device to approximate a −1°C/min decline in temperature.
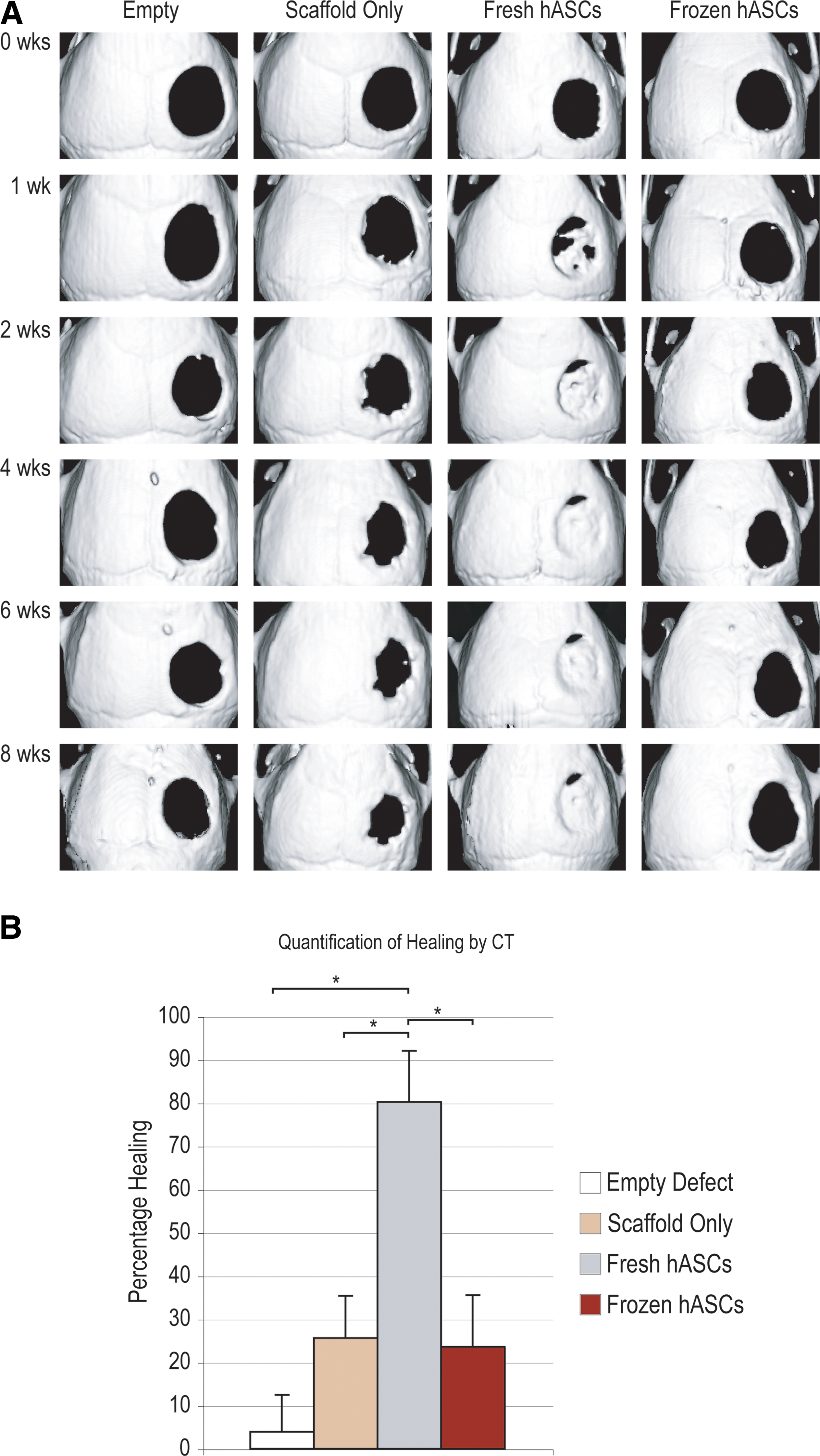
Calvarial defects as shown by microcomputed tomography (CT) analysis. Four millimeters calvarial defects were allowed to heal for up to 8 weeks; serial microCTs were performed.
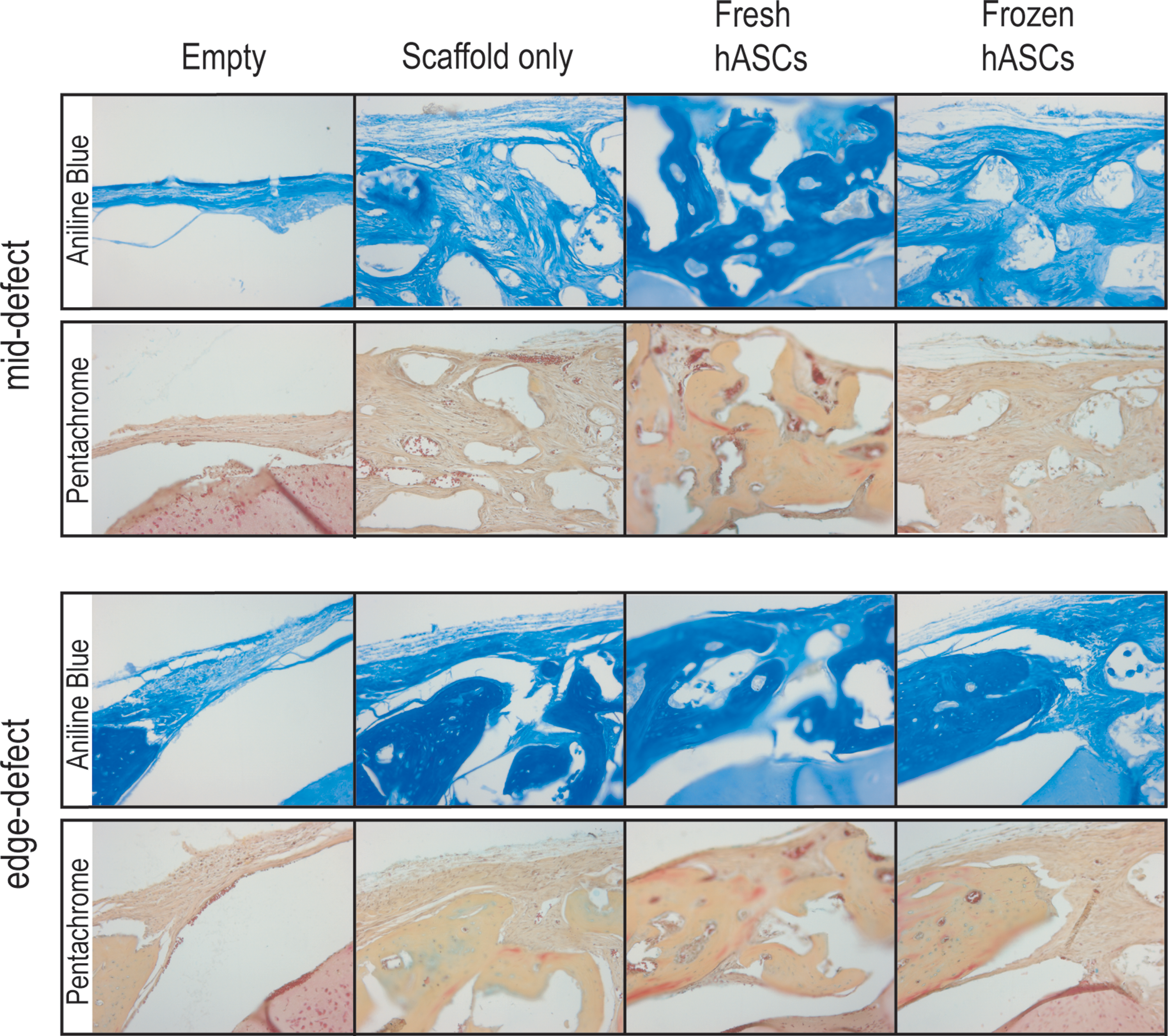
Histology of calvarial defects. Four millimeters calvarial defects were allowed to heal for 4 weeks before histological analysis by aniline blue and pentachrome. Four treatment groups included those defects left empty (empty), those treated with a scaffold but without hASCs (scaffold only), those treated with a fresh hASC-engrafted scaffold (fresh hASCs), and, finally, those treated with a frozen hASC-engrafted scaffold (frozen hASCs). Pictures were taken of the mid-point and edge of the defect site. In aniline blue stains, bone appears dark blue. In contrast, in pentachrome stain, bone appears yellow. Color images available online at
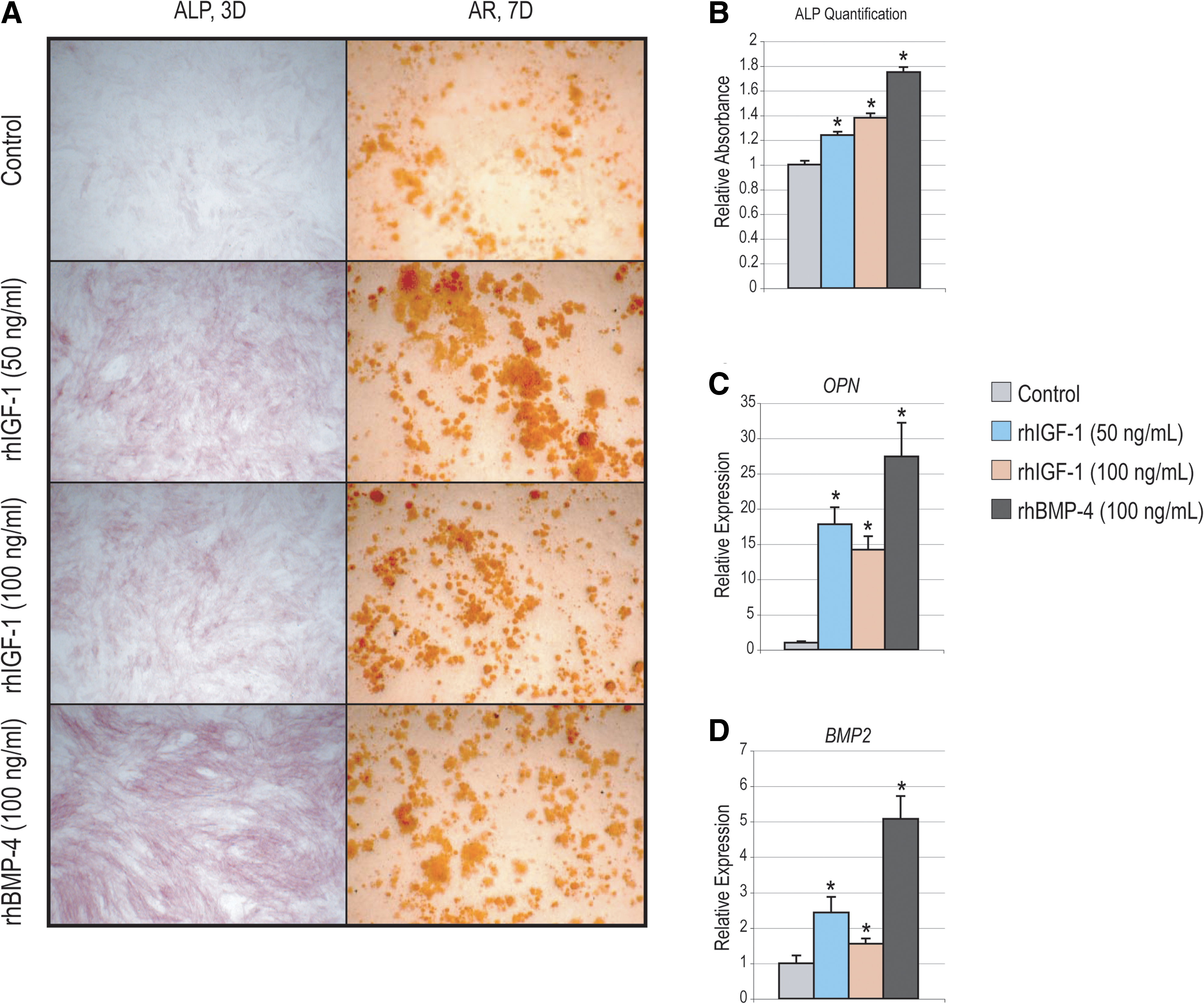
Rescue of frozen hASC osteogenic differentiation.
Surface electron microscopy
To examine cellular morphology, surface electron microscopy (EM) was performed on cells after 48 h in culture. Cells were fixed on glass cover-slips (12 mm circular, 15,000 cells) with 4% paraformaldehyde and 2% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.2), rinsed in the same buffer, and postfixed for 1 h with 1% aqueous OsO4. After dehydration in an ascending ethanol series, samples were critical point dried with liquid CO2 in a Tousimis Autosamdri-814 apparatus (Tousimis), mounted on adhesive carbon films on 15 mm aluminum stubs (Ted Pella), and sputter coated with 100A of Au/Pd using a Denton Desk 11 Sputter Coater. Samples were observed with a Hitachi S-3400N Variable Pressure scanning electron microscopy operated at 15 kV, working distance 7–8 mm, and secondary electron detection under high-vacuum conditions (<1 Pa). Images were captured in TIFF format. Representative images were taken at 100 × and 600 × ; regions of particular interest were then examined and recorded at higher magnifications.
In vitro culture assays
Cell viability was compared between fresh and frozen (post-thaw) ASCs by Trypan blue staining. In the case of fresh ASCs, p0 cells were trypsinized and cell counting with Trypan blue was performed. In the case of frozen ASCs, Trypan blue staining was performed immediately after thawing. Percentage Trypan blue-positive cells was calculated (n = 6 per group). To assess cell attachment, 1 × 105 ASCs were seeded in 6-well plates in 2 mL standard growth medium. After 8 h, cells were counted by hemocytometer of both the unattached cells in the supernatant and attached cells after trypsinization. Percentage attachment was calculated (n = 6 per group).
Proliferation of hASCs was assessed by bromodeoxyuridine (BrdU) incorporation and cell counting as previously described [32]. Cells were grown in 96-well plates seeded at a density of 1,000 cells/well. After 1, 2, and 3 days in culture, BrdU labeling was performed for 8 h. Next, BrdU incorporation was quantified using photometric enzyme-linked immunosorbent assay (Roche Applied Science). For cell counting, 10,000 cells were seeded in 12-well plates and the growth medium was replenished every other day. Cells were counted on 1, 3, 5, and 7 days of growth.
Adipogenic differentiation was assessed as previously described [33]. ASCs were seeded in 6-well plates at a density of 150,000 cells/well. The adipogenic differentiation medium containing Dulbecco's modified Eagle's medium, 10% FBS, 1% penicillin/streptomycin, 10 μg/mL insulin, 1 μM dexamethasone, 0.5 mM methylxanthine, and 200 μM indomethacin was added after cell attachment. After 4 days, the medium was exchanged for 10 μg/mL insulin. Oil red O staining and photometric quantification were performed at 1 week of differentiation [33]. Quantification by measurement of absorbance was performed after leaching with isopropranol. Finally, specific gene expression was examined after 1 week by quantitative real time (RT)-polymerase chain reaction (qRT-PCR) (see Supplementary Table S1, available online at
For osteogenic differentiation, cells were seeded onto 6-well plates at a density of 100,000 cells/well. After attachment, cells were treated with the osteogenic differentiation medium (Dulbecco's modified Eagle's medium, 10% FBS, 100 μg/mL ascorbic acid, 10 mM β-glycerophosphate, and 1% penicillin/streptomycin). The medium was changed every 3 days. ALP staining and quantification was performed at 3 days to assess early osteogenic differentiation as previously described [32]. Briefly, hASCs were fixed with an acetone/citrate solution and stained with a diazonium salt solution composed of fast violet blue salt/naphthol AS-MX phosphate solution. Quantification of ALP activity was normalized to total protein content by measuring the p-nitrophenol formed from the enzymatic hydrolysis of p-nitrophenylphosphate. This quantity was normalized to total protein quantity (Pierce). Alizarin red (AR) staining and photometric quantification was performed at 7 days [34]. Cells were fixed in ethanol and stained with an AR solution. Staining was quantified by extraction with a cetylpyridinium chloride solution and measurement of absorbance. Von Kossa staining was after 7 days of differentiation. Briefly, cells were washed with phosphate-buffered saline (PBS), fixed in formalin, incubated with 1% aqueous silver nitrate under ultraviolet light, and stained with 5% sodium thiosulfate. Finally, gene expression was analyzed and 3 and 7 days of differentiation by qRT-PCR (Supplemental Table S1).
RNA isolation and quantitative real-time PCR
Total RNA was isolated from cells and tissue as previously described [32,35]. After 2 washes in cold, sterile PBS, cells were lysed with a cell scraper. Isolation was performed with the RNeasy Mini Kit (Qiagen Sciences). Reverse transcription was performed with 1 μg RNA using TaqMan Reverse Transcription Reagents (Applied Biosystems). Quantitative real-time PCR was carried out using the Applied Biosystems Prism 7900HT Sequence Detection System and Sybr Green PCR Master Mix (Applied Biosystems). Specific primers for the genes examined were designed based on their PrimerBank sequence and are listed in Supplemental Table S1. The levels of gene expression were determined by normalizing to values of Gapdh and performed in triplicate.
Calvarial defects
Critical-sized (4 mm) calvarial defects were created in the right parietal bone of 60-day-old male CD-1 nude mice using a high-speed dental drill as previously described [12]. A skin incision was made just off the midline, the pericranium was removed, and a full-thickness calvarial defect was created. The dura mater, sagittal, coronal, and lambdoid sutures were left undisturbed.
In preparation for cell engraftment, scaffolds were seeded with hASCs as previously described [12]. About 150,000 hASCs were resuspended in 25 μL of the growth medium, and left for 24 h of incubation. Before engraftment, scaffolds were rinsed in sterile PBS. Animals were divided equally into 4 treatment groups: (1) empty defects, in which a 4 mm defect was created but left empty, (2) scaffold only, in which a scaffold without cells was placed in the defect site, (3) nonfrozen (fresh) hASCs with scaffold, in which hASCs were impregnated in a scaffold, and (4) frozen (post-thaw) hASCs with scaffold (n = 5 per group).
Scaffold creation
Apatite-coated poly(lactic-co-glycolic acid) (PLGA) scaffolds were fabricated from 85/15 PLGA (Birmingham Polymers) by solvent casting and a particulate leaching process [11,12]. Briefly, PLGA/chloroform solutions were mixed with sucrose to obtain 92% porosity (volume fraction), and compressed into thin sheets in a Teflon mold. After freeze-drying overnight, scaffolds were immersed water to dissolve the sucrose, and gently removed. After particulate leaching, scaffolds were disinfected with ethanol and dried under a laminar flow hood.
Microcomputed tomography
Microcomputed tomography was performed, using a high-resolution MicroCAT II™ (ImTek Inc.) small animal imaging system [12,13]. The 2-dimensional projection images were used to reconstruct tomograms with a Feldkamp algorithm, using a commercial software package (Cobra EXXIM; EXXIM Computing Corp.), resulting into a resolution of 80 μm. Three-dimensional reconstructions were generated by MicroView software (GE Healthcare). Percentage healing was calculated relative to computed tomography images taken immediately postoperatively, and with the use of Adobe Photoshop.
Histology
Animals were sacrificed by CO2 asphyxiation and cervical dislocation to confirm radiographic findings. Briefly, tissues were formalin fixed, decalcified with 19% ethylenediaminetetraacetic acid, paraffin embedded, and sectioned at 8 μm width per standard protocols [36]. To assess bone healing, Aniline Blue staining was performed on every 10th section throughout the sample to provide detailed histology of the regenerate. Next, select slides were stained with Pentachrome, in which bone appears bright yellow.
Statistical analysis
Means and standard deviations were calculated from numerical data, as presented in the text, figures, and figure legends. In figures, bar graphs represent means, whereas error bars represent one standard deviation. Statistical analysis was performed using an appropriate 2-tailed Student's t-test or analysis of variance (ANOVA) test as described in the figure legends. For Figs. 2, 3B, and 3C, a Student's t-test was used. For Figs. 5 and 7, a 1-factor ANOVA was used. For Fig. 3D, a 2-factor ANOVA was used (cell population and time point). Inequality of standard deviations was excluded by employing the Levene's test. For all statistical analysis, *P ≤ 0.05 was considered to be significant; an exact value is stated in the text.
Results
Effects of freezing on hASC morphology
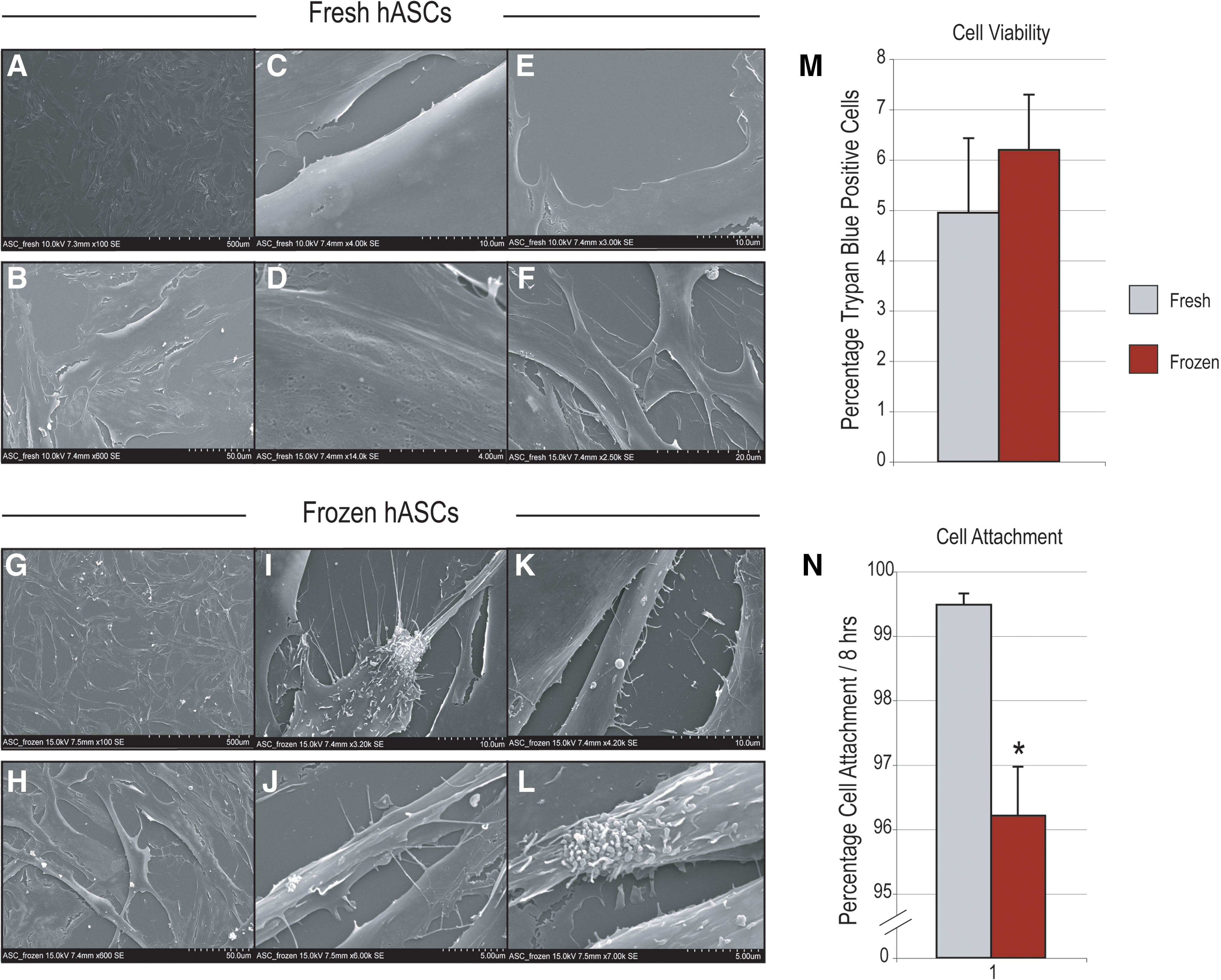
To detail the effects of freezing, cell morphology was first examined by scanning electron microscopy after 2 days in culture (Fig. 1). On the lowest magnification (100 × ), hASCs adopted similar morphologies independent of whether they had undergone freezing (compare “fresh” hASCs Fig. 1A to “frozen” hASCs Fig. 1G). Both populations generally had a fibroblast-like cell morphology, and both were observed to be subconfluent at the time of observation. At 600 × magnification, fresh hASCs were noted to have a flattened morphology, with broad and extended cell boundaries and without significant 3-dimensional rounding of cell edges (Fig. 1B). In addition, most cell edges were smooth and primarily without cellular projections. In contrast, freeze–thaw hASCs at 600 × were observed to be more stellate in appearance as opposed to the flattened and broad appearance of fresh hASCs (Fig. 1H). For example, cell width often fell within 10 μm among the freeze–thaw hASCs. For the most part, cell width remained over 20 μm among fresh hASCs. In addition, the cell edges among freeze–thaw hASCs appeared roughened or sometimes scalloped in comparison to the more smooth edges among fresh hASCs. At higher magnifications, more differences were observed between fresh and frozen hASCs (compared Fig. 1C–F to 1I–L). Cell edges among fresh hASCs were observed to be quite smooth with little contour deformities (see eg, Fig. 1C, E). In remarkable dissimilarity, a number of unusual features were observed among frozen hASCs. These included cell surface/edge projections that at times were finger like (Fig. 1K), stellate (Fig. 1I, J), or even appearing ciliated in nature (Fig. 1L). Thus, significant cell surface features of hASCs were observed to change upon freeze–thaw.
Effects of freezing on hASC viability, attachment, and proliferation
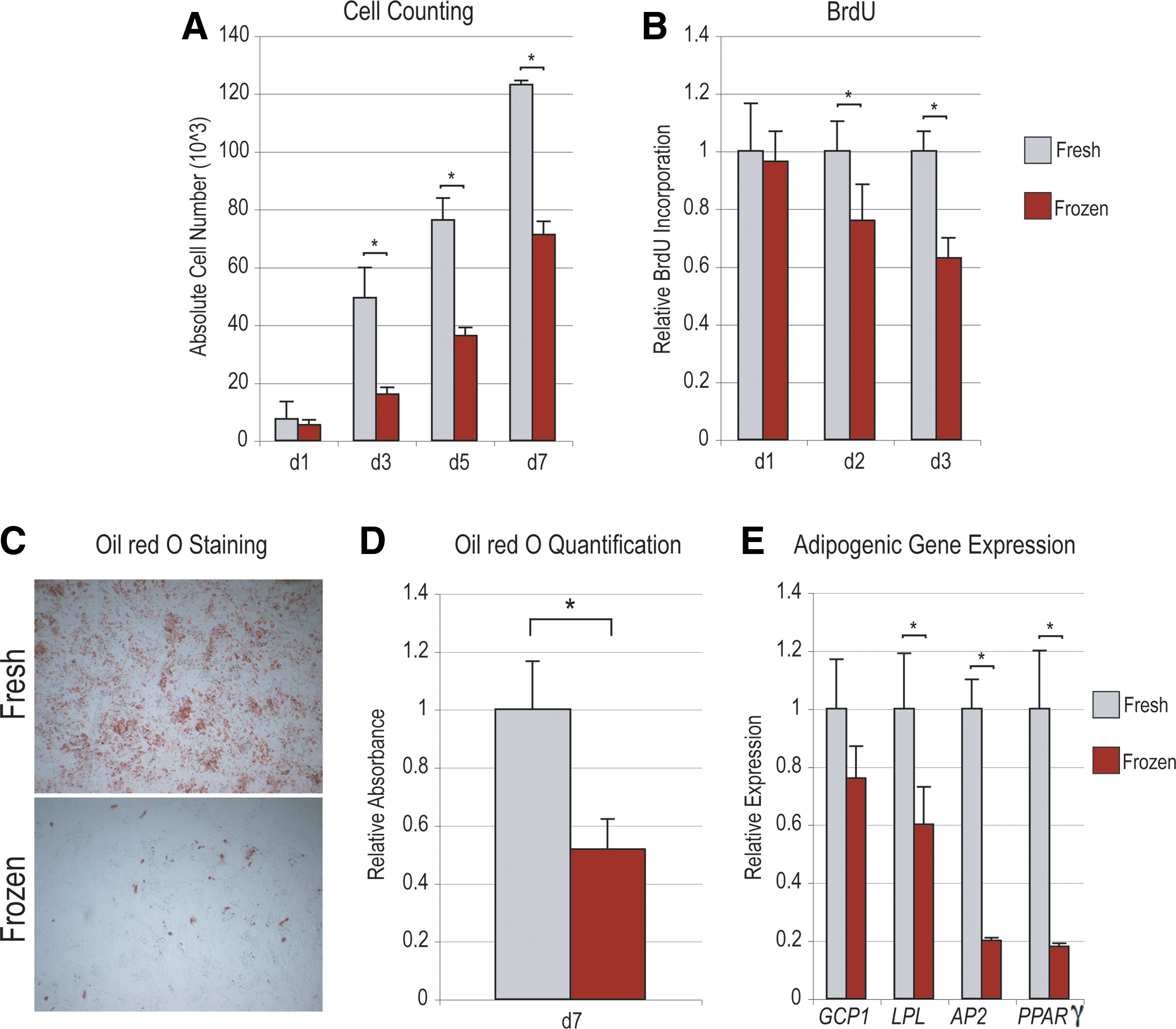
Next, parameters of cellular viability, attachment, and proliferation were compared between “fresh” and “frozen” hASCs (Figs. 1M, N, and 2). Freeze–thaw showed no statistically significant effect on cell viability (Fig. 1M), as assessed by Trypan blue staining immediately post-thaw. Those cells subjected to freezing did show a small but significant impairment in cell attachment: in the case of “fresh” hASCs over 99% cell attachment was observed, as compared with ∼96% attachment among post-thaw hASCs (Fig. 1N). Next, cellular proliferation was assessed by cell counting and BrdU assays (Fig. 2A, B). After equal seeding, cell counting assays were performed over a period of 7 days (Fig. 2A, n = 3). Results showed that fresh hASCs grew in standard culture conditions to a significantly greater degree than frozen hASCs (*P = 0.0056, 0.001, and 0.0001 at 3, 5, and 7 days, respectively). Similar results were observed with BrdU incorporation assays, performed at 1, 2, and 3 days of growth (Fig. 2B, n = 6). A significant increase in BrdU incorporation among fresh relative to frozen hASCs was recorded at 2 and 3 days (*P = 0.0047 and 0.0001, respectively).
Effects of freezing on hASC adipogenic differentiation
Next, adipogenic differentiation of hASCs was performed. Previous observations from our laboratory suggest that over a period of 1 week in culture, significant intracellular lipid accumulation is observable in vitro [33]. Lipid accumulation was compared between hASCs that had undergone a freeze–thaw as compared with control hASCs (fresh) by Oil red O staining (Fig. 2C, D). A significant attenuation of lipid formation was observed among freeze–thaw as compared with fresh hASCs, both by microscopic examination and photometric quantification (*P = 0.0076). To verify a decrease in adipogenic differentiation among frozen hASCs, gene expression was assayed by qRT-PCR (Fig. 2E). All markers examined, including GCP1, LPL, AP2, and PPAR-γ, showed a decrease in transcript abundance among frozen hASCs after 7 days adipogenic differentiation (although this did not reach significance in GCP1). Thus, freeze–thaw was observed to have a deleterious effect on both proliferation and adipogenic differentiation of hASCs in vitro. We next turned to the osteogenic differentiation of hASCs.
Effects of freezing on hASC osteogenic differentiation
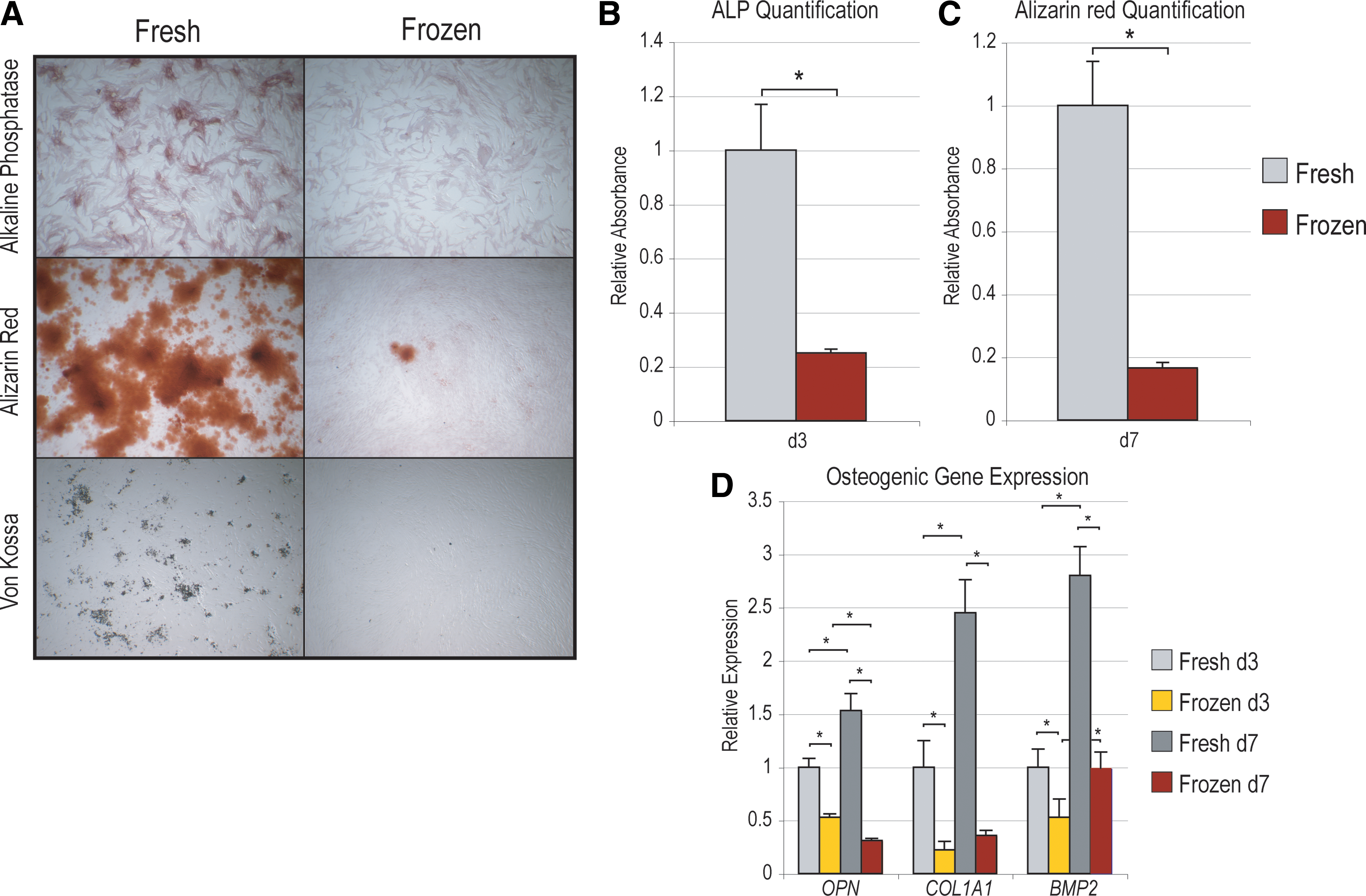
First, hASC osteogenic differentiation was assessed by ALP staining at 3 days differentiation, in which ALP appears purple (Fig. 3A, top). Microscopic examination showed a significant attenuation of ALP staining among freeze–thaw hASCs, although numerous individual cells remained ALP positive. This contrast was even more apparent when bone nodule formation was assessed by AR and Von Kossa staining (Fig. 3A, middle, bottom). While abundant staining was observed under control conditions (fresh hASCs), those ASCs subjected to freeze–thaw showed very sparse mineralization. ALP and AR staining were quantified by absorbance assays, normalized to total protein (Fig. 3B, C), each showing an ∼80% decrease among frozen as compared with fresh hASCs (*P = 0.0016 and 0.0005, respectively). Finally, osteogenic gene expression was assessed by qRT-PCR after 3 and 7 days differentiation, including the extracellular proteins Osteopontin (OPN) and Type I Collagen (COL1A1), and osteogenic cytokine BMP-2 (Fig. 3D). Results showed that each individual marker increased in expression over the time course of differentiation in fresh hASCs (*P < 0.05). However, among frozen hASCs, expression was attenuated among all markers and at all time points (*P < 0.05 for both 3 and 7 days).
Comparison of freezing methods on in vitro ASC parameters
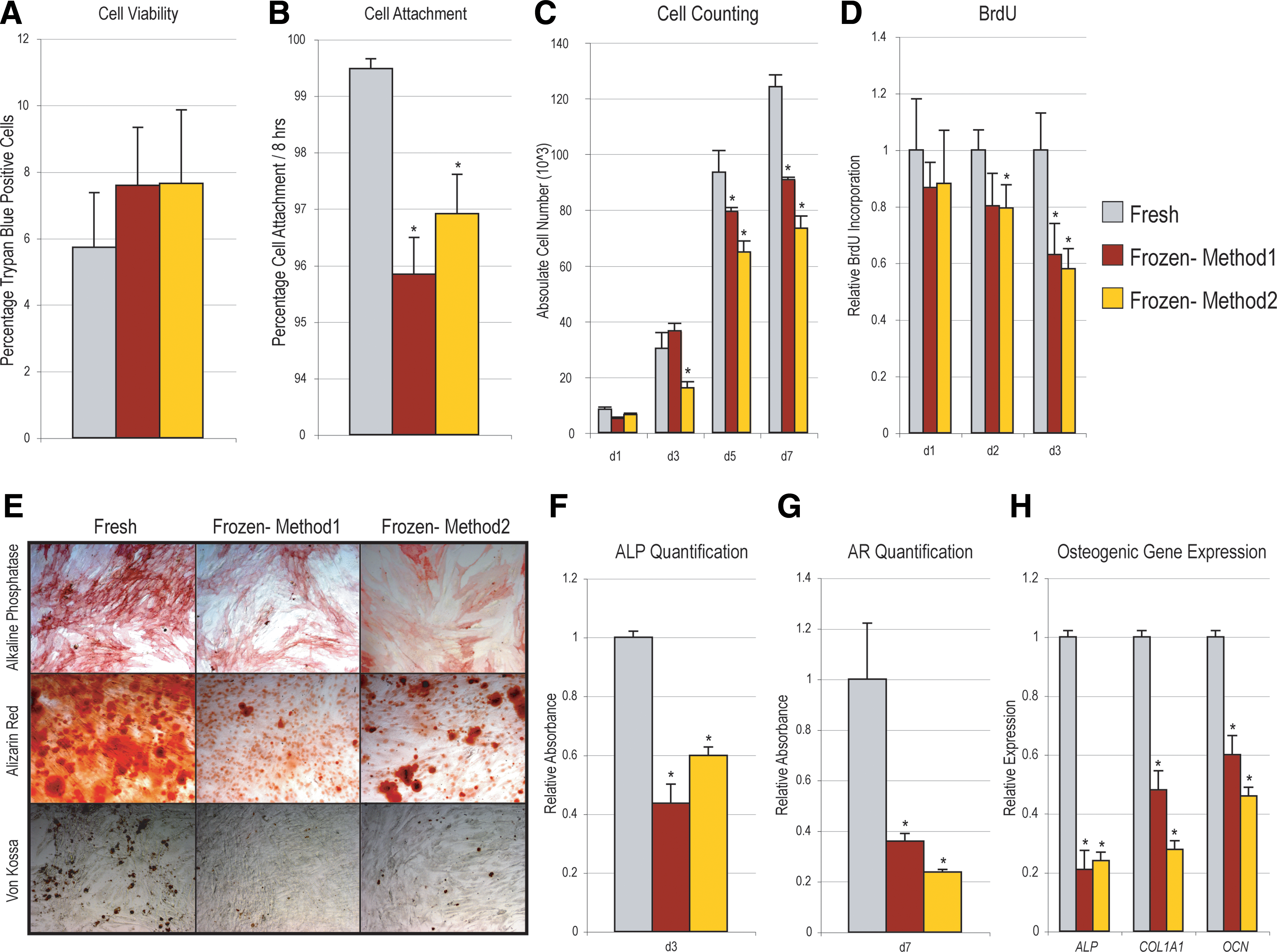
After freeze–thaw, we observed a significant decrease in ASC proliferation and differentiation in vitro. Would this same observation extend to alternative techniques for cell freezing? To answer this, the rate of ASC freezing was more precisely controlled to approximate a −1°C/min cooling rate using a Mr. Frosty device (Fig. 4, see yellow bars, Method 2). In comparison, hASCs from the same patient were frozen per original technique (Method 1), or assayed without freezing (“fresh”). Results showed that either method of cell freezing resulted in similar effects (compare red and yellow bars, Fig. 4). While neither method of freezing significantly impaired cell viability, both resulted in a small but significant decrease in cell attachment (Fig. 4A, B, *P < 0.05). As well, both methods of freezing significantly decreased hASC proliferation and osteogenic differentiation, by all markers examined (Fig. 4C–H, *P < 0.05). Thus, across 2 different methods of hASC freezing, similar deleterious effects were observed.
Effects of freezing on hASC in vivo healing of a calvarial defect
Next, we sought to extend our in vitro observations to a standard model of bone regeneration via the engraftment of hASCs into a critical-sized calvarial defect in an athymic mouse (Fig. 5). As expected of a critical-sized defect, those animals left untreated showed minimal healing at up to 8 weeks postinjury (far left column, Fig. 5A). As a control, defects were treated with hydroxyapatite (HA)-PLGA scaffold only; these defects as well showed little healing, although this was significantly greater than untreated or “empty” defects (middle left column, Fig. 5A). Next, per standard protocol, hASCs were seeded onto hydroxyapatite (HA)-PLGA scaffolds and subsequently engrafted into 4 mm defects (right columns, Fig. 5A). These hASCs were either freshly derived or freeze–thaw as per our in vitro assays (fresh as compared with frozen hASCs). Fresh hASCs showed robust healing of a defect within the first weeks postoperatively (middle right column, Fig. 5). In contrast, however, frozen hASCs showed little osseous healing, even 8 weeks postoperatively (far right column, Fig. 5). These findings were subsequently quantified at the 4 week time point, which verified that freeze–thaw had a significant deleterious effects on the in vivo osteogenic differentiation of hASCs (Fig. 5B). For example, those defects left empty healed by ∼4%. The grafting of a scaffold only increased healing to ∼26%, although this did not reach statistical significance (*P = 0.078 in comparison to empty defects). In marked contrast, fresh hASC-engrafted scaffolds led to an ∼80% healing over just 4 weeks (*P = 0.0034). This was not observed among frozen hASC-engrafted defects, in which the average healing was 24%, a significant decrease in comparison to fresh hASC-engrafted defects (*P = 0.014).
Differences between calvarial defect treatment groups were further analyzed by histological analysis. Images are presented at the edge and center of each treatment type, at 4 weeks postoperatively. Those defects left empty showed were hypocellular and showed very little bone formation (far left, Fig. 6). Those defects engrafted with a scaffold only showed a significantly more cellular defect and with some bony healing, particularly at the edges of the defect (middle left, Fig. 6). In contrast, robust and thick bone regenerate was observed throughout defects (mid-defect and edge-defect) engrafted with fresh hASCs (middle right, Fig. 6). Strikingly, defects engrafted with frozen hASCs more closely resembled the “scaffold only” group, without significant bone formation (far right, Fig. 6). Thus, the process of freeze–thaw was observed to have a deleterious effect on the osteogenic differentiation of hASCs both in vitro and in vivo.
Effects of recombinant IGF-1 and BMP-4 on frozen hASC osteogenic differentiation
We next examined whether known pro-osteogenic cytokines could “rescue” the deleterious effects of freeze–thaw on the osteogenic differentiation of hASCs. In a candidate fashion, we utilized rhIGF-1 (50–100 ng/mL) and rhBMP-4 (100 ng/mL) [19,37]. Concentrations were utilized based on previously data among fresh hASCs. Results showed that frozen hASCs remained responsive to the pro-osteogenic effect of both rhIGF-1 and rhBMP-4 (Fig. 7). This included an upregulation of both ALP after 3 days differentiation and AR staining after 7 days differentiation (Fig. 7A). Quantification of ALP activity revealed a similar and statistically significant increase above baseline in frozen hASCs (Fig. 7B). Likewise gene expression of OPN and BMP2 significantly increased either with addition of rhIGF-1 or rhBMP-4 at 3 days differentiation (Fig. 7C, D).
Discussion
This study expands on what is already known regarding the potential use of ASCs for osseous tissue repair and regeneration. Prior studies have focused on various cellular insults to ASC and their subsequent in vitro properties, including their proliferation and differentiation. These cellular insults ranged broadly, from transient and prolonged hypoxia [38,39], laser irradiation [40], and freeze–thaw [26]. In the case of hypoxia, previous studies from our laboratory have observed that even transient exposure to hypoxia (1% O2) significantly impairs the ability of mouse ASCs to differentiate down an osteogenic lineage [38]. These deleterious effects of hypoxia are not irreversible, however, as we previously observed incubation with various histone deacetylase inhibitors (including valproic acid and sodium butyrate) to rescue the osteogenic differentiation of ASCs exposed to hypoxic insult [4]. In much the same way, we sought here to examine the potentially deleterious effects of freeze–thaw on hASC proliferation and differentiation. Our results suggest that numerous cellular parameters are impacted by freeze–thaw, including a downregulation of osteogenic differentiation. This, however, is ameliorated, or rescued by exposure to recombinant growth factors such as rhBMP-4 or rhIGF-1.
ASCs of human origin have a capacity to undergo rapid osteogenic differentiation in vitro. In our observations, hASCs show bone nodule formation as early as after 1 day in the differentiation medium [41], and show robust mineralization within 1 week in vitro [19]. Our laboratory has previously documented the ability of ASCs derived from human origin to heal the critical defect model [12], in a period of only 4 weeks by radiographic analysis. In the present study we again observed that freshly isolated, undifferentiated hASCs successfully healed a critical-sized cranial defect. Interestingly, however, the same defect when engrafted with frozen hASCs underwent little healing. In fact, the healing observed in a defect with an empty, osteoconductive HA-PLGA scaffold only showed slightly more healing than those engrafted with frozen hASCs. This suggests that not only were freeze–thaw hASCs of no utility in the healing of a mouse calvarial defect, but also they, in fact, may have impeded successful bone regeneration. In our opinion, this observation has real and substantive importance in the future translation of hASCs to skeletal tissue engineering in the human patient. It may be that in the future a 1-stage procedure for derivation of hASCs via liposuction followed by engraftment in a skeletal defect is ideal. This would then obviate the need for cryostorage of ASCs and circumvent any potential deleterious effects.
When comparing the osteogenic differentiation of fresh and frozen hASCs in our study, it is important to recognize that at baseline no cytokines or signaling factors were added to our standard differentiation medium. A handful of cytokines and other factors are known to stimulate osteogenic differentiation in ASCs. These include but are not limited to retinoic acid and Vitamin D [18], BMPs [6,8,15,16,42], and Sonic Hedgehog [21]. In a candidate fashion, we utilized 2 recombinant proteins to potentially rescue the osteogenic differentiation of frozen hASCs: rhIGF-1 and rhBMP-4. IGFs are known mediators of skeletal growth and bone formation [43 –46]. Specifically, IGF-1 has been shown to promote the differentiation of bone cells in both an autocrine and paracrine manner [47,48]. Prior investigators have shown IGF-1 to promote osteogenesis in bone marrow mesenchymal cells in vitro [49]. In our previous study, we observed rhIGF-1 to have a similar osteogenic effect in fresh hASCs, particularly in the presence of platelet-derived growth factor [19]. BMPs, first recognized to induce ectopic bone formation, have been thoroughly studied in the differentiation of ASCs by methodologies, including addition of recombinant protein, addition of neutralizing antibodies, viral over expression, and siRNA [6,15,42,50]. These studies suggest, in summary, that BMP-2 is sufficient to enhance ASC osteogenic differentiation and conversely BMP/BMPR1B signaling necessary for the normal progression of osteodifferentiation to occur. Here we extend this observation to include BMP-4 as well, not surprising as hBMP-2 and hBMP-4 share 85% amino acid sequence homology (R&D Systems). Our results suggest that both rhIGF-1 and rhBMP-4 are suitable molecular stimuli to mitigate the deleterious effects of freeze–thaw on hASC osteogenesis.
One would postulate that the use of such cytokines in addition to hASC engraftment could be easily used to more rapidly heal a critical-sized cranial defect. In fact, the marriage of cell biology, molecular biology, and mechanical engineering may lead to the most benefits in the field of tissue engineering. For example, in our model system, we utilize a multipotent cell type (hASCs), a potent pro-osteogenic stimulus (eg, BMP-4), and an appropriate biomimetic, osteoinductive scaffold (HA-PLGA among others). Various methods may exist for the potential delivery of osteogenic stimuli and cytokines. The relatively unsophisticated subcutaneous injection over a cranial defect site may do. However, a more novel approach may utilize hASCs engrafted on a scaffold that releases growth factors to promote even more robust bone formation. Alternatively, a short treatment period in vitro followed by in vivo engraftment may be just as advantageous without potentially stimulating neo-osteoclastogenesis (as has been observed with both retinoic acid and BMPs) [7,17]. These potential methods of cytokine stimulation will be the focus of future endeavors.
Finally, we acknowledge that limitations to the present study exist toward to the extrapolation to all scenarios. First, only 2 methods of freezing were employed in the study: gradual freezing using either a Styrofoam container or a Mr. Frosty device, but both using the same freezing medium (90% FBS, 10% dimethyl sulfoxide). We chose this for 2 reasons: first, out of simplicity of comparison we sought to compare fresh hASCs of identical human derivation to a frozen population from the same hASC pool; second, we anticipated that those conditions chosen by our laboratory are not dissimilar to those in other basic science laboratories. However, recent advances have shown that other cryoprotective agents are in fact superior in preserving ASC differentiation potential [30,31]. These articles, however, largely do not quantify differences in osteo- or adipogenic differentiation and do not extend their findings to in vivo studies. Therefore, our study may serve either as impetus toward the betterment of procedures of cryostorage of hASCs or alternatively toward the avoidance of freezing hASCs in general. Previous observations from our laboratory and others have found that the osteo- and adipogenic differentiation of ASCs does vary based on patient, age, gender, and anatomic site of derivation [51 –55]. Thus, to minimize potential heterogeneity, all hASCs were consistently derived from young, female patients from the flank and thigh regions only. We anticipate that hASCs derived from dissimilar donors would have a similar cellular reaction to freeze–thaw. We cannot exclude, however, that hASCs from a different demographic would have an altogether different and unexpected reaction to freeze–thaw. Future studies must verify if these data are generalizable to hASCs as a whole.
Conclusions
The freezing of hASCs for storage significantly impacts their biology, both in vitro and in vivo. Most significantly, the ability of ASCs to undergo osteogenic differentiation after freeze–thaw is substantively muted, both in vitro and in vivo. The use of recombinant proteins such as IGF and BMP, however, may be used to mitigate the deleterious effects of the freeze–thaw process.
Footnotes
Acknowledgments
This study was supported by National Institutes of Health, National Institute of Dental and Craniofacial Research grant R21 DE-019274; the Hagey Laboratory for Pediatric Regenerative Medicine and the Oak Foundation to M.T.L.; National Institutes of Health, National Institute of Arthritis and Musculoskeletal and Skin Diseases grant 1F32AR057302-01 to B.L.; and Genentech Foundation Fellowship to A.W.J. We thank Dr. Lydia-Marie Joubert for her expertise and technical assistance.
Author Disclosure Statement
The authors have no financial interest in any of the products, devices, procedures, or anything else connected with this article. There was no internal or external funding received to complete this study. University of Stanford IRB approval was obtained before commencement of the study (IRB # 2188, 9999).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.