
Abstract
Embryonic stem cells seem to have the intriguing capacity to divide indefinitely while retaining their pluripotency. This self-renewal is accomplished by specialized mechanisms of cell-cycle control. In the last few years, several studies have provided evidence for a direct link between cell-cycle regulation and cell-fate decisions in stem cells. In this review, we discuss the peculiarities of embryonic stem cell-cycle control mechanisms, implicate their involvement in cell-fate decisions, and distinguish centrosomes as important players in the self-renewal versus differentiation roulette.
Unique Cell-Cycle Characteristics in Embryonic Stem Cells
P
Cell-Cycle Regulation in Somatic Cells
The cell cycle, a highly organized process of cell growth, DNA replication, chromosome separation, and cell division, is regulated by cyclin-dependent kinases (CDKs). These kinases, among other functions, phosphorylate the retinoblastoma protein (pRB), thereby promoting cell division/cycle entry with progression from G1 to S phase. When unphosphorylated, pRB binds E2F transcription factors and represses E2F target gene expression. Phosphorylation of pRB by CDKs leads to the release of E2Fs and the upregulation of genes that drive cell-cycle progression to S phase (such as cyclin E or CDC25A) and take part in DNA replication (such as thymidine kinase or DNA polymerase alpha) (Fig. 1).

Regulation of G1-phase progression in somatic cells, mESCs, and hESCs. Somatic cells: In response to mitogenic signals, cyclin D is synthesized and activates CDK4/CDK6. Cyclin D-CDK4/CDK6 complexes phosphorylate pRB, releasing E2F factors, which activate the transcription of genes involved in cell-cycle progression, including cyclin E and CDC25A. Subsequently, cyclin E-CDK2 complexes are activated and further phosphorylate pRB, releasing more E2Fs to activate S-phase gene transcription. Cyclin E-CDK2 drives G1/S transition by phosphorylating prereplication complexes and is involved in centrosome duplication. In ESCs, the cell-cycle machinery is, in part, regulated by pathways of self-renewal, which upregulate the expression of positive (eg, cyclin D1 in mESCs) and downregulate expression of negative cell-cycle regulators (eg, p16Ink4a, p21Cip1). Overall, CDK activity in ESCs is much higher than in somatic cells and drives rapid progression through the cell cycle. Dashed line, inactive regulatory pathway; ?, suggested/possible regulation. mESC, mouse embryonic stem cells; hESCs, human embryonic stem cells; CDK, cyclin-dependent kinases; pRB, retinoblastoma protein.
The phosphorylation of pRB is sequential. First, in response to growth factor and/or mitogenic signaling, which induce cyclin D expression, pRB is phosphorylated by cyclin D-CDK4/CDK6 complexes, leading to progression through G1 phase. Induction of E2F-directed cyclin E and CDC25A expression results in the formation and activation of cyclin E-CDK2 complexes, which cause further pRB phosphorylation and drive the G1-S transition. Cyclin A-CDK2 complexes then regulate S- and G2-phase events, and finally, cyclin A/B-CDK1 complexes promote entry into mitosis and control early mitotic events.
For activation, CDKs have to be bound by cyclins, whose levels vary in a cyclical fashion during the cell cycle. Further, CDK4, CDK2, and CDK1 require phosphorylation on Thr172, Thr160, and Thr161, respectively, by CDK-activating kinase (CAK, a complex of CDK7 and cyclin H) [7,8]. The activity of CDK2 and CDK1 is additionally held in check by inhibitory phosphorylation on Thr14/Tyr15 from Wee/Myt1 [9,10], which is relieved by CDC25 phosphatases. Further, CDK4/CDK6 and CDK2/CDK1 complexes are inhibited by proteins of the Ink4 (p15Ink4b, p16Ink4a, p18Ink4c, p19Ink4d) and Cip/Kip (p21Cip1, p27Kip1, p57Kip2) families, respectively (reviewed by Sherr and Roberts [11]).
However, although most cells engage the above-described cell-cycle machinery, multiple knockout studies revealed that most cyclins/CDKs are dispensable for cell-cycle progression (reviewed by Hochegger et al. [12] and Malumbres and Barbacid [13]). These studies led to the current model that cyclins A and B and CDK1 represent the “core” components of the cell-cycle machinery and are absolutely integral elements of the cell-cycle engine [12,14], whereas interphase CDKs and cyclins D and E play essential roles only in the proliferation of specific cell types [12,13]. Especially in some stem-cell types, however, cell-cycle pathways seem to operate in a more rigid and nonredundant fashion [15].
Cell-Cycle Regulation in ESCs
With respect to cell-cycle regulation and structure, ESCs demonstrate substantial differences from somatic cells. ESCs proliferate at a fast rate. Progression through a single cell cycle takes about 8–11 h in mESCs [1 –3], 15–16 h in hESCs [16], and 12–21 h in rhesus monkey ESCs when compared with roughly 24 h for many somatic cell types in tissue culture [17]. The short cell-cycle duration results, for the most part, from a very short G1 phase that lacks a restriction point and a G1 checkpoint. In contrast to somatic cells, ESCs do not require exogenous growth factors to enter the cell division cycle, are not subject to contact inhibition, and do not enter quiescence upon growth factor withdrawal [18,19]. This independence from growth factors (ie, restriction point nonfunctionality) stems from constitutively hyperphosphorylated pRB (Fig. 1). Therefore, E2F target genes are constitutively expressed and ESCs progress rapidly to S phase, spending only a very short time (about 2 h in mESCs) in G1 phase. All interphase cyclins (D1, D3, E, A2) [2,18,20,21] are present in mESCs at comparable levels throughout the cell cycle [2,20], with cyclin levels being significantly elevated compared with somatic cells [21]. mESCs do not express CDK inhibitory proteins of the Ink4 and Cip/Kip families [18,20,22]. Therefore, most CDKs are active throughout the cell cycle in mESCs, with the exception of CDK1-cyclin B, which becomes selectively activated before mitosis [2], and CDK4-cyclin D1 complexes, which exhibit little to no activity in mESCs [20]. The driving force underlying the rapid cell-cycle progression in mESCs is the high and precocious activity of CDK2 [2]. Chemical inhibition of CDK2 activity prolongs cell-cycle duration [2] and slows the progression of mESCs through G1 phase [23]. Overexpression of the CDK2 inhibitor p21Cip1, for example, by downregulation of its negative regulator, the mir-290 cluster, has similar effects [24].
In hESCs, a shortened G1 phase has been attributed to elevated mRNA levels of cyclin D2-CDK4 and low levels of p21CipP1, p27Kip1, and p57Kip2 [16,25,26], which are controlled by miR-372 (p21Cip1) [27] and miR-92b (p57Kip2) [26] (Fig. 1). Also, CDK inhibitors of the Ink4 family are expressed at low levels or not at all [28,29]. Therefore, similar to mESCs, CDKs have very high activity, with CDK2 displaying the overall highest kinase activity in hESCs [30]. Also similar to mESCs, inhibition of CDK2 activity delays the G1-S transition [31] and knockdown of CDK2 leads to G1 arrest in hESCs [30], indicating a crucial role for CDK2 for G1-phase regulation in both mESCs and hESCs [23,30].
Linking Cell-Cycle Regulation to Cell-Fate Decisions in ESCs
Major changes in the expression of cell-cycle regulatory proteins and cell-cycle structure associated with differentiation provided the first hints of a connection between cell-cycle regulation and stem-cell potency. Further evidence came from studies in adult stem cells, particularly neural progenitor cells, which suggested that the length of G1 phase directly impacts cell-fate decisions (self-renewal vs. differentiation). These studies revealed that proliferating and self-renewing neural progenitors have shorter cell cycles than those committed to neuronal differentiation [32]. Inhibition of CDK activity prolonged their cell cycle and induced formation of neurons [33], whereas shortening of the cell cycle prevented neural differentiation [32]. Cyclin D1-CDK4 overexpression studies in neural stem cells have also shown that G1 lengthening is necessary and sufficient to switch neural progenitors from self-renewal to neurogenesis [34].
Recent studies have revealed the existence of similar regulatory connections between cell-cycle structure and self-renewal in both hESCs and mESCs and have shown that inhibition of cell-cycle progression is sufficient to induce differentiation of ESCs. Downregulation of CDK2 activity in hESCs by either chemical inhibition or knockdown led to G1/S-phase delay [31] and G1 arrest [30], respectively, and induced hESC differentiation, as indicated by downregulation of OCT4 [30,31] and expression of markers of extraembryonic lineages [30]. Likewise, CDK2 inhibition prolonged G1 phase and induced differentiation-associated changes in gene expression and morphology in mESCs [23]. Together, these studies suggest a crucial role for CDK2 in ESC self-renewal. On the other hand, it was shown that CDK2 is a nonessential protein as Cdk2-knockout embryos develop normally and CDK2-deficient mice are defective only in meiosis, where CDK2 appears to play an irreplaceable role [35,36]. However, these 2 observations are not necessarily exclusive. Knockout studies create conditions in which, among other mechanisms, certain proteins can substitute for the missing protein (eg, CDK1 is capable of taking over the role of CDK2), and further, ESCs in blastocyst represent a very transient state in early embryogenesis and are primarily destined to differentiate and succumb to multiple (intrinsic and extrinsic) signals regulating their fate; therefore, knocking-out CDK2 might not disturb cell-fate decisions in the developing early embryo.
Further evidence for a connection between cell-cycle regulation and cell-fate decisions comes from a study of CDK2-associated protein 1 (CDK2AP1; p12DOC-1), an inhibitor of G1-S transition that acts through downregulation of CDK2 [37], which revealed its role as a competency factor in mESC differentiation by modulating the phosphorylation level of pRB [38]. In this study, Cdk2ap1−/− mESCs were shown to be resistant to the leukemia inhibitory factor (LIF) withdrawal-induced differentiation that results in altered pRB phosphorylation [38]. The differentiation competency of Cdk2ap1−/− mESCs was restored upon ectopic expression of CDK2AP1 or a nonphosphorylatable pRB mutant [38]. The essentiality of RB family proteins in mESC differentiation was demonstrated in triple knockout mESCs (Rb1−/− , Rbl1−/− , Rbl2−/− ), which were incapable of undergoing proper differentiation [39]. As single- and double-knockout ESCs showed no defect in differentiation, RB family members appear to be able to compensate for one another in this setting [39]. These observations indicate the existence of an intrinsic link between cell-cycle regulation and cell-fate decisions (self-renewal vs. differentiation) in stem cells that is likely to be affected by the length of each phase of the cell cycle and suggest that G1 phase corresponds to a window of increased sensitivity of stem cells to differentiation signals [30].
Alternatively, prolongation of cell-cycle progression in stem cells might be sensed and interpreted as stress, leading to differentiation. In both ESCs [40] and adult stem cells [41], genotoxic stress induces differentiation as an alternative to apoptosis and senescence to maintain genetic stability in the stem-cell pool. Central roles in this “stemness checkpoint” have been attributed to proteins of DNA-damage response, including ataxia-teleangiectasia mutated (ATM) [41], p53 [40], and other proteins (reviewed by Ruzankina et al. [42]). However, the link between DNA-damage response signaling and the self-renewal machinery has not been fully elucidated yet and might differ between distinct stem-cell systems. It might include both direct signaling to self-renewal machinery [40] and cell-fate modulation through changes in cell-cycle regulation [43]: DNA damage-activated p53 induced mESC differentiation by suppression of Nanog expression [40], whereas treatment of hESCs with the p53 activator nutlin resulted in an accumulation of hESCs in G0/G1, associated with an increase in differentiation markers and a decrease in pluripotency markers, through upregulation of the CDK2 inhibitor p21Cip1 [43].
The hypothesis of a link between cell-cycle regulation and cell-fate decisions is further supported by the observation that core pluripotency factors OCT4, SOX2, and NANOG regulate the expression of key cell-cycle regulatory proteins such as CDK1, cyclin D1, CDK6, CDC25A, and CDC7 [29,44]. Evidence of another link between cell-cycle regulation and self-renewal machinery, namely, between E2F and OCT4, came from a study by Chavez and coworkers [45]. This work suggests that E2F may act as a regulatory cofactor for OCT4 at the promoters of OCT4 target genes and that Origin recognition complex subunit 1-like, a direct E2F target involved in DNA replication and mitotic cell-cycle progression, belongs to the core OCT4 regulatory network.
Many more connections likely exist between master cell-cycle regulators and stemness, including regulation via microRNAs [46]. For instance, the c-Myc/E2F-driven miR-17-92 cluster, which controls the G1-S transition, is fundamental for hESC self-renewal and cell proliferation and is downregulated upon hESC differentiation [47]. Another example is the miR-290 cluster that targets multiple cell-cycle regulatory genes, including Cdkn1a (encoding p21Cip1), Rb1, Rbl1, Rbl2, and Lats2 [48]. It is expressed in mESCs, is repressed during mESC differentiation, is undetectable in adult mouse organs [49 –51], and has been recently shown to be a general feature of pluripotent cells [52]. Importantly, LIN28/c-Myc (which regulates miR-17-92) and OCT4/SOX2-regulated miR-302 have been shown to be among a handful of factors necessary and sufficient to convert differentiated cells into induced pluripotent stem cells (iPSCs) [53,54].
Cell-Cycle Regulation and Reprogramming
Discovery of the reprogramming of somatic cells to a pluripotent state (iPSCs) by overexpression of Oct4, Sox2, Klf4, and c-Myc transcription factors [55 –62] has been one of the most exciting discoveries in biology in the last decade. It stimulated further research on mechanisms defining pluripotency and self-renewal and led to examination of the potential roles of those proteins in the process of reprogramming. Multiple studies have focused on modifying the reprogramming protocol to achieve higher efficiency in this rather inefficient process as well as high-quality, genetically stable iPSCs without exogenous expression of protooncogenes.
Recent studies have suggested that cell-cycle regulation is a rate-limiting step in the reprogramming process [63 –69]. It has been shown that loss of p53 function can enhance the efficiency of reprogramming [63 –65,68,69] by the loss of suppressive effects of the p53 target p21Cip1 on cell proliferation [64,65,68]. Further, loss of p53 may facilitate the reprogramming by promoting immortalization [63,70]. In support of this hypothesis, disruption of the Ink4/Arf locus (encoding CDK inhibitors p16Ink4a and p15Ink4b and a positive regulator of p53, p19Arf) has been shown to improve reprogramming efficiency [66,67]. Interestingly, the reprogramming factor KLF4 likely functions (in part) by p53 suppression [71].
Further, LIN28, a positive regulator of cyclin A, cyclin B, and CDK4 [72], enhances reprogramming efficiency [60] by accelerating cell division [68]. Further support for a link between cell-cycle regulation and the early reprogramming process comes from the observation that Rem2 GTPase increases reprogramming efficiency (besides suppression of p53 activity) via increased expression and nuclear localization of cyclin D1 [69] to promote cell-cycle progression. Together these studies imply a possibility of reprogramming somatic cells by imposing ESC-specific cell-cycle features.
DNA-Damage Response
Checkpoint pathways are activated in response to DNA damage to restore genomic integrity and prevent formation of mutations. When DNA damage occurs, these mechanisms halt cell-cycle progression to prevent replication of damaged DNA or passage of damaged chromosomes to progeny and thus avoid mutations and/or genomic instability. DNA damage checkpoint pathways also induce DNA damage repair machinery. If the damage cannot be repaired, cells are instructed to undergo apoptosis. Accumulation of mutations might lead to aberrant cell function, cellular transformation, and tumorigenesis. Major consequences may arise from the disruption of stem-cell function, affecting numbers and/or function of stem cells as well as their differentiated progeny, tissue function, and homeostasis and possibly leading to premature aging or formation of cancer stem cells, which can promote cancer. PSCs of early embryos (as well as their derivatives, ESCs) must possess especially effective DNA-damage response mechanisms because any malfunction of these cells might cause major malformations of the embryo and jeopardize its health and viability.
DNA-Damage Response in Somatic Cells
In somatic cells, DNA damage is sensed by the phosphoinositide 3-kinase–related kinases (ATM), ATM and Rad3-related (ATR), and DNA-dependent protein kinase. Their many substrates mediate cell-cycle arrest in G1, S, or G2 phase as well as DNA repair and cell death [73 –75] in response to various types of DNA damage, of which DNA double-strand breaks (DSBs) are probably the most dangerous. These errors can be induced by exogenous factors such as ionizing radiation (IR) or genotoxic chemicals, including chemotherapeutic drugs, as well as endogenous processes such as chromosome breakage, telomere erosion, replication of single-strand breaks, or action of reactive oxygen species. Activated ATM/ATR proteins phosphorylate multiple substrates, including the signal transducers Chk1/Chk2 and the transcription factor p53 (Fig. 2). Chk1/Chk2 imposes cell-cycle arrest by marking CDC25 phosphatases, positive regulators of CDKs, for proteasomal degradation and by enhancing p53 stability through phosphorylation on Ser20. This phosphorylation interferes with p53 binding to the ubiquitin ligase Mdm2, which shuttles p53 out of the nucleus and targets it for proteasomal degradation. Phosphorylation of other p53 residues enhances its transactivatory and DNA-binding activities, resulting in expression of its downstream targets, including the CDK inhibitor CDKN1A (encoding p21Cip1) or proapoptotic genes (eg, PUMA, BAX). Both the immediate Chk1/Chk2-CDC25A pathway and the delayed p53-p21Cip1 pathway contribute to core G1 checkpoint mechanisms and converge on CDK2 to inhibit its activity and induce G1 arrest.
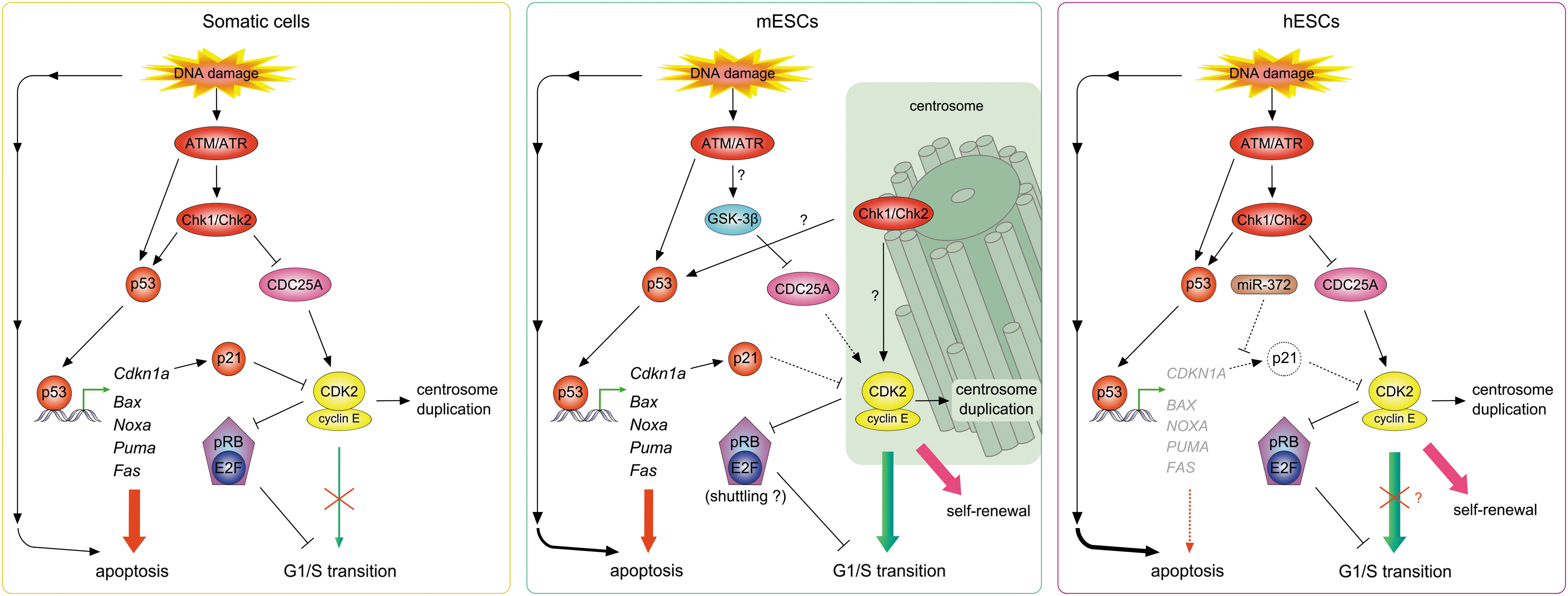
G1 checkpoint pathways in somatic cells, mESCs, and hESCs. Somatic cells: In response to DNA damage induced by IR, ATM/ATR kinases are activated and themselves activate Chk1/Chk2 and p53. Chk1/Chk2 phosphorylate the CDK-activatory phosphatase CDC25A, targeting it for degradation and thus preventing CDK2 activation. Activated p53 induces the expression of proapoptotic (eg, Bax, Noxa) and CDK inhibitory (Cdkn1a) genes, thereby leading to apoptosis and/or cell-cycle arrest, respectively. In mESCs, IR-induced DNA damage is sensed by ATM/ATR kinases, but Chk1/Chk2 are sequestered on centrosomes and are not involved in CDC25A degradation. CDC25A is degraded because of a signal mediated by GSK-3β. p53 is activated in response to DNA damage and induces expression of p21Cip1 and proapoptotic targets; mESCs with damaged DNA die also by apoptosis independent from p53-mediated transcription. Neither upregulation of p21Cip1 nor downregulation of CDC25A affects the activity of CDK2, likely because of its localization to centrosomes. Sustaining CDK2 activity, which might be critical for maintenance of self-renewal, results in G1/S transition in the presence of DNA damage. hESCs activate G1 checkpoint pathways after DNA damage similarly to somatic cells. However, p53 targets are not induced and hESCs with damaged DNA die by induction of the p53 mitochondrial pathway of apoptosis. p21Cip1 levels are not upregulated, in part because of miR-372 activity against p21 mRNA. The issue of hESC G1 arrest in response to DNA damage remains unclear; some studies reported a lack of G1 arrest after IR, whereas others revealed G1 prolongation after UV-induced DNA damage in hESCs. Dashed line, inactive regulatory pathway; ?, suggested/possible regulation; ATM, ataxia-teleangiectasia mutated; IR, ionizing radiation.
DNA-Damage Response in ESCs
Despite the essential role of the G1 checkpoint in maintaining genomic stability, ESCs do not arrest in G1 after DNA damage [76 –79]. Instead, ESCs rely on S and G2/M checkpoints or undergo apoptosis. Why do ESCs prefer to enter S phase with damaged DNA or undergo cell death over stopping and repairing DNA in G1 phase? There must be a good reason for this strategy.
From the classical point of view, the absence of a G1 checkpoint in ESCs is beneficial for the ESC population as a whole by facilitating removal of ESCs with damaged DNA, thus avoiding mutations that might arise from inaccurate DNA repair and maintaining pristine genetic information in ESCs [80]. Consistent with this hypothesis, ESCs are hypersensitive to DNA damage [81,82]. However, mutations through inaccurate DNA repair can also arise in later cell-cycle phases. Why might the G1 checkpoint be inactivated while all others, including the S and G2/M checkpoints, remain intact? What are the mechanisms that selectively inactivate the G1 checkpoint but sustain the functionality of other DNA damage checkpoints in ESCs?
Numerous studies have attempted to reveal the mechanisms underlying G1 checkpoint nonfunctionality in ESCs. Initial observations suggested that in mESCs, p53 is localized to the cytoplasm and cannot be translocated into the nucleus [76] (Table 1). Therefore, neither Cdkn1a nor proapoptotic genes are induced and mESCs do not arrest in G1 or undergo p53-dependent apoptosis in response to DNA damage [76]. However, another study suggested that p53 is functional and revealed that p53-mediated downregulation of Nanog expression is a novel mechanism for maintenance of genetic stability in mESCs, which induces their differentiation into other cell types [40]. Nonfunctionality of p53 has been further challenged by a recent study showing p53 to be activated and Cdkn1a expression induced after IR in mESCs [83] (Fig. 2). However, despite increased levels of the CDK2 inhibitor p21Cip1, mESCs escape G1 arrest after DNA damage. What mechanism might explain this phenomenon?
The table summarizes mostly results from immunolocalization studies.
Abbreviations: mESC, mouse embryonic stem cells; hESC, human embryonic stem cells; N/A, data not available.
Studies on p53 role in hESCs have been rather inconsistent. p53 was shown to be nuclear in hESCs [79,84 –86] (Table 1), but its impact on target genes has remained controversial. Some studies have reported an inability of p53 to induce expression of its target genes [84], whereas others revealed robust upregulation of p53 targets (including PUMA and CDKN1A) in response to DNA damage [25,78,85,86]. However, despite induction of p21Cip1 mRNA levels, p21Cip1 protein levels do not increase after DNA damage in hESCs [78,86], probably because of negative regulation of p21Cip1 mRNA translation by miR-372 [27] (Fig. 2).
With respect to the parallel G1 checkpoint pathway employing the Chk1/Chk2-CDC25A cascade, in 2 mESC studies, Chk2 was reported to be sequestered at centrosomes (Table 1) and separated from its substrate, CDC25A, and therefore, unable to mediate its degradation with subsequent G1 arrest [80,83]. However, these studies reported different phosphorylation patterns of Chk proteins: Chk2 was constitutively phosphorylated in one of the studies, whereas in the other its phosphorylation was induced by DNA damage. Koledova and colleagues also reported constitutive phosphorylation of Chk1 on Ser345 [83], which might be consistent with the centrosomal sequestration of Chk1 observed in their study, as Chk1-Ser345 phosphorylation has been suggested to be important for localization of Chk1 on centrosomes [87]. More importantly, CDC25A levels were sustained after IR in the study by Hong and Stambrook [80], but Koledova and colleagues found robust CDC25A degradation mediated by glycogen synthase kinase 3β (GSK-3β) [83]. It is difficult to reconcile the results of these 2 studies. Differences in mESC lines might account for some of the observed differences; however, it is important to note that the observations of Koledova et al. were made in 2 divergent mESC lines, an inbred and a hybrid line, thus substantially limiting bias from cell line variability.
For comparison, in hESCs, Chk1/Chk2 were reported to be localized to the nucleus [79,86] and activated in response to DNA damage like in somatic cells [78,79,86]. However, after IR, hESCs escaped from G1 and arrested only at the G2/M boundary [78,79]. On the other hand, after UV irradiation, a G1 delay, associated with CDC25A degradation and CDK2 activity downregulation, was observed in hESCs [86].
Interestingly, neither downregulation of CDC25A levels nor upregulation of p21Cip1 led to a G1 arrest in mESCs because CDK2 activity was not abrogated [83]. An important role for DNA damage-refractory CDK2 activity in driving rapid G1/S-phase progression after DNA damage in mESCs was shown by chemical inhibition of CDK2, which slowed down mESC progression from G1 to S after IR [83].
What might make CDK2 unresponsive to changing levels of regulators CDC25A and p21Cip1? Immunolocalization studies have implicated CDK2's ESC-specific localization; in these cells, CDK2 predominantly localizes to centrosomes, whereas CDC25A localizes to cytoplasm and nuclei and was not detected at centrosomes [83] (Table 1). Thus, centrosomal CDK2 might be sheltered from varying levels of CDC25A and probably also from p21Cip1 [83]. Alternatively, levels of p21Cip1 might be insufficient to buffer high levels of CDK2 in mESCs because equimolar concentrations of p21Cip1 and cyclin-CDK2 complexes are required for efficient CDK2 inhibition [88]. The observation of a large CDK2 pool at mESC centrosomes has raised many questions. Among them, the possible mechanism of centrosomal CDK2-mediated G1-phase regulation is the most intriguing, although conclusive evidence for its existence remains elusive. We hypothesize that centrosomal CDK2 (possibly in cooperation with the cytoplasmic pool of CDK2) might regulate the activity of relevant cell-cycle (eg, pRB-E2F) and cell-fate/self-renewal (eg, SOX2) [89] regulatory proteins that shuttle between the cytoplasm and nucleus. The role of centrosomal pools of Chk1 and Chk2 in G1/S-phase regulation, if any, is also not understood. Are they sequestered to centrosomes to avoid interacting with CDC25A, as suggested by Hong and Stambrook [80], or do they actually play an active role in maintaining centrosomal CDK2 activity? Keeping in mind that even with Chk1/Chk2 at centrosomes, CDC25A is targeted for degradation by GSK-3β [83] and that centrosomal Chk1 regulates centrosomal CDK1 activity during unperturbed cell cycles as well as in response to DNA damage in somatic cells [90,91], it is more probable that the latter is the case.
In somatic cells, DNA damage induces a Chk1-dependent amplification of centrosomes that leads to lethal multipolar mitoses [92]. This mechanism might act as an additional checkpoint when earlier checkpoints fail or are not functional, for example, in ESCs, which are naturally devoid of a G1 checkpoint. Based on the CDK2 requirement for centrosomal amplification [93 –96] and the recent finding that DNA damage causes Chk1-dependent activation of CDK2 [97], we suggest that colocalization of CDK2 and Chk1 on centrosomes might allow for fast and effective centrosome amplification after DNA damage in mESCs, with subsequent efficient elimination of damaged cells from the stem-cell pool via mitotic catastrophe. Further, Chk1-dependent activating phosphorylation of CDK2 after DNA damage might contribute to sustained total CDK2 activity after DNA damage in mESCs.
In view of CDK2's role in ESC self-renewal [23,30], maintaining CDK2 activity after DNA damage might be important to sustain pluripotency and avoid differentiation in ESCs [83]. In context of this hypothesis, it would be interesting to investigate whether downregulation of CDK2 activity and the G1 delay observed in UV-irradiated hESCs [86] is associated with differentiation.
New evidence for tight cooperation between DNA damage and cell-cycle regulatory mechanisms in cell-fate decisions comes from studies in muscle stem cells, in which DSBs naturally occur during differentiation [98 –100]. These DSBs are targeted to specific genomic loci, including critical differentiation regulatory genes such as Cdkn1a, to induce their expression [101]. It is tempting to speculate that this process may be involved in the development of other tissues as well.
Centrosomes as Cell-Cycle Regulatory Centers
By implicating centrosomes as critical shelters for CDK2 from activated G1 checkpoint pathways in mESCs [83], we suggest a novel role for centrosomes in the coordination of fate decisions in stem cells and support the hypothesis of centrosomes as coordination centers of cell-cycle regulatory events. Most of our conclusions are drawn from results of immunolocalization studies, which revealed major differences with respect to the localization of some cell-cycle regulatory and DNA-damage response proteins in mESCs, hESCs, and somatic cells (summarized in Table 1). In the future, additional studies including centrosomal fractionation followed by protein analysis and genetic manipulations preventing protein targeting to centrosomes will help to better understand the significance and potential roles of the centrosomal localization of these proteins in mESCs.
Centrosomes are microtubule-organizing centers in animal cells. During G1 phase, there is only one centrosome (consisting of 2 centrioles and pericentriolar material), but it is replicated during S phase. The 2 centrosomes form a bipolar mitotic spindle during mitosis, thereby allowing for accurate chromosome segregation and cytokinesis. Aberrations in centrosome number and/or function have been associated with genomic instability and tumorigenesis. However, whether these centrosomal aberrations are a cause [102 –104] or a consequence of malignant transformation remains unclear [105 –107].
Several studies have proposed a role for centrosomes in the regulation of G1-S and G2-M transitions and DNA damage checkpoint signaling because multiple proteins involved in cell-cycle control and the DNA-damage response are found to localize to centrosomes (for a review, see Doxsey et al. [108]). Consequently, a new concept of centrosomes as cell-cycle regulatory centers suggests that centrosomes might function as “scaffolds” to promote interactions between various regulatory components during the cell cycle [108 –110].
Studies of karyoplasts (acentrosomal cells) revealed that an activity associated with core centrosomal structures is required for cell-cycle progression from G1 to S phase, as ablation of centrosomes in late G2 phase led to an arrest at the G1/S boundary of the next cell cycle [111,112]. This activity might be conferred to cyclin E because its localization to centrosomes is required to promote S-phase entry in a CDK2-independent manner [113]. Further, alterations in centrosome composition caused by knockdown of discrete centrosomal proteins can induce p38-p53-p21Cip1–dependent G1 arrest [114].
The first hint of a role for centrosomes in mitotic entry came from work in oocytes showing that injection of centrosomes can induce progression into mitosis in G2-arrested starfish oocytes [115] and activate maturation-promoting factor (MPF; cyclin B-CDK1) and accelerate mitotic entry in Xenopus eggs [116]. Subsequent work in somatic cells demonstrated the presence of CDK1 [117,118] and cyclin B [119] at centrosomes. More recently, activated cyclin B-CDK1 complexes were shown to initially appear on centrosomes during prophase [120]. This activation is regulated by Chk1, which localizes to centrosomes during interphase and shields centrosomal CDK1 from unscheduled activation by cytoplasmic CDC25B [90]. In response to DNA damage, Chk1 accumulates at centrosomes and induces G2/M cell-cycle arrest [91]. Also, a subset of Chk2 molecules localizes to centrosomes and the mitotic apparatus, suggesting a role for Chk2 in the regulation of mitosis [90,121]. Together, these observations indicate that centrosomes are sites where cell-cycle decisions and checkpoint reactions take place.
Centrosomal Decisions in Self-Renewal
The limited information available for mESCs suggests that centrosomes serve as sites for sequestration of checkpoint components (Chk1, Chk2) [80,83] and as shelters for cell-cycle regulatory proteins (CDK2) [83] to prevent reactions that might limit self-renewal (Fig. 3). It would be interesting to investigate whether hESCs use similar mechanisms to escape from G1 arrest after DNA damage, that is, whether CDK2 localizes predominantly to centrosomes in these cells as well. Because mESCs and hESCs differ in cell-cycle regulation and have been suggested to represent different pluripotent states, equivalent to inner cell mass epiblast progenitors and the early postimplantation epiblast, respectively [122], the outcome of such research might be surprising.
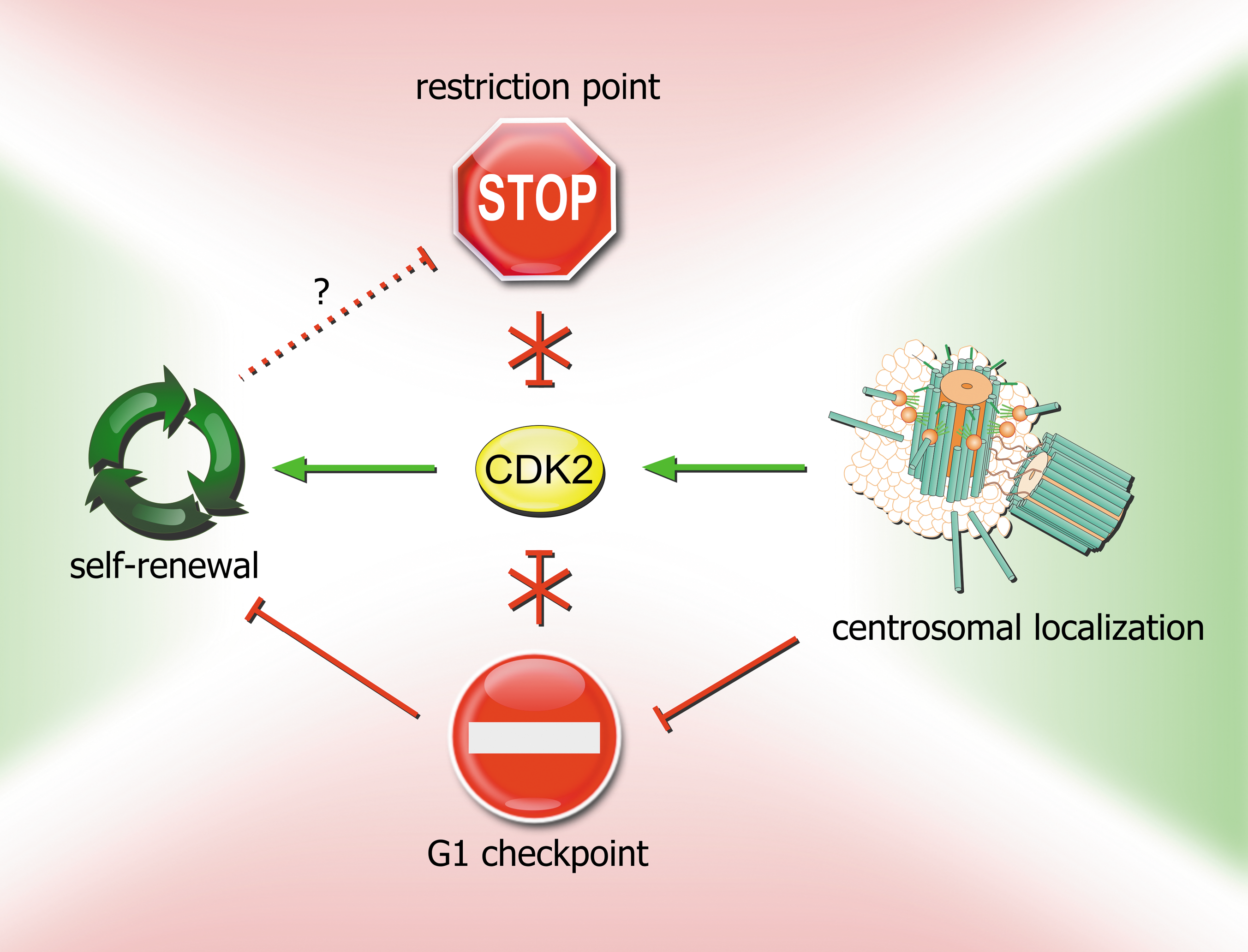
Regulation of CDK2 activity by the G1 checkpoint, restriction point, and localization to centrosomes in the context of self-renewal in mESCs. In somatic cells, CDK2 activity is negatively regulated by the restriction point and the G1 checkpoint. However, because of CDK2's role in self-renewal, these mechanisms are inactivated in mESCs. G1 checkpoint activity is partially abrogated by sequestration of Chk1/Chk2 proteins to centrosomes, which separates them from their substrates, and partially by CDK2 localization to centrosomes, where it is protected from other activated G1 checkpoint pathways (p53–p21; GSK-3β–CDC25A). Mechanisms of self-renewal may also be involved in abrogation of the restriction point (dashed line).
Another only recently appreciated function of centrosomes is in the regulation of asymmetric stem-cell division. Although centrosomes are not essential in many somatic cell types, there is compelling evidence that centrosomes are required for the efficient asymmetric division of stem cells. Well-studied examples can be found in early Caenorhabditis elegans and Xenopus embryos, Drosophila neuroblasts [104,123,124], and Drosophila male germline stem cells (GSCs) [125 –127], in which perturbations of centrosome function or number compromise asymmetric division [128,129]. In some cases, such as in Drosophila male GSCs, the asymmetric outcome of the cell division is specified by asymmetric placement of daughter cells into microenvironments that provide extrinsic signals that direct cells to different fates [130,131]. In others, such as in Drosophila neuroblasts, stem cells undergo asymmetric division based on differential segregation of intrinsic fate determinants [132,133]. Relationships between spindle orientation and symmetricity of stem-cell division have been also observed in stem cells from mammalian skin [134], muscle [135], and neuronal stem cells [136,137], suggesting a broad involvement of centrosomes (through spindle orientation) in the accomplishment of asymmetric stem-cell divisions across species [125,137 –140].
Interestingly, Drosophila male GSCs [125] and mouse radial glia progenitors [137] have been shown to preferentially inherit and retain the old mother centrosome, and the asymmetric behavior of old and new mother centrosomes explains the asymmetric outcome of stem-cell divisions. This observation suggests that an “immortal centrosome,” retained in the stem cell, could help to determine stem-cell fate and raises the possibility that preferential centrosome inheritance is a conserved stem-cell characteristic [141].
On the other hand, asymmetrically inherited centrosomes in stem cells could merely be innocent bystanders that are differentially segregated because of their role in spindle orientation. In Drosophila male GSCs, the old mother centrosome has a greater microtubule nucleating capacity than the newer daughter centrosome and is associated with robust microtubules, some of which extend to adherens junctions and connect the GSCs to hub cells [125]. Therefore, the firmly tethered mother centrosome might ensure proper orientation of the mitotic spindle during stem-cell division, stem cell-specific inheritance of the mother centrosome, and stem-cell maintenance signals from hub cells.
Alternatively, centrosomes could contribute to stem-cell maintenance and daughter-cell programming by partitioning organelles and signaling molecules asymmetrically between the stem-cell and its more differentiated progeny through recycling endosomes, for example, in the developing Drosophila nervous system [142], or by regulation of degradation of cell-fate determinants [143,144]. Also, centrosomes could serve as carriers of developmental information. One study has revealed asymmetric inheritance of a fate-determining mRNA that associates with only one centrosome during early embryonic development in a mollusk [145]. Further, in mammalian cells, the mother centriole organizes the primary cilium, a sensory organelle [146] responsive to chemical and mechanical signals outside the cell with an essential role in a number of signal transduction pathways, including Hedgehog [147 –152], Wnt [153], and platelet-derived growth factor signaling [154] (for review, see Veland et al. [155] and Satir et al. [156]). Centriole duplication at G1/S results in 2 centrosomes, one with an older mother centriole and one with a new mother centriole, that are segregated in mitosis. Because the cell receiving the older, mature mother centriole usually grows a primary cilium first [157], the asynchrony in cilium formation could differentially influence the ability of the 2 daughter cells to respond to environmental signals and thereby their behavior and fate specification [137,158]. hESCs possess primary cilia with functional Hedgehog signaling machinery [159]. Recently, it has been shown that in a subpopulation of cultured hESCs, OCT4, SOX2, and NANOG localize to the primary cilium [160] where they might undergo processing analogous to that suggested for the Gli proteins in Hedgehog signaling [155].
Increasing evidence suggests that centrosomes are key coordinators of multiple cellular signaling pathways and cell-fate decisions in stem cells with crucial roles during development and in tissue homeostasis. Future experiments investigating this remarkable organelle and its behavior during stem-cell division will hopefully reveal a unifying mechanistic principle of differentiation.
ESCs as a Model for Embryonic Pathways: A Handy Tool or an Artifact?
An intriguing issue is to what extent observations made in ESCs in vitro correspond to the in vivo situation of cells in early embryos. Transfer of stem cells from the embryo to the culture dish subjects cells to selective pressure from their new environment, and ESCs adapt to culture conditions through self-transformation events involving epigenetic and karyotypic changes [161,162].
One of the stress factors imposed on embryo-derived stem cells is oxygen. ESCs are routinely cultured under normoxic conditions, although it is well known that the mammalian reproductive tract, the native environment for preimplantation embryos, is hypoxic [163]. Stem cells, including ESCs, might benefit from residing in hypoxic niches where oxidative DNA damage may be less frequent [164]. This hypothesis is supported by the observation of alterations in mitochondrial DNA in late-passage hESC lines cultured under high oxygen tension [165]. Also, hESCs were shown to prefer hypoxia for maintenance of a highly proliferative, pluripotent state [166 –168]. However, only short-term hypoxia increased mESC proliferation (through upregulation of cell-cycle regulatory proteins such as CDK2) [169]. Long-term hypoxia, on the other hand, inhibited self-renewal and led to mESC differentiation [170]. The full extent to which ESCs differ from their developmental counterparts in embryos remains elusive. Nevertheless, in the absence of better systems, ESCs remain the most suitable tool for modeling processes in cells of early embryos as well as cancer stem cells.
Concluding Remarks
In ESCs, high CDK2 activity drives rapid progression through G1 phase to minimize potential exposure to differentiation signals and maintain the self-renewing state. Because downregulation of CDK2 activity is sufficient to induce stem-cell differentiation, mechanisms negatively regulating CDK2 activity in somatic cells, including the restriction point and the G1 checkpoint, are deactivated in ESCs. Another level of protection of CDK2 activity in ESCs might be its specific localization to centrosomes, at least in mESCs, where it is sheltered from activated G1 checkpoint pathways. Chk1 and Chk2 proteins are also sequestered to centrosomes in mESCs to separate them from their substrates and possibly to increase CDK2 protection. Therefore, in the cell-fate game, centrosomes appear to play a mediatory role, providing an interface where crucial cell-cycle and cell-fate decisions can take place.
Footnotes
Acknowledgments
This work was supported by the Ministry of Education, Youth, and Sport, Czech Republic (grants 2B06077 and MSM 6198959205 to V.D.), and by funds of the Deutsche Krebshilfe (108560) and Deutsche Jose Carreras Leukämie Stiftung (DJCLS R 06/04) awarded to A.K.
Author Disclosure Statement
No competing financial interests exist.