
Abstract
Mesenchymal stem cells (MSCs) are adult stem cells that hold great promise in the field of regenerative medicine. They can be isolated from almost any tissue of the body and display, after expansion, very similar properties and minor differences, probably due to their microenvironment of origin. Expansion in vitro can be obtained in cytokine-free, serum-enriched media, as well as in serum-free, basic fibroblast growth factor–enriched media. A detailed immunophenotypic analysis is required to test the purity of the preparation, but no unique distinguishing marker has been described as yet. Functional assays, that is, differentiation studies in vitro, are needed to prove multilineage differentiation of expanded cells, and demonstration of pluripotency is necessary to identify most immature precursors. MSCs show powerful immunomodulative properties toward most of the cells of the immune system: this strengthens the theoretical rationale for their use also in an allogeneic setting across the major histocompatibility complex (MHC) immunological barriers. Systemic intravenous injection and local use have been tried: after systemic injection, MSCs show a high degree of chemotaxis based on pro-inflammatory cytokines, and localize at inflamed and neoplastic tissues; local regeneration has been improved using synthetic, as well as organic scaffolds. On the other hand, inadequate heterotopic in vivo differentiation and neoplastic transformation are potential risks of this form of cell therapy, even if evidence of this sort has been collected only from studies in mice, and generally after prolonged in vitro expansion. This review tries to provide a detailed technical overview of the methods used for human bone-marrow (BM)-derived and adipose-tissue (AT)-derived MSC isolation, in vitro expansion, and characterization for tissue repair. We chose to use BM-MSCs as a model to describe techniques that have been used for MSC isolation and expansion from very different sources, and AT-MSCs as an example of a reliable and increasingly common alternative source.
Introduction
M
Despite this, a consensus on the definition and properties of MSCs after in vitro expansion has been recently reached by the International Society for Cellular Therapy [11]: a specific immunophenotype, ex vivo plastic-adherent growth, and multilineage differentiation are defined as the minimal prerequisites needed. Given the high level of heterogeneity existing in isolated MSCs [12], the term “stem cells” should be limited on this basis to cases with proven pluripotency [11].
It may be difficult for the unexperienced scientist to enter the ever-growing field of research on MSCs without previous knowledge: to help new enthusiasts in their initial steps, we tried to summarize a detailed technical overview of the methods used for human MSC isolation, expansion, and characterization for tissue repair, using BM-derived MSCs, the first to be isolated and thoroughly evaluated, as a model. We based on both personal experience and the available literature: whenever unspecified, the suggested protocols and methods are among the ones currently in use in our laboratories.
Isolation
Bone marrow
MSCs have been first isolated from the BM. The ease of collection and the relatively high frequency of MSCs [1/104–1/105 BM-mononuclear cells (MNCs)] [1,2,13] make it still a commonly used source of MSCs. BM is usually collected by aspiration from the iliac crest or sternum through a Jamshidi needle. Small volumes (<4 mL) are preferred, to prevent hemodilution: vertebrae have provided an additional potential site of collection, while femoral heads are a further alternative, even though they do not always seem reliable sources [14].
Thanks to their density (1.073 g/dL), MSCs are isolated in the mononuclear ring of a density gradient centrifugation (1.077 g/dL) [14]. Centrifugation is carried out at 600 g for 20–30 min with no brakes, and the ring of BM-MNCs is collected manually with a Pasteur pipette and then washed twice with phosphate-buffered saline (PBS) (centrifuged at 300 g for 10 min, at room temperature) [13,14].
After resuspension in a growth medium (GM), cells are counted in an hemocytometer using a vital color—for example, Acridine Orange/Bromure Ethidium—and plated at a density of 1.2 × 106 BM-MNCs/cm2 [passage 0 (P0)] [15
–18]. Other authors have suggested a slightly lower cell density (8 × 105) for initial plating [19]; a technical study testing different media proved alpha modified Eagle's medium (α-MEM) + Glutamax + fetal bovine serum (FBS) 10% and α-MEM +
After 72 h the plates should be washed with PBS and the GM entirely changed [15]. Adherent cells organize in small clusters of spindle-shaped cells within 7–10 days. Subsequent changes of GM, usually twice a week, should be limited to 40%–50% of total volume.
Adipose tissue and other sources
MSCs have been isolated from almost every tissue of the body [4,5], including lymphoid organs (thymus and spleen) [21], subcutaneous fat [22,23], periodontal ligament [24], scalp tissue [25], endometrium and menstrual blood [26], peripheral blood [6], umbilical cord blood [27] and Wharton's jelly [28,29], as well as fetal tissues [30]. The amount of material needed depends on the abundance of MSCs (eg, 40–50 mL in the case of subcutaneous lipoaspirate) [22].
In almost all cases, careful surgical removal of the connective tissue surrounding the parenchyma is needed as a first step, followed by enzymatic digestion, made by a solution of collagenase type II (0.075% in PBS or HBSS, 30 min at 37°C, shaken at 120–150 rpm) [22,23] or equivalent protease (eg, collagenase type I 0.1% + bovine serum 1%, 60 min at 37°C, in continuous agitation) [31,32]. The cell-enriched supernatant is then collected and the enzyme neutralized by adding GM in a 1:2 ratio. In the case of subcutaneous fat, after centrifugation (200 g, 10 min at room temperature) the supernatant will contain low-density floating adipocytes, whereas the pellet will contain the vascular-stromal fraction [22,23,31,33]. The pellet should then be washed 3–4 times in PBS. Contaminating erythrocytes, which are most evident in the case of splenic tissue, are then lysed using 160 mM NH4Cl solution (10 min at room temperature), and the cellular suspension is passed through a cell strainer of 70 μm (100 μm in the case of subcutaneous fat) to remove small tissue debris and cellular aggregates. The final pellet is resuspended in GM and the cells are counted and plated. Initial density depends on the abundance of MSCs in the tissue: 10 × 106 cells/cm2 for cells isolated from the thymus or lymphonodes, 5 × 106 cells/cm2 from the spleen, and 1 × 106 cells/cm2 in the case of subcutaneous fat [21].
After, at 72 h the plates are washed with PBS and GM entirely changed. As previously stated, subsequent changes, twice a week, should be limited to 40%–50% of total volume, to preserve medium conditioned by soluble factors released by MSCs. After 6 days in culture, adipose-derived stem cells usually reach early confluence, and need to be detached and replated at lower density (1.8–3.1 × 103 cells/cm2) to improve the purity of the preparation [22,23,33]. Usually, 3–5 weeks of in vitro culture is sufficient to generate a homogeneous cell population.
Even though MSCs share common features regardless of their source, there is evidence of a role of different specific microenvironments in determining small differences in their immunophenotype (eg, CD34 expression early after isolation) and in vitro differentiation [13,31,32].
Expansion
Adhesion to the substrate
As previously stated, early separation of adherent elements is carried out in the first 72 h of culture. After 7–14 days of culture, clusters of spindle-shaped, fibroblast-like adherent cells can be detected on the plastic surface of the flask [1 –3] (Fig. 1); although this feature suggests the clonogenic potential of cells in the cultured population, it does not permit the quantification of clonogenicity, which requires limiting dilution methods.
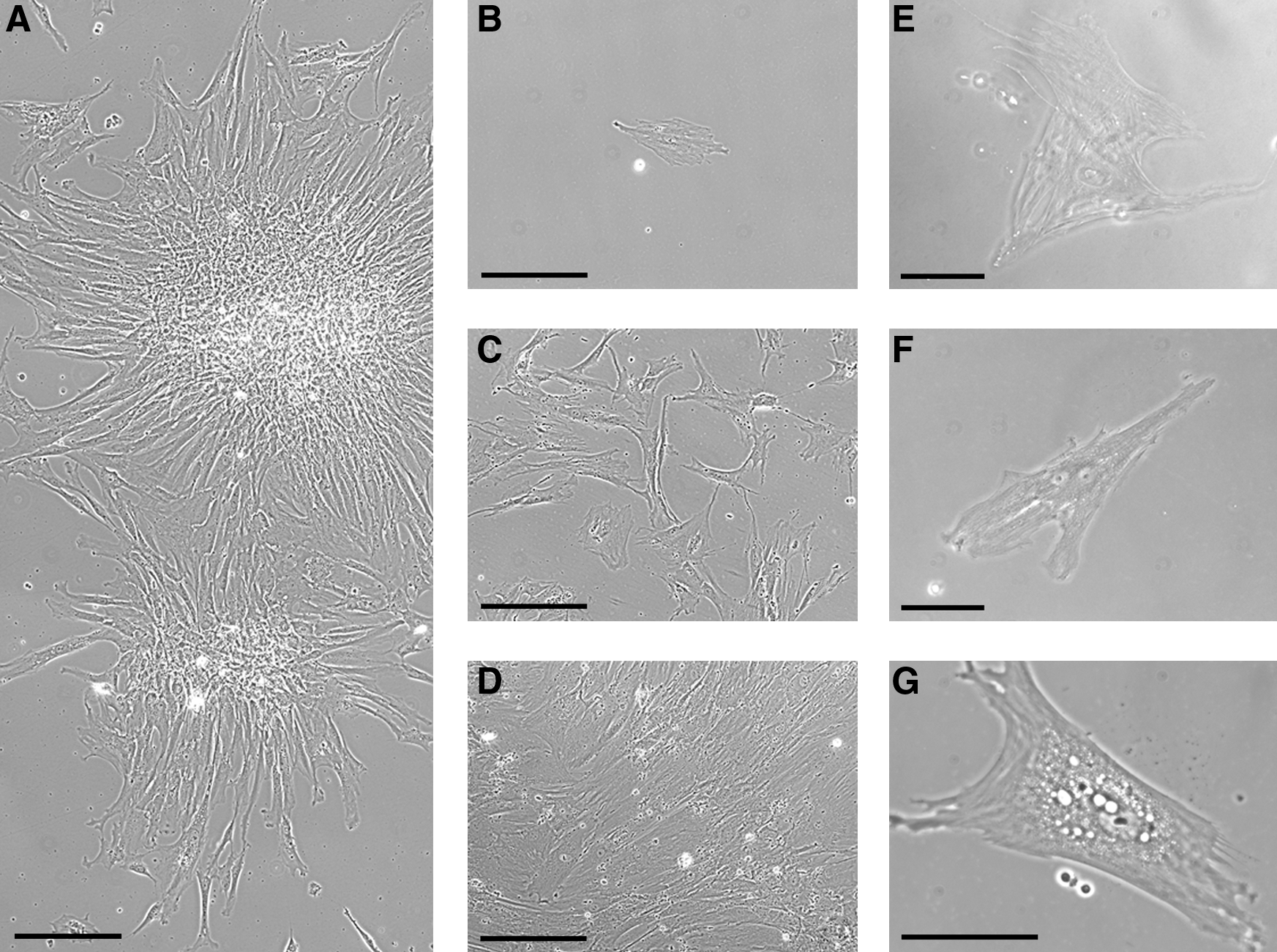
MSC culture in vitro. (
Normally, <0.01% of BM adherent cells are ultimately MSCs [3,13]. Therefore, homogeneity can be achieved only after prolonged culture, usually after 2–3 weeks of culture (P3–P4) [3,15,16].
Role of GM
MSCs expand in basic GM, usually in the absence of cytokines, with the addition of 10%–20% fetal calf serum (FCS)/FBS [3,14
–16,19,20,34
–36] to provide essential proteins for cell adhesion and growth and to prevent spontaneous differentiation. The addition of antibiotics is often useful (penicillin 100 U/mL + streptomycin 100 μg/mL) [15,16]. α-MEM and Dulbecco's MEM (DMEM) are mostly used [20]. Composition may vary, from high-glucose DMEM supplemented by 15% FCS [15,16] to low-glucose DMEM + 10% FBS [14,17,18], to α-MEM + 10% FBS [19], to DMEM/F-12 Ham's medium + 10% FBS [31,32]. A technical study has been conducted comparing different media and protocols to expand BM-MSCs from the same donors [20]: α-MEM + Glutamax + FBS 10% and α-MEM +
The minimal growth conditions allow for the selection of cultured cells and preserve their uncommitted status. In fact, early immunophenotypical analysis can usually detect contaminating lineage-committed cells, such as hematopoietic precursors, resting lymphocytes, monocytes, and macrophages [13,36]. Preferential in vitro expansion of BM-MSCs is therefore based on three key properties of MSCs: their ability to adhere to plastic surfaces, their ability to continuously grow in poor media, and their high sensitivity to trypsinization as compared with monocytes [14]; therefore, to increase homogeneity after replating especially at earlier passages, trypsinization should never exceed 5–7 min at 37°C. When cultures reach near-confluence (70%–80%), cell-contact growth inhibition should be prevented by detaching, for example, by means of prewarmed 0.05% trypsin–ethylenediaminetetraacetic acid solution, and replating the cells at a lower density (from 1 × 104 to 8 × 104 cells/cm2) [15 –19,21]. This additionally enhances the preferential expansion of MSCs.
Cytokine-induced growth
MSCs are highly sensitive to basic fibroblast growth factor (bFGF, FGF-2), which increases MSC expansion and maintains their uncommitted state [37]. FGF-2 has also been used to replace bovine/fetal serum in FBS-free media [38]. Similarly, other related cytokines increase MSC proliferation and prevents their differentiation, such as FGF-4 [39] and heparin-binding epidermal growth factor–like growth factor (through binding to human EGFR related receptor tyrosine kinase [HER-1]) [15]. On the other hand, there is at least some evidence that FGF-2 may alter in vivo generation of bone [40], and when combined with other cytokines [ie, FGF-2 5 μg/mL + bone morphogenetic protein-2 (BMP-2) 2 μg/mL], FGF-2 promoted osteoblastic differentiation and bone growth when used for lumbar arthrodesis [41]. On the contrary, at higher doses (≥5 ng/mL) FGF-2 appears to induce neuronal differentiation [42] and possibly enhance MSC immunosuppressive ability [20].
MSCs express fetal liver kinase (FLK1) [43] and vascular endothelial growth factor receptor 2 (VEGF-R2) and VEGF-R3 [44]. Moreover, the vascular fate of MSCs can be modulated indirectly, through platelet-derived growth factor receptors (PDGF-R) that regulate MSC migration and proliferation [45].
The search for serum-free media
The appropriate medium is critical to obtain a sufficient amount of MSCs, while retaining all their differentiation potential, and keeping a safety level in accordance with clinical needs. The use of serum-free media containing only well-defined factors is one of the main goals for future developments in MSC expansion. The first step in culturing still requires proteins already present in the medium or exogenously added, to make native MSCs adhere to their support. Culture in total serum deprivation (basic medium without cytokines or growth factors) allows the establishment of an MSC subpopulation that shows features of early progenitors with increased expression of embryonic stem cell genes [46]. However, serum-free media usually require the addition of growth factors (eg, PDGF, FGF-2) [47] to substain growth. A good expansion can be obtained by using serum substitutes (eg, ULTROSER®) [48]; however, these media still contain serum components that may not be highly purified, well defined, or characterized.
The use of serum-free media is important also for the clinical safety: the use of FCS/FBS has raised some concerns, due to the theoretical transmissions of prions and potentially unidentified zoonoses with the cells, as well as the possibility to induce immune reactions by the host. A possible approach to avoid such risks, although with some practical problems, would require autologous or allogeneic human serum for in vitro expansion: in one study, autologous serum has proved to be superior to both FCS and allogeneic serum in inducing long-term proliferation and expansion of MSCs [49].
MSC expansion for clinical use
The production of MSCs for clinical intervention needs to comply with good manufacturing practice (GMP) to ensure the final delivery of a safe, reproducible, and efficient “cell-drug.” All steps of the process must be defined, from the source for isolation to culture methods, to the procedures, materials and methods used for cell culture, and quality controls. Even if MSCs are expandable from virtually all tissues [10], to date the preferred source remains the BM. In fact, hundreds of millions MSCs can be expanded in vitro starting from 10 to 20 mL of BM aspirate, although cell yield may vary depending on age and condition of the donor [35,50] and on the expansion techniques [20]. Some other tissues, such as trabecular bone [51], cord blood, or amniotic membrane [52] could have clinical interest in the future. Moreover, adipose tissue (AT) may represent an important alternative to easily obtain a large number of MSCs [23]. The parameters of the culture process must be optimized to reach GMP goals; the first critical parameter is the plating density, which could be involved in the maintenance of early progenitors [35]. Additionally, the time in culture may also change the quality of MSCs. In humans, after 3 weeks and 12–15 population doublings, MSCs decrease their proliferation rate and progressively loose their multipotency [53].
At present, a real serum-free medium is currently not available for clinical-grade MSCs. The main alternative to FCS/FBS-supplemented medium is the use of human serum enriched with platelet growth factors, namely, platelet lysate [54,55], which increases safety by excluding xenogeneic proteins. Totally closed systems for MSC cultures for the production of large amounts of MSCs are currently under development [56]. To ensure that MSCs expanded in different culture conditions retain all the required characteristics, careful quality controls of the processed MSCs must be established: these controls must not only analyze the microbiological safety and phenotype of the cells by flow cytometry, but also critically test the genetic stability of expanded cells.
Cryopreservation
Cryopreservation is carried out in a cell preservation fluid consisting of FBS + 20% dimethylsulfoxide (DMSO; Me2SO), which should be added to the cells in GM in a 1:1 ratio, to reach a final concentration of 106 cells/mL in 10% DMSO [57 –60]. Similar media are effective also for AT-MSCs [31], while commercial preparations based on the same principles are also available [61]. Cells should be frozen with a slow and progressive drop in temperature (−1°C/min). This can be achieved by passing the cells at increasingly lower temperature [58], by means of a freezing box containing room-temperature isopropylic alcohol, or, more accurately, by using automated freezing devices based on liquid nitrogen in closed circuits [59,61,62]. The latter proved superior to conventional slow freezing and 2 different vitrification protocols in terms of preservation of viability and growth after thawing of MSCs isolated from fetal liver [62]. The cells are then rapidly stored at −196°C in liquid nitrogen vapors. Freezing is generally thought to diminish cell growth capability after thawing, even though some studies, cultivating both frozen and unfrozen freshly isolated MSCs in parallel, proved the persistence of comparable cell growth after cryopreservation [57,63]. Differentiation of MSCs is scarcely affected by cryopreservation [57,59,63].
The thawing medium should contain high FBS concentration: this can be achieved by rapidly thawing the cryovials in a 37°C water bath while adding fresh culture media in doubling volumes [58], or by increasing FBS concentration in GM. We use a thawing medium made of 60% FBS + 40% GM, which should be prewarmed at 37°C and added immediately to the cells in a 10:1 ratio right after initial thawing in the water bath, to dilute DMSO as rapidly as possible and re-establish chemical homeostasis; after centrifugation, the pellet is plated in normal GM, and a complete change of GM is made after 24 h.
Immunophenotype
MSC immunophenotypic characterization
No unique single marker has been found for MSCs so far. Therefore, the combination of markers necessary to identify a homogeneous cell population should include CD105, CD73, CD90, CD44, CD29 (all expressed by MSCs), and CD34, CD45, CD11c, CD14, CD31/PECAM-1, and other endothelial markers (all not expressed by MSCs) [3,13,31,32,64]. Although most MSCs, including BM-MSCs and AT-MSCs, are negative for CD117 (c-kit) [13,31], this marker has been shown to be expressed by MSCs isolated from Wharton's jelly [29]. Adipose-derived MSCs are CD34pos at the first stage of culture, and CD34 expression decreases on the cells over time [13,65], while never becoming completely abolished [31]. MSCs do not express the costimulatory molecules CD80 (B7-1), CD86 (B7-2), CD40, or CD40L, even after interferon gamma (IFN-γ) stimulation [16,66]. An example of immunophenotypic characterization of expanded BM-MSCs is provided in Fig. 2. Although hematopoietic precursors are expected to fully differentiate after initial passages (P3–P4), MSCs are capable of maintaining long-term hematopoietic precursors [67]. In one study [19] a significant contamination by blood precursors (up to 15%) was present at earlier passages (P1), and more common in MSCs expanded from young adult donors as compared with pediatric donors (15.45% vs. 3.66% at P1). These hematopoietic colonies were then lost during culture, and accounted for 0.5% and 4.32% of the entire cell population, respectively, at P10 [19].
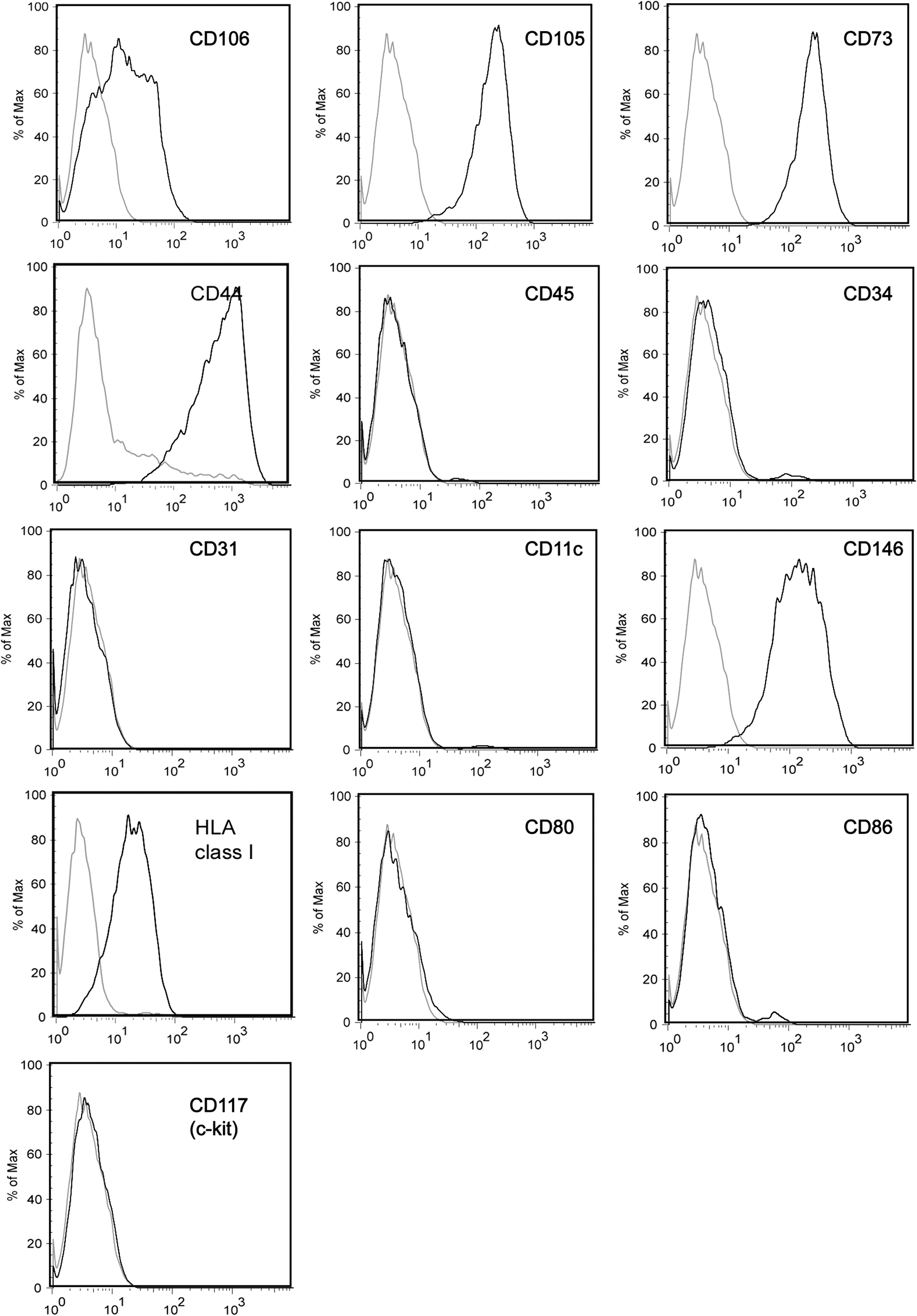
Immunophenotype of human BM-MSCs (P3) expanded in vitro. Expression of specific markers (black) as compared with isotype controls (gray).
Immunophenotype of MSCs is heterogeneous and dynamic: slight differences exist between MSCs expanded in vitro and their putative in vivo counterpart, as detected by a panel of commonly expressed mesenchymal markers right after tissue sampling. This may be also due to the difficulty of unequivocally detecting the in vivo counterpart of expanded MSCs, which might be different in relation to different sources [68]. Other changes in the immunophenotype also happen during culture: for instance, in BM-MSCs CD29 seems to upregulate [69]; CD146 [40] and CD271 [12,70] gradually disappear during in vitro culture. Similarly, studies comparing the immunophenotype of freshly isolated human AT-derived cells from the stromal vascular fraction (SVF) and serial-passaged AT-MSCs show a progressive increase of mesenchymal markers (CD13, CD29, CD44, CD63, CD73, CD90, and CD166) during culture; as previously noted, CD34 expression is maximal on SVF cells right after tissue digestion and at P1, while persisting at lower levels also at later passages (P4); the same is true for CD146, a marker of pericytes [31,32]. Although at low levels, endothelial markers, such as CD31, CD144, or VE-cadherin, remain detectable in SVF cells and in AT-MSCs throughout the culture period [31]. Sometimes, changes in immunophenotype can be related to a significant switch in behavior of the cells (eg, the loss of CD90 seems to be linked to neoplastic transformation of MSCs) [71], or to the effect of specific cytokines in the medium (eg, IFN-γ can upregulate major histocompatibility complex-II [MHC-II]) [11]. In most cases, cultures tend to become more homogeneous as they progress over time [1,2,31].
In any case, immunophenotype alone cannot specifically distinguish between MSCs and more differentiated mesenchymal progeny; functional assays, demonstrating clonogenicity and in vitro multilineage differentiation, are always needed to complement the immunophenotyping and to fully characterize the expanded cell population.
Use of immunophenotypic sorting to ameliorate MSC isolation
Several markers have been tested to improve the efficiency of MSC isolation and expansion, mostly in the case of BM-MSCs. None of them have provided definitive advantages as compared with isolation from whole BM-MNCs. However, the negative selection of committed lineages by sorting with a cocktail of markers of differentiation can significantly increase the rate of MSCs isolated from a sample by retaining the late-adhering MSCs that are normally lost after the initial change of the medium. In specific conditions, immunomagnetic sorting based on CD105 improves the isolation efficiency when MSCs are expected to be very rare, for example, in the samples obtained by washing the empty BM collection bags [72].
Initial selection based on STRO-1 increases 10–100-folds the formation of colony-forming units fibroblasts (CFU-F) according to the separation method (immunomagnetic vs. dual color cell sorting) and the coexpression of other markers [73]. In other cases, however, no difference in CFU-F number has been noted [74].
CD133 was used for positive selection in 1 study by Tondreau and colleagues [75]. Isolated adherent cells expressed the embryonal transcription factor Oct-4, showed pluripotent differentiation potential, and were endowed with higher proliferative potential than those from the CD133neg population. Interestingly, they appeared CD73 pos and CD105 pos, and negative for CD14, CD34, CD45, and HLA-DR. CD49a (α1 integrin subunit) is another important marker of native MSCs, as all the multipotential CFU-F are included in the CD49apos fraction of BM-MNCs, and a double selection using CD49a and CD133 can be effectively used to amplify the number of early progenitors [76].
CD146 has been suggested as a highly specific marker of MSCs, as only CD146 pos MSCs were capable of supporting hematopoietic repopulation of a subcutaneously transplanted bone chip by CD34 pos hematopoietic stem cells (HSCs) and their progeny [40]. Further, CD146 also identifies STRO-1neg cells with MSC features from human endometrium [77]. Similarly, CD200 has been used to purify native MSCs at very high efficiency [78]. CD271 was also used as a selective marker of MSCs, and cells isolated from a CD271pos population showed higher clonogenicity and lower hematopoietic contamination than MSCs, either isolated from whole BM-MNCs or from CD105pos cells [79]. Recently, CD271 has been proposed as one of the most specific markers for BM-derived MSCs and used as the gold standard to compare putative new identifiers [80].
A potential side effect of sorting is the activation of unknown molecular pathways due to cross-linking of surface antigens. To avoid this, aptamers have been recently proposed as alternative sorting tools. These single-stranded DNA or RNA molecules can fold into a 3-dimensional structure and bind to a variety of targets, including peptides, enzymes, and various cell surface receptors, with high affinity and no immunogenicity [81]. Aptamers can be up to 100 bases long; they have been custom-generated from combinatorial libraries through an in vitro selection process and have also been recently applied in MSC isolation with consistent improvement in the frequency of clonogenic MSCs in the expanded population. However, the target of the aptamers remains unclear [70].
Despite these potential advantages, any kind of selection increases the potential risk of isolating a biologically distinct subpopulation; it has been recently shown in a population of MSCs derived from CD271pos, MSCA-1pos, and CD56pos (39D5 epitope) cells that MSCs tend to differentiate into chondrocytes and pancreatic-like islets [82].
MSCs and their in vivo counterpart
Although the phenotype of cultured MSCs is well known, the phenotype of in vivo, or native, MSCs still remains elusive. New data show that MSCs seem to be located within the vicinity of vessel walls: CD146 is a marker of multipotent native MSCs; these latter also express other molecules typical of perivascular cells [40]. It has been argued by some authors that all the cells with properties of MSCs derive from pericytes [10]: when cultivated using the same conditions routinely used for MSCs, whatever their tissue origin, pericytes grow, display an MSC phenotype, and are capable of differentiating into osteoblastic, chondrocytic, and adipocytic lineages. In the case of BM-MNCs, enrichment in native MSCs may be obtained by cell selection according to different surface antigens, such as STRO-1, CD146, CD49a, CD200, or CD271. However, expression of some of them, for example, STRO-1, CD146, and CD200, is downregulated during culture [78].
All considered, one may question whether MSCs are indeed a unique and homogeneous population; to this aim, the embryonal development and the tissue origin of MSCs need to be considered. For a long time, MSCs were considered a unique cell population originating from the mesoderm. Actually, it has been recently demonstrated that a first wave of MSCs arises from neuroepithelium during embryogenesis [68]. These cells are transiently found during ontogenesis and their real persistence after birth as part of the MSC pool is currently unknown. Their origin could partially explain the different differentiation behavior of MSCs expanded from different tissues. Alternatively, MSCs may be subjected to the influence of specific microenvironments, thus explaining the preferential differentiation profiles. For instance, while BM-MSCs do not easily differentiate into cardiomyocytes, adipose-derived MSCs show a greater tendency to develop into endothelial and cardiac cells both in vivo and in vitro [83,84].
Functional Assays
Self-renewal and clonogenicity (CFU-F assay)
CFU-F assays are made at best by single-cell seeding after FACS [1]. Alternatively, it is possible to plate at limiting dilution, for example, 1 × 106 cells in a T25 flask (4 × 104/cm2), and then counting cell clusters with >50 cells as colonies at day 10 [18,85]. The possibility to obtain clones at every passage is a necessary condition to claim the maintenance in culture of uncommitted, self-renewing progenitor cells; however, a more detailed analysis of their ability to self-renew and to differentiate into multiple lineages at the clonal level is needed to actually demonstrate the persistence/expansion of such cells in vitro [1,2].
Differentiation in vitro
Although multipotent in vitro differentiation is a fundamental property of MSCs [11], this is actually true only for a minority of expanded MSCs [86]; in vitro culture leads to a substantial loss of multipotentiality, due to cellular senescence [87], and the real differentiation of MSCs highly depends on the tissue source [88]. For instance, adipose-derived MSCs tend to be less sensitive to osteoblastic differentiation, and are prone to differentiate more easily into adipocytes; stromal cells isolated from BM are capable of supporting hematopoiesis in vivo upon de novo bone formation [40], whereas dental pulp stromal cells give rise to dentin rather than bone in vivo [24]. Moreover, MSCs expanded from antigen-sorted populations may show a tendency toward specific lineages [82]. Finally, plating density also influences differentiation; for instance, a plating density around 104 cells/cm2 at 70%–80% confluence is suggested for adipoblastic and osteoblastic differentiation [34,89] (Table 1).
Abbreviations: agc1, aggrecan1; ALP, alkaline phosphatase; bFGF, basic fibroblast growth factor; BMP-2, bone morphogenetic protein-2; BSA, bovine serum albumin; Col II a1, type II collagen; Col IX a1, type IX collagen; Col X a1, type X collagen; DMEM, Dulbecco's modified Eagle's medium; DMS, dexamethasone; DUSP1, dual-specific phosphatase-1; EGF, epidermal growth factor; FABP4, fatty acid-binding protein 4; FBS, fetal bovine serum; IBMX, 3-isobutyl-L-methylxantine; IBSP, integrin-binding bone sialoprotein; LPL, lipoprotein lipase; MSCs, mesenchymal stem cells; PDGF, platelet-derived growth factor; PPARγ2, peroxisome proliferative activated receptor γ2; RUNX2, runt-related transcription factor 2; SPC, sphingosyl-phosphoryl-choline; TGF-β1, transforming growth factor β1; IMDM, Iscove's modified Dulbecco's medium; IHC, immunohistochemistry.
Signs of induction are detectable a few hours after stimulation, in the case of morphological changes in the cell shape and mRNA expression of specific genes, or after several days, in the case of the accumulation of neutral lipid vacuoles (Fig. 3), de novo protein production or positivity to specific cytochemical stainings (Table 2). However, in vitro multipotent differentiation does not imply by definition the effective in vivo differentiation in the same lineages: a few studies demonstrated long-term and functional in vivo differentiation of MSCs; in addition, neural differentiation in vivo of MSCs is still debated [90].

In vitro multipotent differentiation of MSCs. (
Abbreviation: PG, propylene glycol; Fast Green FCF, C37H34N2Na2O10S3, C.I. 42053, E143; FR, fast red solution; IHC, immunohistochemistry.
Notes on adipogenic differentiation.
3-isobutyl-L-methylxanthine (IBMX) (Table 1) is either a competitive nonselective phosphodiesterase inhibitor [91] and a nonselective adenosine receptor antagonist [92]. Its final action on cell metabolism is to raise intracellular cyclic adenosine monophosphate (cAMP), activating protein-kinase A. IBMX has shown to promote the conversion of fibroblasts into adipocytes, apparently with no effect on cell proliferation [93]. Its importance in inducing early adipogenic differentiation has been also established on a preadipocytic cell line, 3T3-L1, where the addition of IBMX and dexamethasone to the medium leads to a significant increase in the production of two species of chondroitin 4-sulfate proteoglycans (CSPG-I and -II) and heparan sulfate proteoglycan [94], and promotes their differentiation into adipocytes. Subsequently, it has become part of induction media of most differentiation media used also for MSCs [95]. Its action may be negligible once activation of the signaling pathways involved in adipogenic commitment has been established, especially in the presence of insulin [96].
For an early evaluation of differentiation, the analysis of de novo mRNA expression of genes related to adipocytic differentiation, can be performed. The accumulation of neutral lipid vacuoles usually does not become visible before 4–5 days from the start of the induction and may require 2–3 weeks to occur (Fig. 3).
Suggested genes typical of adipogenic differentiation include peroxisome proliferative activated receptor γ2 (PPAR-γ2), fatty acid-binding protein 4 (FABP4), and lipoprotein lipase (LPL) [97] (Table 1).
Notes on osteogenic differentiation.
Von Kossa staining reveals calcium salts using substitution with oxidized (black) silver nitrate [98 –100] (Table 2; Fig. 3). After the addition of the silver nitrate solution, the sample should be exposed to the light of an electric bulb for 30–60 min. Caution is advisable when examining black areas, because of the occurrence of false positive signals.
Genes of osteocytic commitment include alkaline phosphatase (ALP), runt-related transcription factor 2 (RUNX2/Cbfa1/Pebp2αA), integrin-binding bone sialoprotein (IBSP), osteocalcin (BGLAP), osteopontin (SPP1), osteoprotegerin (tnfrsfIIb-OPN), and dual-specific phosphatase 1 (DUSP1) [97] (Table 1).
Notes on chondrogenic differentiation.
Chondrogenic differentiation is usually performed with cell pellets. Although several authors prefer DMEM with low glucose for chondrogenic differentiation [15,97,101], media with high-glucose concentrations have been used as well: as pointed out by Mackay and colleagues, high-glucose concentration may help increase the micromass pellets before chondrogenic differentiation [102] (Table 1).
At the start of the induction, 1 mL of the induction medium without transforming growth factor (TGFβ1) is added to the top for the first 24 h; then, the supernatant is replaced with the induction medium containing TGF-β1 for the next 2–3 weeks.
Evaluation of chondrogenic differentiation is carried out by staining with Toluidine-blue, Alcian-blue, or Safranin-O [98 –100]. The first 2 methods are probably more indicated to highlight proteoglycan [103]; by contrast, Toluidine blue has been found in some cases less accurate than Safranin O in the detection of cartilage glycosaminoglycans due to its stoichiometric staining characteristics [104] (Table 2 and Fig. 3).
Gene expression analysis should include aggrecan1 (agc1), collagen type II (Col II a1), type IX (Col IX a1), and type X (Col X a1) [97].
Cardiomyogenic differentiation
After 24-h exposure to 5-azacytidine (3 μM), an unspecific demethylating agent, MSCs can acquire in 1 week markers for myocyte and smooth muscle cells, an elongated morphology and sarcomeric striations; after 2 weeks, these cells show reciprocal connections and can begin spontaneous beating [105] (Table 1). Sarcomeres can be observed by phalloidin staining, myocytic proteins, and transcription factors by immunohistochemistry and reverse transcriptase (RT)-polymerase chain reaction (PCR). Analysis should include Nkx2.5, GATA-4, MEF-2, α-sarcomeric actinin, α-sarcomeric actin, and β-myosin heavy chain.
Alternative approaches for myogenic differentiation are based on the treatment with BMP-2 and FGF-4 [106]; insulin, dexamethasone, and ascorbic acid [107]; or coculture with adult cardiomyocytes [108]. MSCs also appear to differentiate into myocytes spontaneously, although at a very low rate [83].
Differentiation into smooth muscle cells
Differentiation of MSCs into smooth muscle cells seems to depend on TGF-β1 signaling, either directly [109] or after stimulation with TGF-β3 or
Neural differentiation
MSCs can be induced to differentiate into neural lineages (Table 1): in one method [42] MSCs were grown at low density (3,000 cells/cm2) on poly-lysine-coated plates [use 10 μg/mL poly(lysine) in PBS overnight] for 7 days; the amplification medium comprised low-glucose DMEM, 10% FCS, glutamine 2 mM, and bFGF 25 ng/mL.
In another approach [111,112] MSCs were incubated with 5 ng/mL bFGF for 24 h, followed by the complete substitution of the medium for 2–16 h with DMEM, N2 supplement, butylated-hydroxyanisole, KCl, valproic acid, and forskolin. With either methods, MSCs show a dramatic change in morphology after 24–48 h, and they develop elongated neurite-like branches and axon-like structures, significantly increase their basal nestin expression (from 50.3% ± 8.2% to 94.5% ± 3.4%), and start to express neuronal and glial markers, such as NF-L, β3-tub [38], PMP-22, GFAP, and NeuN [111]. Functional assays show the appearance on the cells of functional neural receptors and pharmacologically sensitive voltage-dependent calcium channels [42]. However, with both differentiating media MSCs revert to basic conditions when induction is suspended. In contrast, coculture with Schwann cells seems to lead the cells to express an irreversible neural phenotype [111].
Senescence and transformation
Senescence, defined as irreversible growth arrest, is the ultimate physiological condition of human MSCs expanded ex vivo. It is characterized by the gradual disappearance of telomerase in the cells, decreased telomeric length, and increasing appearance of p16INK4a-positive MSCs, a marker for upregulation of signal pathways related to senescence and apoptosis in several stem cell types [113]. Senescent MSCs accumulate internal fatty vacuoles, lower their growth rate, and eventually stop growing and become apoptotic. The absence of senescence phenomena after prolonged expansion suggests the neoplastic transformation of MSCs, which should be tested by different approaches (in particular, [114]): Cytogenetic analysis: normally at the end of the first (P1) and second (P2) passages, by means of standard karyotyping according to the International System for human Cytogenetics Nomenclature, and fluorescent in situ hybridization according to standard protocols [114]. Expression of genes involved in transformation: assessment by means of real-time RT-PCR of c-myc, p21, p53, and p16
INK4a gene expression and the presence of functional full-length hTERT transcript, and the integrity of Ink4a/ARF locus by PCR analysis of p16 exons 2 and 4 [114]. Culture in soft agar and β-galactosidase (β-Gal) expression: in vitro testing of anchorage-independent colony development is carried out by culturing MSCs in soft agar at 2 time points, end of P3 and end of last passage. Senescence of cultivated MSCs may be studied by β-Gal staining using specific senescence detection kits [114]. In vivo tumorigenesis in mice: female SCID mice are sublethally γ-irradiated and infused intravenously with 106 MSCs; animals are monitored for 8 weeks and then sacrificed for organ analysis [114].
Through these different approaches, it has been recently shown that clinical-grade human MSCs expanded in vitro for tissue engineering or immunoregulatory purposes show poor susceptibility for neoplastic transformation [114], although 2 previous studies had described the capacity of human MSCs to accumulate chromosomal instability and give rise to carcinoma in immunocompromised mice after long-term culture [115,116]. Tarte and colleagues have shown that some transient and donor-dependent recurring aneuploidy may be detected in vitro, independently of the culture process. However, MSCs with or without chromosomal alterations show progressive growth arrest and enter senescence without evidence of transformation either in vitro or in vivo [114].
Experimental Evidence for Potential Use of MSCs for Tissue Repair
A proper expansion and the use of a large amount of homogeneous MSCs are often not sufficient to obtain a significant improvement in a clinical setting. The use of MSCs alone has not led to clear-cut results in terms of tissue regeneration yet. MSCs were initially used in osteoarthritis, with poor results [117]. More promising results were obtained in a mouse model of osteogenesis imperfecta, where intravenously injected MSCs engrafted and gave origin to de novo osteogenesis [118]. Further, the use of MSCs in three affected children led to new lamellary bone formation and improved laboratory and clinical findings [119]. However, the effective role of MSCs in inducing de novo osteogenesis has been questioned; in fact, the conditioning regimen used to achieve BM transplantation might have directly favored this phenomenon.
Similarly, the inoculation of human MSCs in a mouse model of Duchenne's muscular dystrophy (mdx mouse) was associated with scarse evidence of formation of new functionally active myoblasts and of significant sarcolemmatic expression of dystrophin [120].
Thus, it has become clear that the microenviroment in which MSCs are transplanted (ie, growth factors and local cellular interactions) plays a pivotal role in determining both MSC biology (survival, proliferation, and specific differentiation) and eventually a clinical measurable improvement. For instance, in tendon repair MSCs alone led to a partial recovery of tendineal function (20%; 37% at 12 months in a model of lesions of Achilles' tendon); nevertheless, in this study ectopic bone formation was detected in 30% of cases [121]. Growth factors [117], biomaterials like poly-
In cardiac regeneration, there are several animal studies pointing to effective improvement of cardiac function after intramyocardial injection or intracoronary infusion of MSCs, either by transdifferentiation [129] or by positive paracrine effects exerted by the cells on various targets, including resident cardiac progenitors [130]. In fact, even if MSC-derived myocytes connect with preexisting residual myocardium through connexin-43 GAP-junctions in several animal models [8,129], the significance and the long-term in vivo persistence of MSC-derived cardiomyocytes in humans is still under debate [130]. Two clinical trials have been concluded, so far, with positive results on very short follow-up (3–6 months) [131,132], whereas several others are on-going. Another potential approach would involve in vitro predifferentiation of MSCs into cardiomyocyte (CMCs): after exposure to 5-azacytidine, MSCs react to electrical currents [105] and to adrenergic and cholinergic stimuli; in mice, they integrate in preexisting tissue and survive for up to 4 weeks [133]. Nevertheless, no clinical trial using these cells has been conducted so far.
MSCs have also been used to engineer transplantable immunocompatible heart valves starting from decellularized homograft [134] or xenografts [135]. Currently, in vitro engineering using decellularized porcine valves seem somehow more promising, as residual cellularity and calcification of the valves before MSC implantation can hamper subsequent tissue formation and function. Still, like in bone engineering, moderate stress, for example, the one provided by cyclic flexure or laminar flow artificially reproduced in some types of bioreactors, seems important to drive tissue formation [136].
In vivo neural differentiation of MSCs is even more problematic. Adipose-derived MSCs can migrate through the hemato-encephalic barrier after adhesion to the endothelium by VLA-4 [112], and they can differentiate into neuroglial lineages after intraventricular in utero injection in rats [137]. They provide a certain level of functional recovery in animal models of Parkinson and hypoxic-ischemic neural damage [138]. To date, however, there is no evidence about the neural regenerative potential of MSCs in vivo in humans.
In summary, the question is: to fully characterize MSCs expanded in vitro we have to carry out all the functional assays that have been previously described; but are we really sure that in vivo differentiation of MSCs is absolutely required to achieve positive effects in treated patients? On the basis of the current experimental evidence, the answer could be no: only a few studies managed to demonstrate the long-term persistence of MSC-derived differentiated cells. Paracrine effects by a large number of biologically relevant molecules and cytokines produced by MSCs have been often advocated to explain the functional benefits achieved in animal models and treated patients [119,120,130,138]. It is likely that in most cases MSCs may simply help tissue repair without directly contributing to it, by acting on resident progenitor cells or boosting recipient tissue activity.
In other words, we are capable of thoroughly characterizing what we recognize as MSCs, but nowadays we have only partial knowledge about the precise mechanisms by which MSC-based therapy seems to work.
Potential Risks of the Use of MSCs in Clinical Trials
MSCs have been successfully used in phase-II and -III clinical trials for the prevention and treatment of graft-versus-host disease after allogeneic HSC transplantation, and they have a number of promising applications in regenerative medicine, especially in the orthopedic clinical setting. However, their short- and long-term safety needs to be addressed, not only in preclinical animal models, but also with a long-term follow-up of MSC-treated patients. For instance, the engraftment of MSCs in a specific tissue may result in inadequate differentiation, as shown by the appearance of undesired ectopic bone within the heart of MSC-treated animals in a mouse model of myocardial infarction [139]. This type of unexpected differentiation needs to be further investigated in different relevant animal models before starting large-scale human trials. Further, the possible neoplastic transformation during prolonged MSC expansion must be always taken into consideration, as previously discussed. Its occurrence was demonstrated during long-term culture of adipose-derived MSCs: the cells acquired chromosomal abnormalities, then became capable of growing persistently, and finally expressed human telomerase and formed tumors when injected in immunocompromised mice [140]. Actually, neoplastic transformation of MSCs seems to be generally a long process that requires sequential steps [141]; as recently shown, it does not occur even in long-term cultures [114, 142]; however, as long-term-expanded MSCs show progressive growth arrest and enter senescence [114], it is always preferable not to use these cells for clinical use.
Finally, it has to be reminded that theoretically MSCs injected into patients might promote the growth of preexistent tumors, even without evidence of their direct neoplastic transformation [143]; this event is related either to the immunosuppressive activity of MSCs or the production of pro-angiogenic soluble factors, for example, angiopoietin 1 and VEGF [144]. However, preclinical experimental data are not clear-cut: in some cases (Kaposi sarcoma) MSCs inhibited tumor growth [145], while they led to faster tumor development when coinjected with melanoma cells [146]. Therefore, the careful pretransplant evaluation and long-term follow-up of all patients treated with MSCs are required.
Conclusions
After Friedenstein et al.'s article in 1976 [1] and almost 20 years of relatively poor interest, MSCs have experienced a Renaissance period in the last decade of the 20th century, as the improvement of isolation and expansion methods for stem cells permitted to obtain large numbers of cells from small starting samples. Thus, MSCs have now entered the clinical setting. Large international consortia have been formed to develop effective and reproducible protocols for MSC isolation and expansion, while meeting high standards of clinical safety.
Considering the increasing interest toward regenerative medicine based on MSCs and the heterogeneity of isolation, expansion, and differentiation protocols employed by different groups worldwide, a sort of handbook concerning biological features, immunophenotype, culture techniques, functional assays, and quality controls used for BM-MSCs and AT-MSCs (most commonly used so far) may be of potential interest for new scientists entering this intriguing field.
Footnotes
Acknowledgments
The authors would like to thank Dr. Veronica Lisi, Ph.D., for providing the examples of in vitro multilineage differentiation of MSCs, and Dr. Anabel Varela Carver, Ph.D., for critically reviewing the manuscript. This work was supported by a grant from the 7th Framework Program of the European Commission (CASCADE, no 223236; HEALTH-F5-2009-223236) and by a grant from Fondazione CARIVERONA (Bando 2008 - Tecnologie al servizio della salute: “Utilizzo delle capacità rigenerative e immunoregolatorie delle cellule staminali mesenchimali per il trattamento di patologie ossee, neurologiche e cardiologiche”).
Author Disclosure Statement
F.M.: no competing financial interests exist.
L.S.: no competing financial interests exist.
M.K.: no competing financial interests exist.