
Abstract
A combined analysis of data from a series of literature studies can lead to more reliable results than that based on a single study. A common problem in performing combined analyses of literature microarray gene expression data is that the original raw data are not always available and not always easy to combine in one analysis. We propose an approach that does not require analyzing original raw data, but instead takes literature gene sets derived from (supplementary) tables as input and uses gene co-occurrence in these sets for mapping a co-regulation network. An algorithm for this method was applied to a collection of literature-derived gene sets related to embryonic stem cell (ESC) differentiation. In the resulting network, genes involved in similar biological processes or expressed at similar time points during differentiation were found to cluster together. Using this information, we identified 43 genes not previously associated with cardiac ESC differentiation for which we were able to assign a putative novel biological function. For 6 of these genes (Apobec2, Cth, Ptges, Rrad, Zfp57, and 2410146L05Rik), literature data on mouse knockout phenotypes support their putative function. Three other genes (Rcor2, Zfp503, and Hspb3) are part of major pathways within the network and therefore likely mechanistically relevant candidate genes. We anticipate that these 43 genes can help to improve the understanding of the molecular events underlying ESC differentiation. Moreover, the approach introduced here can be more widely applied to identify possible novel gene functions in biological processes.
Introduction
C
We propose to use published microarray data in a different way, omitting normalization and statistical issues between different studies. We do this by looking for jointly regulated genes based on co-occurrence in individual experimental gene sets. A gene set is here defined as a list of genes that are described as meeting the same regulation criteria in a given literature study, such as being significantly upregulated, downregulated, or being clustered together based on having similar expression patterns. In fact, most microarray study publications provide (supplementary) tables with gene lists that meet criteria suitable for the underlying study. In many cases, a gene set will correspond directly to such a table, sometimes to table subsections that have different headers. If 2 genes occur together in a gene set in one publication, there is a reasonable chance that they are relevant to the process and also that they are to a certain extent coregulated. If this is reported in multiple studies, both these chances become increasingly likely. Therefore, by analyzing a sufficiently large number of gene sets, it becomes possible to determine with high confidence which genes are commonly regulated and additionally share coregulation. The latter information can be used to construct a coregulation network, which can provide additional insights into the regulatory processes involved [7,8]. More specifically, if a gene with a hitherto unknown function is coregulated with several genes that all are known to be involved in the same biological process, this gene then can be assumed to play a role in that process [9 –11]. Further, the number of connections (edges) that a gene has in a network can serve as an indicator for its importance in the network process [12].
In this study, we will show the applicability of our method using 3 different gene set collections. One of these collections, containing gene sets derived from studies on embryonic stem cell (ESC) differentiation, will be analyzed in more detail with a view to cardiomyocyte differentiation and its application in toxicity testing.
During embryonic development, the heart is one of the first tissues to be formed and to acquire functionality. The cellular and molecular events involved in early cardiac development are highly conserved between different species (see review in [13,14]). Cardiac progenitor cells originate in the mesodermal germ layer and ultimately differentiate to cardiomyocytes via a cascade of development steps. Although molecular analysis of cardiac development has already identified several genes involved in (part of) this complex differentiation process, much is still unknown. ESC models are often used as a practical model system to study in vitro cardiomyocyte differentiation (reviewed in [15]). Several gene expression studies using this model system have been published, which allows for data from several related studies to be analyzed in combination.
We applied our method to construct and analyze a coregulation network of ESC differentiation, and used this network to derive groups of coregulated and functionally related genes. Using this approach, we identified several novel genes involved in the cardiac cell differentiation process.
Methods
Libraries
The first gene set collection used is the c2.cgp collection, which was downloaded from the MSigDB Web site (
We previously described the third gene set collection, which is the ESC differentiation collection [17,18] (and references sited therein). This collection contains gene sets derived from 42 microarray studies describing gene expression changes during the differentiation of ESC to several types of tissue, with a certain emphasis on cardiomyocytes. During the collecting process, gene sets were included if the underlying study was considered relevant based on mutual agreement by a developmental toxicologist and bioinformatician. To avoid redundancy, gene sets obtained using the Anni textmining tool were excluded for the present study as textmining is implicitly using literature data. The resulting gene set collection used contained 167 gene sets and a total number of 15,196 UGIs.
Coregulation mapping
Gene coregulation mapping was determined using an algorithm written in statistical software environment R (
The resulting coregulation network was further observed using Gene Ontology (GO) functional annotations, or gene expression data from earlier studies [5,17]. The presence of significantly densely connected clusters within the total network was determined using the Cytoscape plugin MCODE [20] using default settings.
Identification of novel genes involved in ESC cardiac differentiation
For the genes in the ESC differentiation coregulation network mapping, Gene Ontology functional annotation and enrichment was determined using the Cytoscape plugin BiNGO [21] in combination with the DAVID Web application (david.abcc.ncifcrf.gov) [22]. Genes were considered already associated with ESC differentiation if they were assigned to one of the following Gene Ontology terms: development processes (including stem cell, organ, heart, or muscle development), cell differentiation, cell proliferation, extracellular matrix (including collagen), contractile fiber, muscle system process (including muscle contraction), and heart contraction. Further, genes for which the textmining tool Anni [23] found concept weights >10−5 for the above-mentioned terms, or the concepts “stem cells,” “gastrulation,” or “ectoderm/endoderm/mesoderm formation” were considered not novel for stem cell differentiation. The concept weight is a measure Anni uses to indicate the degree of literature association of one term with another. We found that when distributions for a larger number of concept weights are combined, the distribution approaches its background level at concept weights around 10−5, and therefore concept weights below this value are considered as not indicative for literature association. For the genes not functionally annotated by Gene Ontology or Anni, those that were part of an MCODE cluster enriched for one of the above functions were considered candidates for such a function.
Results
Method development
To show the applicability of our approach and to optimize the criteria used, we applied our algorithm to 2 gene set collections that are different in nature. The first of these is the MSigDB c2.cgp collection [16], which contains 1,186 gene sets related to a wide range of chemical and genetic perturbations, and, second, the lung-inflammation collection, with 90 gene sets, which is smaller and more focused on acute lung inflammation models. For both gene set collections we found that biologically meaningful coregulation networks could be obtained (Fig. 1A, B). For the c2.cgp collection, various coregulation networks of varying sizes were found. Several of these consisted of genes with a common related functionality such as cell division or immunological response (Fig. 1A). For the lung-inflammation collection, besides some smaller networks, a large and dense network of commonly inflammation-induced genes containing 120 genes was found (Fig. 1B). This network includes 66 immune response genes, 61 of which overlap with the 100 immune response genes found previously as part of a full-scale meta-analysis [5]. Moreover, this network contains all of the 23 “core inflammation response” genes found in the previous meta-analysis.
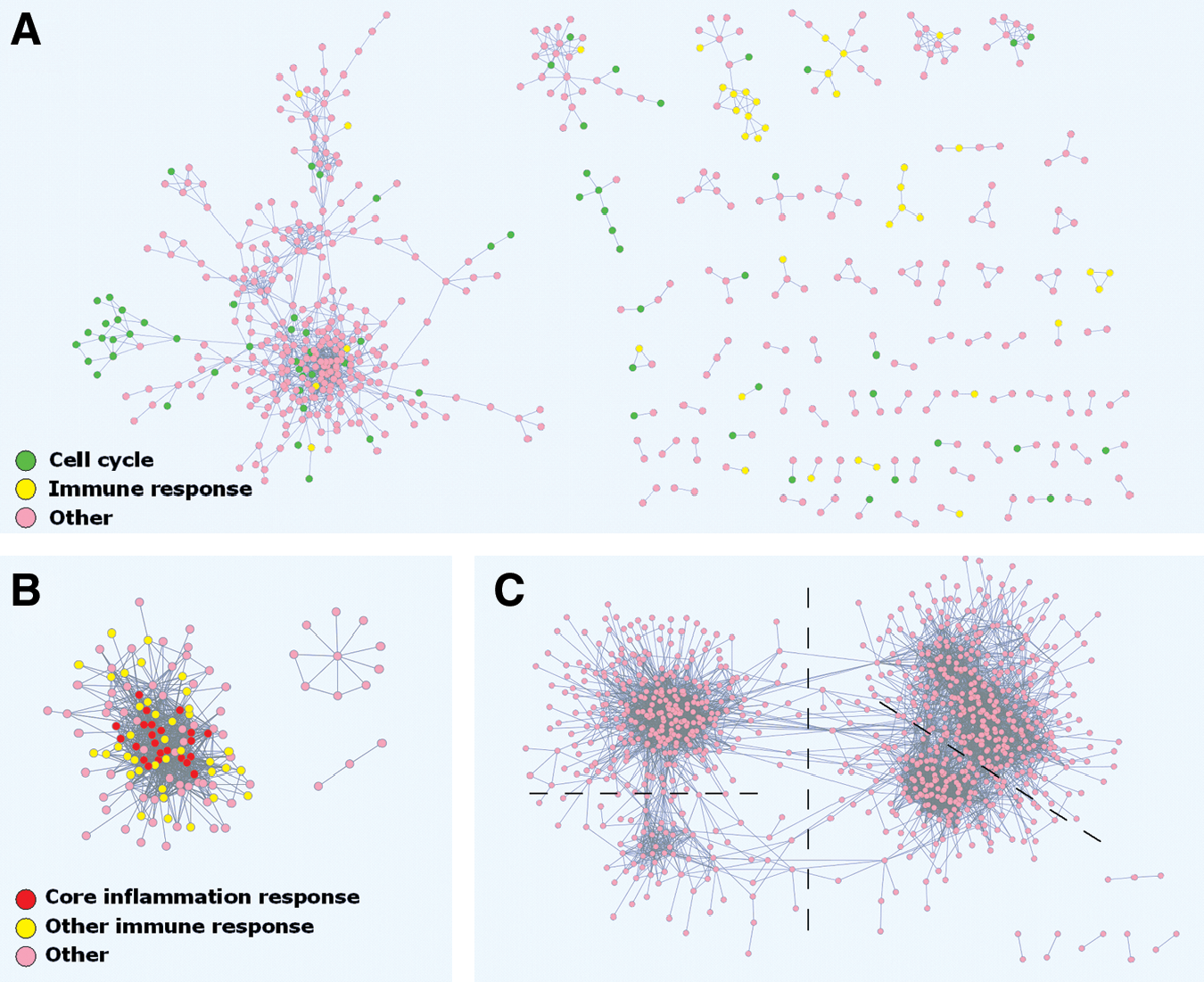
Network observations for 3 gene set collections.
ESC differentiation mapping
When the algorithm was applied to the ESC differentiation gene set collection, a large and densely interconnected network with 927 genes and 7,402 edges was obtained (Fig. 1C). The network appeared to have 2 major sub-networks, at the left- and right-hand side, respectively. This finding was found to be robust for various stringency settings, as was the observation that both major sub-networks comprised 2 minor sub-networks. The overall network was significantly enriched for several GO-terms related to development, differentiation, and muscle function. In addition, enrichment for several other functional categories was found, especially transcription factors, cell proliferation, small heat shock proteins, and several types of metabolite transport. However, we found that known cardiomyocyte marker genes were located exclusively in the lower right-hand side of the network, whereas stem cell pluripotency markers were located only in the top left. This suggests that these 2 areas correspond to different processes or phases in stem cell differentiation. Both the functional and timing aspects were examined further.
By using the MCODE plugin, we identified 21 clusters within the overall network that are significantly densely connected. The size of these clusters ranged from 4 to 39 genes, with an overall total of 304 genes. Several of these clusters were enriched for specific GO terms. Clusters with such similar GO term enrichments appeared together in the network graph, and (by making small adjustments to the MCODE cluster size threshold parameter) it was found that they were mutually strongly connected. Taken together, 3 cluster groups could be distinguished (Fig. 2A; Tables 1
–3; Supplementary Table S1, available online at
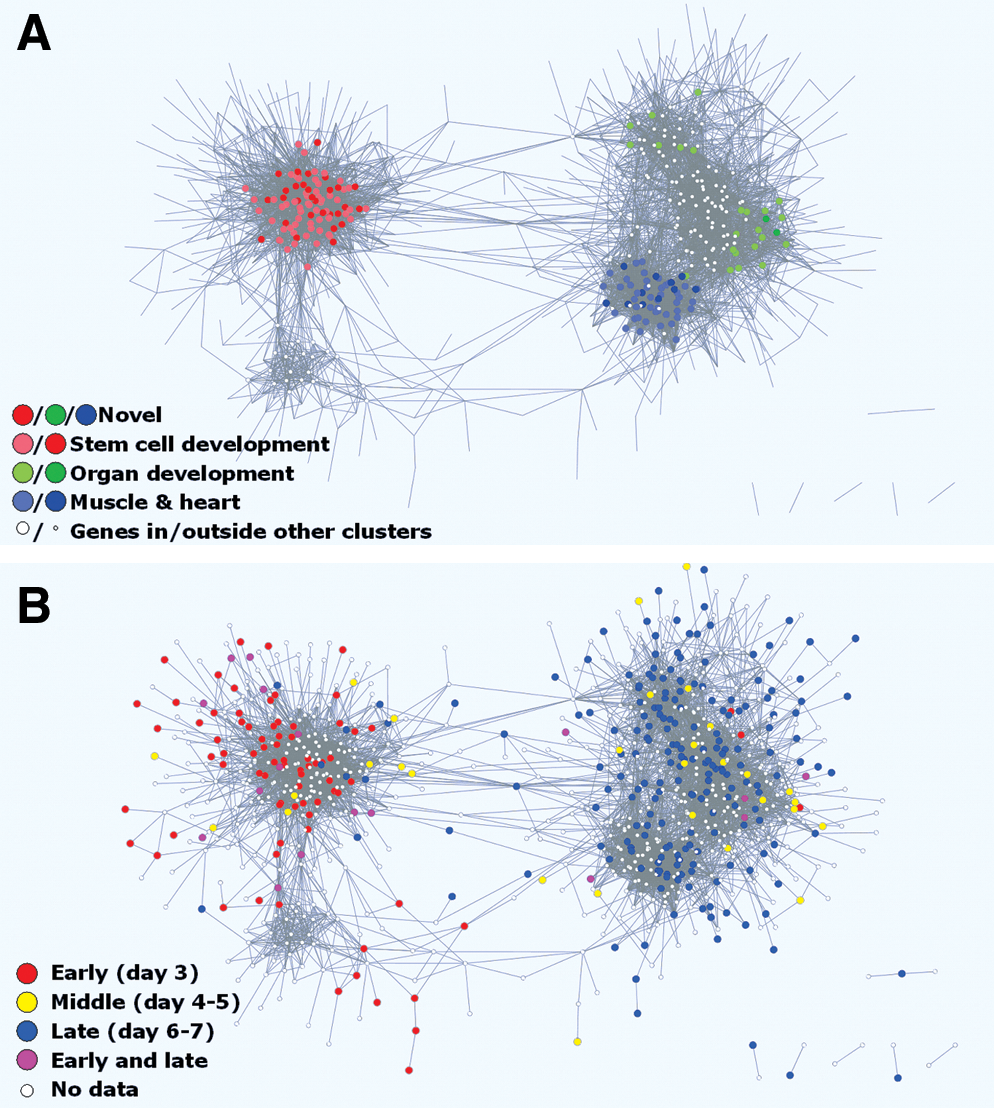
Embryonic stem cell cardiomyocyte differentiation coregulation network.
To observe the differentiation process in an alternative manner, we used data from a recent study in which we described gene expression changes during ESC cardiac muscle cell differentiation [17]. This study was not included in the ESC differentiation gene set collection and therefore provided independent data. For the genes for which differential expression was found, we used different colors to indicate at which differentiation phase these genes had their highest expression. This observation indicated that the left and right side of the network can be associated with early (undifferentiated stem cells) and late (more differentiated) time points, respectively (Fig. 2B). This is in agreement with the different functionalities based on GO term enrichment. Genes expressed during intermediate time points did not group together, but appeared in either of the 2 network sides.
When these 2 observation approaches were combined again, we found that out of the 167 genes in the 3 cluster groups (Fig. 2A, Tables 1 –3), independent data from the van Dartel [17] study showed differential expression for 49 of these genes (29%, compared to 6% for the whole-genome data). Of these, 18 early expressed genes were all found in the stem cell development group (group 1). Of the 4 mid-phase expressed genes, 2 were found in the stem cell development group and 2 in the organ development group (group 2). Of the 26 late expressed genes, 2 were found in the stem cell differentiation group, 4 in the organ development group, and 20 in the muscle/heart function group (group 3). Finally, one gene with both early and late high expression was found in the stem cell development group.
Novel ESC cardiac differentiation genes
Using Gene Ontology for functional annotation of the genes in the ESC development network, we found that, of the 927 genes, 521 could be associated with GO processes relevant for ESC differentiation, and 418 with related Anni textmining concepts. Leaving out these genes left 327 genes not yet described to be involved in ESC differentiation, 165 and 162 of which were located at the left- and right-hand side of the network, respectively. Of these novel genes, 43 were also part of a significantly dense MCODE cluster with a functional enrichment for a GO term related to ESC or cardiac differentiation, making them candidate genes for having a corresponding function assigned to them. These 43 genes are listed in Table 4 and indicated (in dark red, dark green, or dark blue) in Fig. 2. Concerning the overlap with other pathways found previously, among these novel genes there were no genes involved in cell proliferation, there were 3 transcription factors (Rcor2, Zfp57, and Zfp503), and one heat shock protein (Hspb3).
Discussion
Since its introduction, microarray technology has grown to become used in over 6,000 PubMed publications per year. The growth in the number of studies published has been followed by the development and application of various methods for combined or meta-analysis (reviewed in [24]). However, there are several practical issues related to such analyses of gene expression data, and one major hurdle is the limited public availability of the complete raw or normalized data sets used in literature studies. Several approaches to combined or meta-analyses have been published in the last few years, and although this field is still developing, it becomes apparent that no single best method exists as the suitability of an approach will depend on the nature and quality of available data. In this study we propose an approach that is not based on the actual re-analysis of original data, but on the co-occurrence of genes based on processing of gene sets derived from these data. One of the rationales behind our approach is the need for a combined analysis that allows inclusion of large numbers of literature microarray study results, even if the full original data are not (or not easily) available. Our algorithm for network construction allows the inclusion of almost any published study and further does not require elaborate data processing or new software implementations; R and Cytoscape are freely available and already familiar to most bioinformaticians. The algorithm used by us is available as Supplementary Data (available online at
The 3 underlying concepts for our approach have each already been described on their own. First, coexpression or coregulation networks [8] have become a commonly applied method to infer novel gene functionality [7,9 –11]. Second, algorithms for determining gene co-occurrence in literature publications have been applied in textmining applications [25 –28]. However, whereas textmining is mostly based on literature abstracts or database description fields, our approach specifically searches in (supplementary) tables, allowing the use of information that would otherwise have been overlooked. Third, employing gene sets rather than full microarray data on the one hand or literature abstracts on the other has been described in several applications [29 –32]. These approaches, however, look for statistically significant overlap between lists, or to an experimental gene set supplied by the user. Therefore, the combination of these methods provides a novel integrated type of approach that should be able to incorporate some of the advantages of each of the underlying methods. A potential weakness of our approach is that the quality of the gene set collection taken as the starting point for this method will depend on the quality and relevance of the input data. We had to rely on the analyses done by the authors of the underlying studies, including the assessment of the reviewers and editors responsible for the process of peer review.
As part of the algorithm development, our approach was tested on 2 gene set collections, which were selected to be different in nature. On the basis of the multipurpose c2.cgp collection, a large number of small networks were identified, including cell division and immunological response networks (Fig. 1A). The coregulation network for the lung-inflammation gene set collection found a large network, of mainly immunological genes (Fig. 1B), which comprises the majority of the immunological genes that were found regulated by a previous full meta-analysis [5]. Moreover, it contains all of the 23 “core inflammation response” genes found previously [5]. These findings illustrate the usefulness of our approach.
For these 2 gene set collections, the examples given were obtained using stringency settings for which the number of genes in the network description file is around 5% of the number of UGIs in the collection, and these settings were chosen as the default stringency settings. However, comparable results were obtained when this fraction was between 1% and 10% (data not shown). This indicates that the results are robust against small changes in stringency. This was also found for the ESC differentiation coregulation network mapping. Moreover, to ensure that the method does not lead to false-positive results, the c2.cgp and lung inflammation were scrambled; that is, the genes were randomly relocated across the gene sets. Applying the algorithm to these sets did not result in a network even for the lowest stringency level (data not shown), confirming that the method is robust against random false-positive findings.
Applying our algorithm to the ESC differentiation gene set collection with 15,196 UGIs results in a network with 927 genes mostly located within 2 major sub-networks that correspond to early and late differentiation processes stages, respectively. Combining GO term functional enrichment to MCODE clusters within the network reveals that genes involved in similar biological processes are significantly densely connected, which is in agreement with the premise that such genes are coregulated. Moreover, this analysis shows that early differentiation events, such as loss of pluripotency, are concentrated in a cluster group at the left side of the network, whereas the right side of the network contains 2 cluster groups involved in later differentiation phases, one of which is specifically related to muscle and heart function (Fig. 2A). The early–late division is supported by overlaying gene expression data from an independent study on ESC cardiomyocyte differentiation time series [17] (Fig. 2B). Genes expressed during intermediate time points in that study did not form a separate sub-network, but instead appeared within either of the major sub-networks. This can partially be ascribed to the focus of most literature studies on the differences between undifferentiated and fully differentiated cells, leaving intermediate time point genes underrepresented in the combined gene set collection. Further, whereas pluripotent stem cells or differentiated cardiomyocytes are both stable situations regarding gene expression, the intermediate phase can be described as consecutive waves of gene expression corresponding to sequential transient activation of differentiation programs [17,33,34]. Due to their transient expression, such genes are more difficult to identify than those involved in the initial and final phase of the differentiation process and they may therefore have been described in smaller numbers [17,34]. Both these factors will lead to a relatively smaller number of coregulation edges among intermediate phase genes, which influences the Cytoscape observation as its mapping algorithm is developed for grouping together connected genes rather than observing data as a time series. Nevertheless, the additional use of the MCODE plugin results in 3 functional cluster groups that are visually separated and each correspond to a particular differentiation phase.
At the lower left side of Fig. 2A, there is a small sub-network containing a number of genes involved in development, proliferation, and pluripotency. These genes could have been expected to occur in the larger sub-network in the upper left side of this figure. The presence of a separate sub-network might indicate a possible common factor influencing the (co-)expression of these genes. However, as the main associated processes are still comparable to that of its larger neighbor except that the functional annotation enrichment of this smaller sub-network is not significant, the relevance of this sub-network should not be overinterpreted. The same can be said for the 6 groups of 2 or 3 genes each in the very lower right of the network observation. Their occurrence as separate groups is not robust to small changes in the algorithm settings used, and they can be considered as falling just short of being connected to the main network.
The 3 functional groups in the network contain over 60 genes with a GO annotation organ development as an umbrella term, or more specific terms regarding development of a specific organ or tissue. Apart from those involved in heart or muscle, there was also functional enrichment for the following terms: blood vessel development, ear development, eye development, gland development, immune system development, lung development, nervous system development, and skeletal development, as well as some of their daughter terms. Although several genes are annotated to the development of more than one type of organ, muscle and heart (-specific) development-related genes are significantly found in the third group (Table 3), and the second group (Table 2) significantly contains genes (specifically) involved in the development of other organs. Because genes expressed during the intermediate and late time points in the van Dartel [17] study are found in the sub-networks around the second as well as the third group, this indicates that during culture conditions favoring cardiomyocyte differentiation, also other types of tissues are formed.
Other functional analyses showed that the overall network not only contains various development- and heart/muscle-associated genes, but in addition genes involved in cellular proliferation, transcription, and also several members of the alpha crystallin/small heat shock protein family. The first of these processes can easily be understood as the starting point of stem cell differentiation in a culture of proliferating cells. Likewise, transcription factors are necessary to trigger and regulate the developmental changes that occur between proliferating stem cells and fully differentiated cells, which can explain why these genes are mainly found in the development-related first and second group, but hardly in the muscle function-related third group. In contrast, this third group contains a number of small heat shock proteins and the structurally related alpha crystallin B. Several members of this chaperonin family have already been associated with muscle contraction or development, and as Hspb3 has not been associated with such a role yet, this might indicate a novel function for this protein. However, although a common relevance of chaperonins in heart function is conceivable, extending this association to Hspb3 would require additional study and verification, as there is evidence that in heart Cryab and Hspb2 act through different and distinct mechanisms within cardiomyocytes [35].
Of the 927 genes in the network, 327 (35%) have been described in multiple gene lists, but have not yet been linked to a related developmental process in Gene Ontology or by literature association. Although in most of the studies the genes are only part of a bulk of differentially expressed genes, this combined study shows that there is good evidence to describe them as novel genes involved in ESC differentiation. Further, based on their position in the network, it can already be assumed whether they are mainly expressed at early or late differentiation stages. For 43 of these novel genes, there is even more specific evidence, namely, that they are part of densely connected cluster groups within the overall network that are enriched for a relevant biological function.
To determine the in vivo relevance of these 43 genes, we searched the literature as well as the Mouse Genome Informatics (
This indicates that the other genes in Table 4 also provide interesting candidates for further studies regarding ESC cardiac differentiation. Among these, Hspb3 and 2 transcription factors (Rcor2 and Zfp503) are part of pathways enriched within the network, which makes them the mechanistically most conceivable starting points. We compared expression data for these genes across multiple tissues and cell types by means of the BioGPS Web site (
In addition to mechanistic studies of stem cell differentiation, we expect the newly found genes to be potentially useful in applied test systems using ESC differentiation as a model to identify developmental-toxic properties of compounds, such as the ESC test (EST) [44]. The implementation of molecular biological approaches in such models may help to improve the prediction accuracy of the model [17,18]. Further, it may help in understanding the biology of the model, which is useful for the definition of its applicability domain. Thus, a further knowledge of genes involved in cardiomyocyte differentiation as well as their regulation can help to further optimize such test models for developmental toxicity.
Footnotes
Acknowledgments
This study was supported by grant MFA6809 from the Dutch technology society foundation STW and by grant no. 050-06-510 from the Netherlands Genomics Initiative/Netherlands Organization for Scientific Research (NWO).
Author Disclosure Statement
The authors declare that no competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.