
Abstract
We recently reported that concentrated conditioned medium (CdM) from human CD133-derived bone marrow progenitor cells (CD133 CdM) was neuroprotective after stroke. Here we identify stromal-derived factor 1 alpha (SDF-1) as a potential neuroprotective candidate in CD133 CdM by interrogating the transcriptional responses of CD133-derived multipotent stromal cells (CD133dMSCs) after cell injection into the ischemic brain. Human SDF-1 mRNA was upregulated 79-fold by CD133dMSCs when injected into the stroke peri-infarct area compared with cells injected into the uninjured parenchyma of sham-operated animals. In cell protection assays, we replaced the typical growth medium in mouse neural progenitor cell (mNPC) cultures with serum-free CD133 CdM immediately before exposure to hypoxia (1% oxygen) for 48 h. CD133 CdM significantly increased the survival of mNPCs during hypoxia exposure and growth factor withdrawal. To determine whether MSC-secreted SDF-1 influenced mNPC survival, we used lentiviral short hairpin RNA against SDF1 (shSDF-1) to knockdown SDF-1 expression in CD133dMSCs. The CdM generated from shSDF-1-treated cells had a 94% decrease in secreted SDF-1 and was significantly less protective for mNPCs when compared with control CdM from CD133dMSCs transduced with scrambled short hairpin RNA. Pharmacological inhibition of the 2 known SDF-1 receptors, CXCR4 and CXCR7, revealed that only CXCR7 activity was functionally linked to survival signaling in mNPCs during hypoxia exposure. Treatment of mNPCs with CD133 CdM and CXCR7 inhibitor decreased mNPC viability by 36.5% ± 12.8% and decreased cell number by 21% ± 6.7% compared with dimethyl sulfoxide treated controls. These data indicate that SDF-1 is a key neuroprotective cytokine secreted by CD133dMSCs that protects mNPCs through CXCR7.
Introduction
A
Changes in MSC secretion after tissue injury may indicate a response for repair by paracrine action [12 –14]. Thus, proteins/peptides that increase in expression and secretion when MSCs are exposed to ischemic environments may represent neuroprotective factors that could reduce injury from stroke. Here we demonstrate that adult human bone-marrow-derived MSCs enriched by the CD133 epitope (CD133-derived MSCs, CD133dMSCs) robustly increased stromal-derived factor 1 alpha (SDF-1) mRNA levels in response to the ischemic environment of stroke. CD133dMSCs secreted physiologically relevant levels of SDF-1 ex vivo that protected mouse neural progenitor cells (mNPCs) from hypoxia-mediated death. Of therapeutic interest, SDF-1 survival signaling was mediated through CXCR7 and not CXCR4.
Methods
Isolation and preparation of MSCs
CD133dMSCs were isolated and prepared as previously described [5]. CD133dMSCs were cultured in Nunclon Delta–coated 150 cm2 dishes (Nunc; Thermo Fisher Scientific, Rochester, NY) with the complete culture medium (CCM) containing alpha minimum essential medium (α-MEM; Invitrogen, Carlsbad, CA), 20% fetal bovine serum (lot selected for rapid growth of human MSCs (hMSCs; Atlanta Biologicals, Lawrenceville, GA), 100 U/mL penicillin, 100 μg/mL streptomycin, and 2 mM
Lentiviral transduction of CD133dMSCs
For cell tracking in vivo, CD133dMSCs were transduced with lentivirus to express green fluorescent protein (GFP) as previously reported [15]. For short hairpin RNA (shRNA) knockdowns, CD133dMSCs were transduced with puromycin-selectable lentivectors expressing GFP (SHC005V MISSION eGFP shRNA Control Transduction Particles), scrambled (nontargeting) shRNA (SHC002V) or sequence-specific shRNA complementary to SDF-1 (MISSION shRNA Transduction particles TRCN0000003311 and TRCN0000003312) (Sigma-Aldrich, St. Louis, MO). Transduced CD133dMSCs were selected by growth in CCM containing 2 μg/mL puromycin for 3 days, followed by expansion in CCM with 1 μg/mL puromycin for 2 weeks.
CD133dMSC CdM production
CdMs for enzyme-linked immunosorbent assays (ELISAs) and mNPC bioassays were produced and concentrated as previously described [5]. We thawed 1× and 10× CdMs once at 37°C immediately before application to neural cultures to minimize deactivation of bioactive proteins. Protein concentrations of CdMs were measured by Bradford dye-binding assay (Bio-Rad, Hercules, CA). The protein concentrations of CdMs from cells that received short hairpin RNA specific to SDF-1 (shSDF-1) knockdown and cells that received scrambled non-specific short hairpin RNA (shScram, control) did not differ (1 × shSDF-1, 101.6 ± 14.1 μg/mL; 1 × shScram, 92.3 ± 19.2 μg/mL).
Isolation and treatment of mNPCs
GFP mNPCs were isolated as previously described [5]. mNPCs were grown in the neural stem cell (NSC) growth medium (GM) containing Neurobasal A (Invitrogen), 10 ng/mL epidermal growth factor (EGF), 10 ng/mL basic fibroblast growth factor, 2 μg/mL heparin, 1× B27 supplement (Invitrogen), 2 mM
Mouse middle cerebral artery ligation model
All animal work was approved by the University of Vermont College of Medicines' Office of Animal Care in accordance with American Association for Accreditation of Laboratory Animal Care and National Institutes of Health guidelines. Twenty-four C57/Bl6 mice were anesthetized with isoflurane (1%–5%, to effect) and body temperature was maintained by heating pad. Permanent distal middle cerebral artery (MCA) ligation was performed as previously reported [5]. For 12 mice, the MCA was encircled with 10-0 monofilament nylon suture using a surgical needle and ligated. For the 12 mice that underwent sham surgery, the MCA was encircled with nylon suture as above, but not ligated. GFP-positive CD133dMSCs (125,000 cells resuspended in 5 μL of α-MEM) were injected 1 day after MCA ligation with a 30 gauge floating needle to the inferior aspect of the peri-infarct area. Sham-operated control animals were injected with the same number of GFP CD133dMSCs from the same cultures and into the analogous brain region. Animals were euthanized at 48 h after cell injection and brains were removed for total RNA extraction.
Total RNA isolation and real-time polymerase chain reaction for human-specific transcripts
Brains from sham-operated mice and those with stroke were cut into coronal sections with a scalpel and tissue block (Zivic Miller, Portersville, PA). The upper cerebral quadrants containing the infarct zone and injected cells (or analogous area in shams) were cut from the coronal sections and homogenized in a guanidine-based RNA extraction buffer (PowerGen 125; Fisher Scientific, Pittsburg, PA). Approximately 100–200 mg of tissue was homogenized in 1 mL of buffer. The extraction buffer consisted of 4 M guanidine thiocyanate (47.26 g), sodium citrate (0.30 g), anhydrous citric acid (0.35 g), N-lauryl sarcosine (0.5 g), and 1% beta-mercaptoethanol, dissolved into 100 mL (final volume) of de-ionized water. Two molar mass sodium acetate (2M, pH 4.0) was then added to the homogenates (1/10th of total volume). Total RNA was separated from DNA and protein by standard phenol/chloroform/isoamyl alcohol extraction (pH 4.3). The resulting RNA was DNase treated (TurboDNase; Ambion Inc., Austin, TX).
RNA was reverse transcribed into cDNA (Superscript III; Invitrogen). The resulting cDNA (300 ng per sample) was used for real-time polymerase chain reaction (PCR; 7500 Fast Cycler; Applied Biosystems, Foster City, CA). Because RNA samples contained both mouse brain RNA and human RNA from the injected CD133dMSCs, we used commercially validated human-specific primers and Fam-Tam probes (TaqMan® Gene Expression Assays; Applied Biosystems). In addition, mouse cDNA generated from control brain samples (not containing human cells) was used to confirm the human specificity of primer sets. These controls were performed for all real-time PCR along with no template controls (water). Cycle thresholds (CTs) were calculated for each transcript (auto baseline function, ABI 7500 fast PRISM software), and then normalized to human-specific glyceraldehyde 3-phosphate dehydrogenase levels determined from the same samples. Relative quantifications for transcript levels were calculated by the ΔΔCT method and statistical significance was determined by Student's t-test (2-tailed, type 3) that compared ΔCT values between sham and stroke animals [16]. All real-time PCR were performed in duplicate.
Reverse transcriptase-polymerase chain reaction
mNPCs were lifted with trypsin EDTA (0.25% trypsin/1 mM ethylenediaminetetraacetic acid; Mediatech) from laminin/poly-
Immunocytochemistry
After differentiation, neurospheres were fixed with 4% paraformaldehyde for 15 min at room temperature and washed with phosphate-buffered saline (PBS) 3 times. Cells were blocked with blocking buffer (5% goat serum, 0.4% triton X-100 in PBS) for 1.5 h, and then incubated in primary antibodies diluted in blocking buffer: beta-III tubulin (1:500, PRB-435P, Covance, Princeton, NJ), glial fibrillary acidic protein (GFAP) (1:1,000, Z0334; Dako, Glostrup, Denmark), and O4 (1:40, O7139; Sigma, St. Louis, MO). After 3 PBS washes, cells were incubated in secondary antisera (Alexa Fluor 488 Alexa Fluor 350 or Alexa Fluor 594, 1:500) for 1 h at room temperature. After 3 PBS washes, the coverglasses were counter-stained and mounted (Citifluor Antifadent Mounting Medium AF1; Electron Microscopy Sciences, Hatfield, PA).
GFP mNPCs were grown in the absence of extracellular matrix and triturated in trypsin EDTA (Mediatech) to dissociate neurospheres into single-cell suspensions. mNPCs were plated at 20,000 cells/well into 24-well plates (Nunc; Thermo Fisher Scientific) containing laminin/poly-
Enzyme-linked immunosorbent assays
Sandwich ELISAs for SDF-1, interleukin 6 (IL-6), and hepatocyte growth factor (HGF) were performed according to the manufacturer's instructions (R&D Systems, Minneapolis, MN). Poly-horseradish peroxidase-streptavidin (Endogen, 1:2,000; Pierce, Rockford, IL) was used in all assays for biotinylated antibody detection (1-Step™ ABTS; Pierce). Absorbance measurements for all samples and standards were performed in triplicate at 450 nm wavelength (Synergy HT; BioTek Instruments, Inc., Winooski, VT).
Results
CD133dMSCs increase SDF-1 mRNA levels at the stroke penumbra
We sought to determine how CD133dMSCs alter growth factor and cytokine expression when placed in the ischemic environment of the brain after stroke. We performed stroke surgeries in C57/Bl6 mice and injected CD133dMSCs into the inferior aspect of the peri-infarct area 1 day later. By epifluorescence microscopy, GFP-labeled CD133dMSCs were observed throughout the needle track just inferior to the autofluorescent stroke infarct core 48 h after cell injection (Fig. 1A). Brain tissue encompassing the human cells was removed and analyzed by human-specific real-time PCR assays. We examined gene expression for proteins we had previously established to be secreted from CD133dMSCs after nutrient deprivation and hypoxia exposure (simulated ischemia) for 48 h [5]. In the present study, gene expression for cells injected into mice with stroke was compared with that of cells injected into the nonstroke brain parenchyma of sham-operated mice (no ligation of the MCA) at 48 h after transplantation.
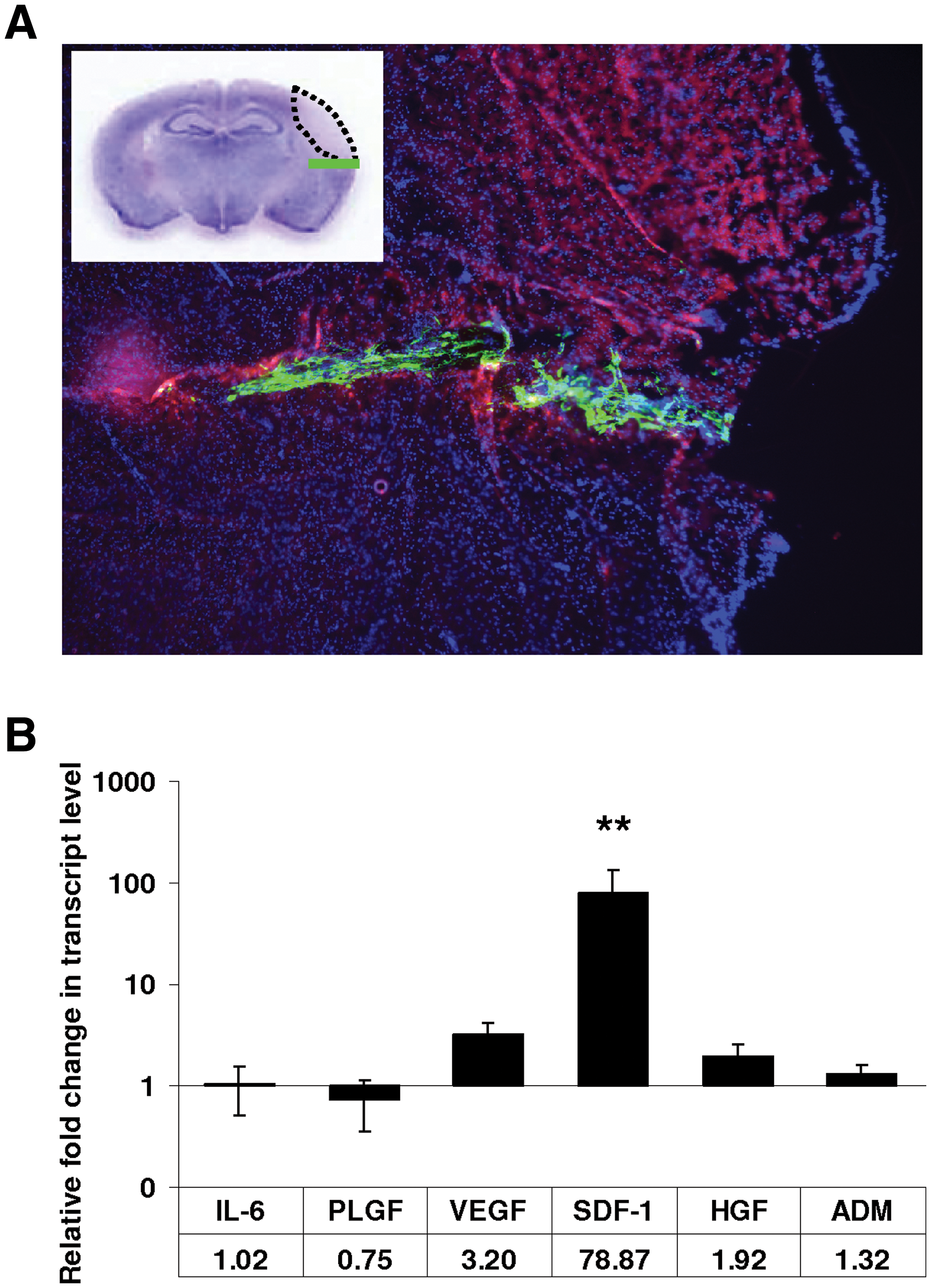
CD133-derived multipotent stromal cells (CD133dMSCs) increased their stromal-derived factor 1 alpha (SDF-1) mRNA levels at the stroke peri-infarct area.
Of the genes tested, only mRNA for SDF-1 was significantly upregulated by CD133dMSCs (78.9-fold, stroke n = 12, sham n = 10, P = 0.008; Fig. 1B). Although the mRNA levels for vascular endothelial growth factor increased by 3.2-fold, this response was not statistically significant (P = 0.068). The mRNA levels for the other secreted factors did not change significantly. As a result, we continued to investigate the role of SDF-1 as a stem cell-derived neuroprotective agent ex vivo. CD133dMSCs that were serum-starved and exposed to hypoxic culture conditions for 48 h also increased SDF-1 mRNA levels compared with control cultures (2.15-fold, P = 0.0014) (data not shown).
Multipotent mNPCs express both SDF-1 receptors
mNPCs were grown as spheres and then differentiated for 7 days (Fig. 2A). Triple staining immunocytochemistry was performed to indicate the formation of neurons (Beta-III tubulin), astrocytes (GFAP), and oligodendrocytes (O4) (Fig. 2A). Staining clearly demonstrated the presence of the 3 committed lineages derived from the mNSCs.
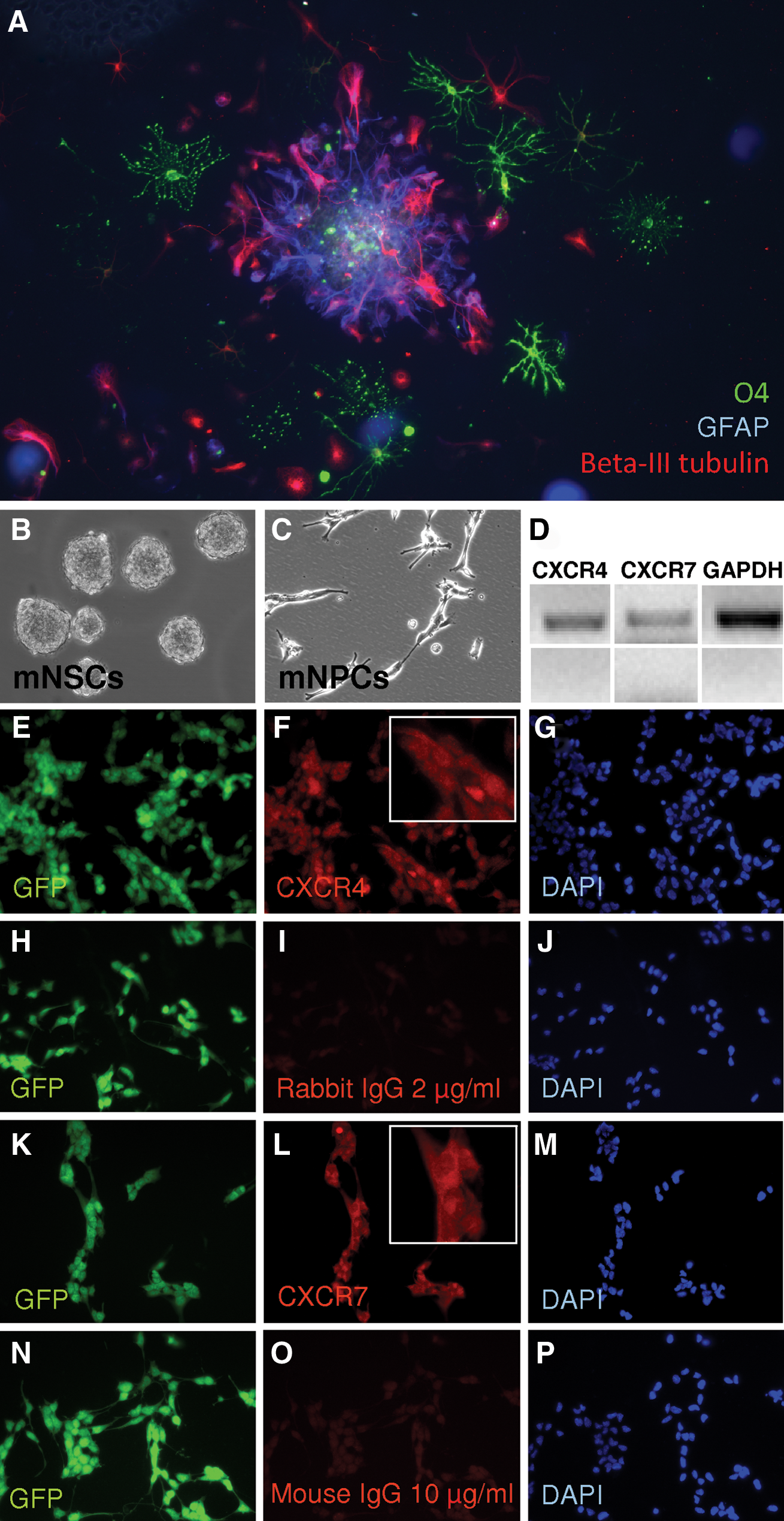
Expression of CXCR4 and CXCR7 by mouse neural progenitor cells (mNPCs).
To determine expression of SDF-1 receptors in the mNPCs, we performed reverse transcriptase (RT)-PCR assays and immunocytochemistry for CXCR4 and CXCR7. Both mRNAs were detected, and mNPCs grown as a monolayer in NSC GM stained positive for both receptors (Fig. 2B–P).
Lentiviral shRNA expression to deplete SDF-1 secretion from CD133dMSCs
Lentiviral transduction of CD133dMSCs with shRNA specific to SDF-1 reduced the levels of secreted SDF-1 by 94% (shSDF-1 CdM) compared with secretion from cells transduced with nontargeting shRNA (scrambled shRNA, shScram CdM) or no lentivector (ELISAs, P < 0.001, Fig. 3). To control for possible off-target effects of shRNA expression on other secreted proteins, we performed ELISAs for IL-6 and HGF. The levels of IL-6 and HGF contained in shSDF-1 CdM and shScram CdM did not differ (IL-6: shSDF-1, 365 ± 31 pg/mL vs. shScram 202 ± 81 pg/mL, P = 0.183; HGF: shSDF-1, 1,287 ± 99 pg/mL vs. shScram 1,672 ± 163 pg/mL, P = 0.128). Subsequently, CdMs generated from the transduced cells were used in ex vivo bioassays.
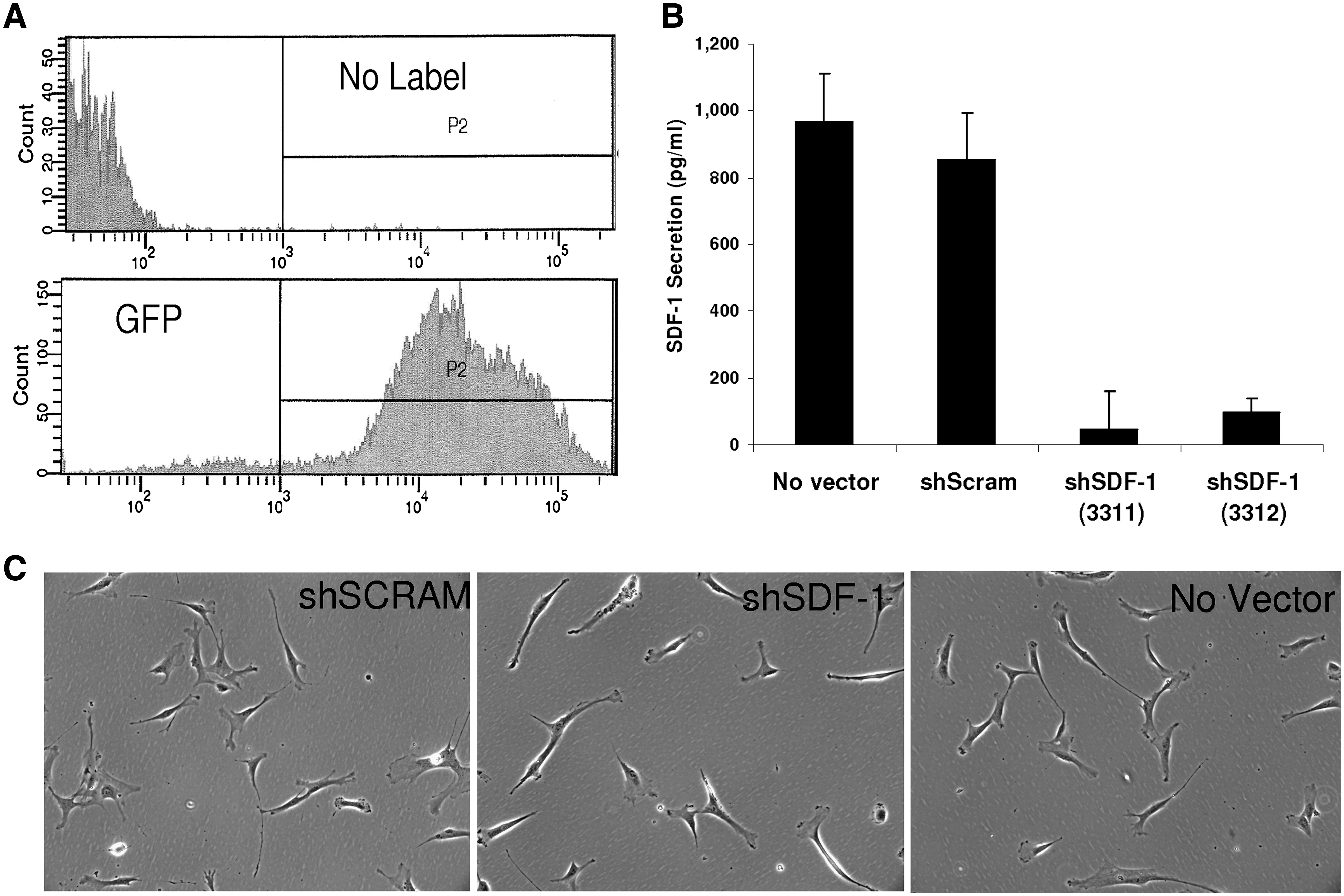
Knockdown of SDF-1 secretion in CD133dMSCs by lentiviral short hairpin RNA (shRNA). CD133dMSCs were transduced with puromycin-selectable lentivectors expressing GFP (transduction control), scrambled (nonspecific) shRNA, or sequence-specific shRNA (against SDF-1).
SDF-1 knockdown diminished the neuroprotective effect of CD133 CdM
mNPCs were challenged by hypoxia exposure (1% O2) and growth factor withdrawal by replacement of NSC GM with serum-free α-MEM (SFM), shSDF-1 CdM (10×), or shScram CdM (10×), and cultured for 48 h. shSDF-1 CdM had a significantly reduced capacity for neural cell protection compared with shScram CdM (62.3% ± 7.4% vs. 107.8% ± 5.1%, P = 0.0016, GM standardized to 100%; Fig. 4A). The level of mNPC protection conferred by shSDF-1 CdM was still greater from that provided by SFM (62.3% ± 7.5% vs. 38.3% ± 10.7%, P = 0.039).
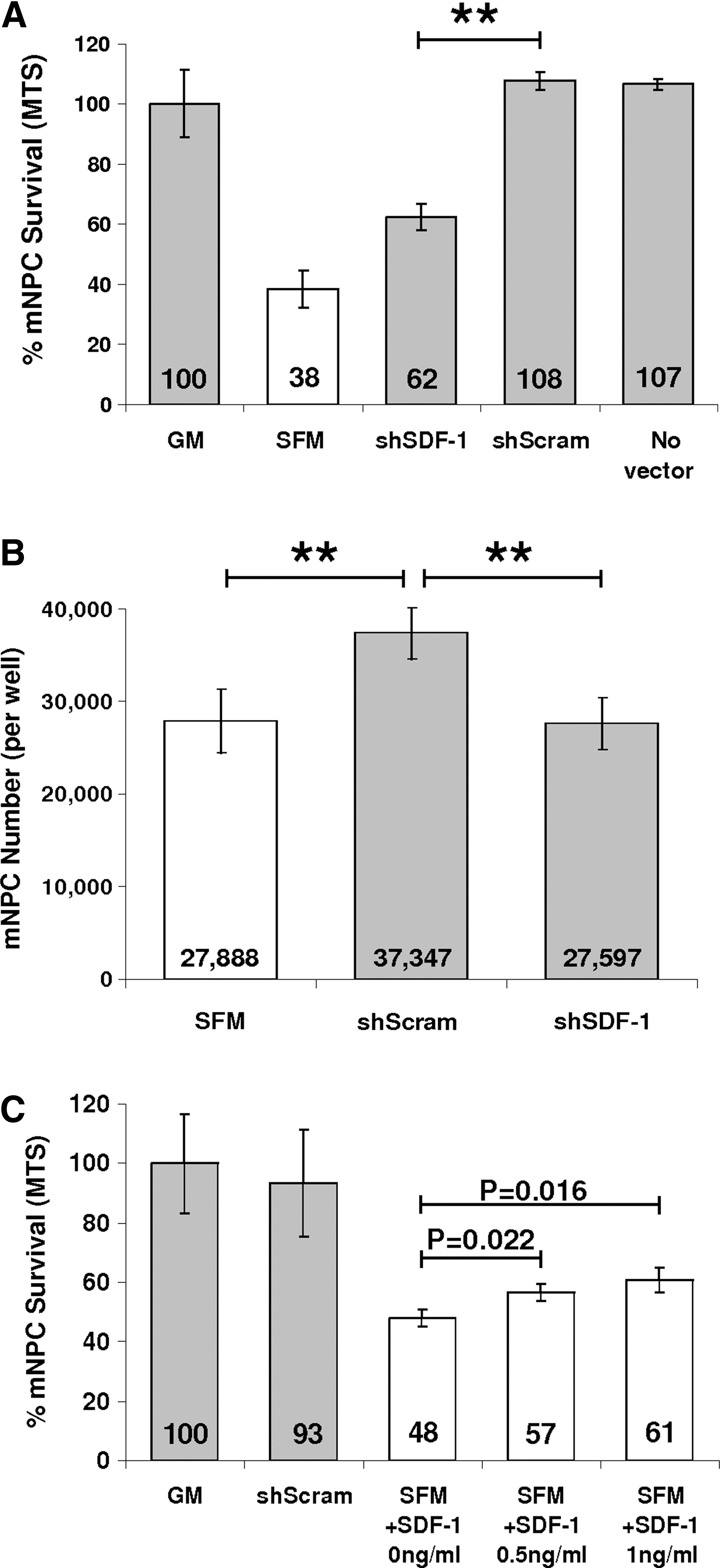
SDF-1 knockdown diminishes the protective effect of the conditioned medium (CdM).
By phase microscopy, the cells incubated in shScram CdM were less numerous, larger, and appeared to be more differentiated; they had more cellular processes than did mNPCs cultured in GM (data not shown). The MTS assay we used to assess cell survival measured cell metabolic viability by dehydrogenase enzyme activity, which may be altered by the state of mNPC differentiation. To more thoroughly ascertain the effects of the different CdMs, in parallel assays we determined mNPC number by dye-labeling of nucleic acid content (CyQUANT assay). Similar to the results obtained with MTS, mNPC number was greater after incubation in shScram CdM compared with culture in shSDF-1 CdM or SFM (shScram: 37,347 vs. shSDF-1: 27,597, P = 0.0024; and shScram: 37,347 vs. SFM: 27,888, P = 0.0056). Cell numbers did not differ between cultures that received shSDF-1 CdM and SFM (P = 0.899) (Fig. 4B).
We next tested whether addition of recombinant SDF-1 to shSDF-1 CdM could restore its ability to protect mNPCs. The amount of SDF-1 added to shSDF-1 CdM made its SDF-1 concentration equivalent to that of shScram CdM (8 ng/mL, as assayed by ELISA; see Fig. 3B). After the addition of recombinant SDF-1, the level of protection conferred by shSDF-1 CdM was no longer statistically different from that of shScram CdM (93.7% ± 7.9% vs. 100% ± 7.6%, P = 0.381, data not shown). Further, addition of recombinant SDF-1 to SFM containing 20 μg/mL heparin increased mNPC viability in a dose-responsive manner (0.5 ng/mL, P = 0.022, 1 ng/mL, P = 0.016; Fig. 4C). Interestingly, shSDF-1 CdM and shScram CdM provided similar levels of neuroprotection during growth factor withdrawal in normoxic conditions (21% O2), suggesting that secreted SDF-1 in CD133 CdM was important for mNPC survival signaling specifically in the context of hypoxia (data not shown).
CXCR7 provides the neuroprotection conferred by SDF-1 in CD133 CdM
To determine the relative importance of CXCR4 and CXCR7 in mediating mNPC survival, we tested inhibitors of CXCR4 (AMD3100) and CXCR7 (CCX771) in our cell protection assay. mNSC GM was replaced with shScram CdM that contained either inhibitor or an equal amount of DMSO (vehicle control). To control for toxicity, CCX771 was added to mNPCs incubated in SFM alone. Contrary to our expectation that addition of AMD3100 to shScram CdM would decrease cell survival, by MTS assay the inhibitor instead increased cell survival by 22.7% ± 6.7% (P = 0.0157) (Fig. 5A). In contrast, addition of the CXCR7 inhibitor CCX771 to shScram CdM decreased mNPC viability by 36.5% ± 12.8% compared with shScram control (P = 0.021) and decreased cell number by 21% ± 6.7% (P = 0.049) (Fig. 5B and C). In toxicity controls, CCX771 did not significantly decrease mNPC viability or cell number when added to SFM (Fig. 5B and C).
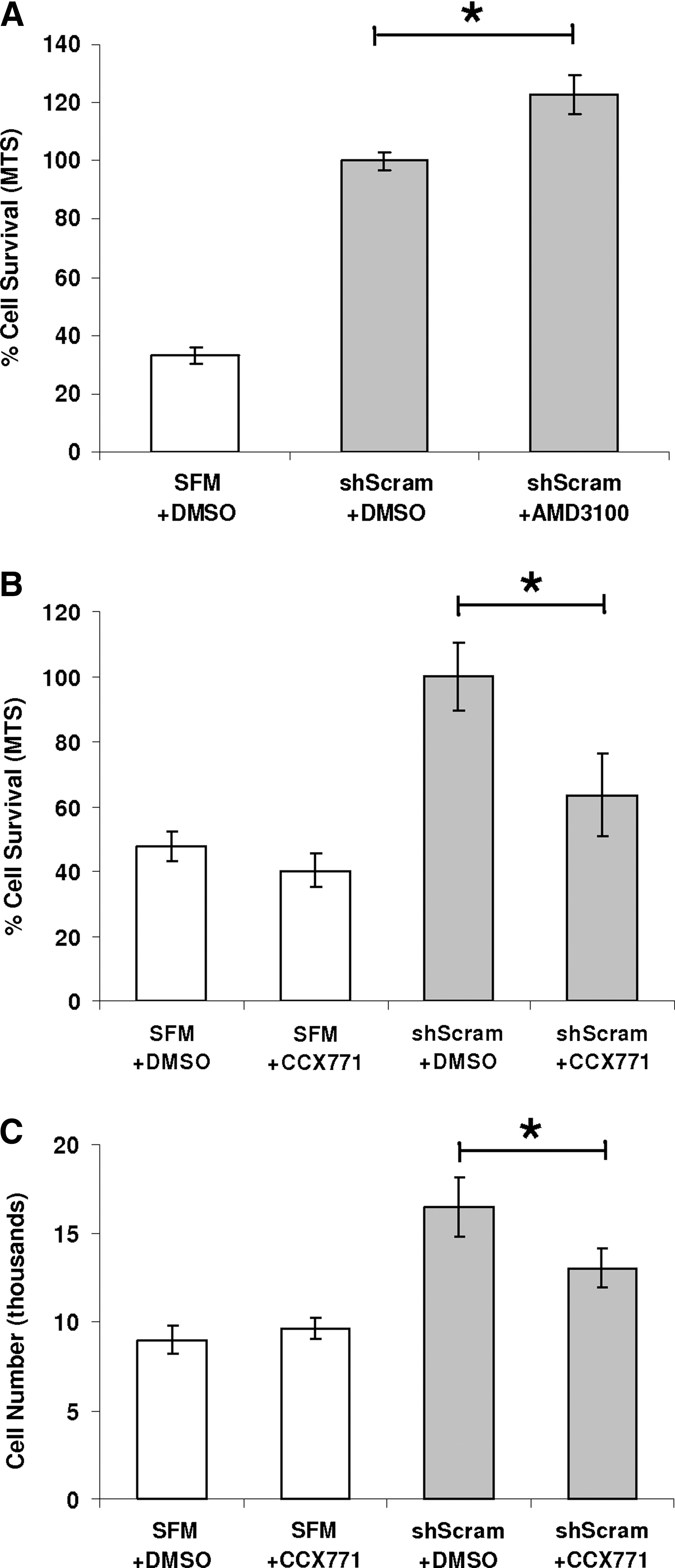
Selective inhibition of CXCR7 but not CXCR4 diminishes the neuroprotection conferred by shScram CdM.
Discussion
We found that CD133dMSCs significantly increased expression of SDF-1 mRNA when transplanted into brain tissue injured by stroke. To our knowledge, this study is the first to analyze the transcriptional behavior of hMSCs within the stroke environment. CD133dMSCs increased SDF-1 mRNA levels ∼80-fold in ischemic versus control brains after stroke. In contrast, cultured CD133dMSC SDF-1 transcript levels increased only 2.15-fold after growth factor withdrawal and an equal amount of time in hypoxia (1% O2), indicating that other variables in vivo affect SDF-1 expression by CD133dMSCs. Importantly, the inflammation and/or injury caused by the needle during cell injection into the brain occurred in both sham-operated mice and those with stroke. Therefore, the changes in SDF-1 gene expression we detected were specific to the environment of cerebral ischemia.
Through cell protection assays in culture we showed that SDF-1 is one of the neuroprotective factors in CD133 CdM. SDF-1/CXCL12 is a chemokine with attractant effects on cells that harbor the fusin/CXCR4 receptor, notably hematopoietic cells and uncommitted neural cells. The SDF-1/CXCR4 axis has been shown to induce hematopoietic cell mobilization and migration [17], as well as adhesion [18], engraftment [19], trans-endothelial migration [20], retention in the bone marrow [21], and the modulation of hematopoiesis [22]. Since many leukocytes commonly possess CXCR4, SDF-1 also displays proinflammatory properties, mobilizing cells of the innate immune response to the ischemic brain [23]. The presence of CXCR4 on embryonic and fetal neural cells is critical for the anatomical development of the mammalian central nervous system [24]. After cerebral ischemia in adults, CXCR4 has been shown to mediate NSC/NPC migration to SDF-1-rich sites in the brain [25 –27].
SDF-1 signaling has antiapoptotic effects in stem/progenitor cells from the bone marrow [28], vascular endothelium [29], kidney [30], pancreas [31], and the inner cell mass of embryos [32]. SDF-1-based survival signaling was recently shown for primary cortical neurons [27], and its ability to protect primary NPCs from hypoxic injury was demonstrated here. CXCR7 is a second and recently deorphanized SDF-1 receptor that appears to mediate antiapoptotic SDF-1 signaling [30].
We report here that CXCR7 and not CXCR4 promoted the survival of mNPCs. Further, instead of reducing mNPC survival during hypoxia exposure, CXCR4 antagonism (AMD3100) increased it. Of note, AMD3100 was recently reported to act as an agonist of CXCR7 [33]. CXCR7 expression in the brain has been shown to increase after stroke [34]. It will be of particular interest to determine whether a selective agonist of CXCR7 can provide neuroprotection after stroke.
Footnotes
Acknowledgments
We thank Thomas Buttolph, University of Vermont, Burlington, VT, for real-time PCR instrument support. We thank Dr. Thomas J. Schall of ChemoCentryx, Inc., for providing the CXCR7 inhibitor and antibody used in this study. This work was supported by grants from the National Institutes of Health: NHLBI HL085210 (J.L.S.) and NIH/NCRR P20 RR016435 (Parsons R, COBRE PI, J.L.S., PI project 3).
Author Disclosure Statement
No competing financial interests exist.