
Abstract
The LIM homeodomain transcription factor 1b (Lmx1b) is a key factor in the specification of the serotonergic neurotransmitter phenotype. Here, we explored the capacity of Lmx1b to direct differentiation of mouse embryonic stem (mES) cells into serotonergic neurons. mES cells stably expressing human Lmx1b were generated by lentiviral vector infection. Clones expressing Lmx1b at a low level showed increased neurogenesis and elevated production of neurons expressing serotonin, serotonin transporter, tryptophan hydroxylase 2, and transcription factor Pet1, the landmarks of serotonergic differentiation. To explore the role of Lmx1b in the specification of the serotonin neurotransmission phenotype further, a conditional system making use of a floxed inducible vector targeted into the ROSA26 locus and a hormone-dependent Cre recombinase was engineered. This novel strategy was tested with the reporter gene encoding human placental alkaline phosphatase, and demonstrated its capacity to drive transgene expression in nestin+ neural progenitors (NPs) and in Tuj1+ neurons. When it was applied to inducible expression of human Lmx1b, it resulted in elevated expression of serotonergic markers. Treatment of neural precursors with the floor plate signal Sonic hedgehog further enhanced differentiation of Lmx1b-overexpressing NPs into neurons expressing 5-HT, serotonin transporter, tryptophan hydroxylase 2, and Pet1, when compared with Lmx1b-nonexpressing progenitors. Together, our results demonstrate the capacity of Lmx1b to specify a serotonin neurotransmitter phenotype when overexpressed in mES cell-derived NPs.
Introduction
E
Serotonergic (5-HT) neurons reveal a close ontogenetic relationship to midbrain dopaminergic (DA) neurons as both neuronal subtypes derived from ventral neuroepithelial progenitors located on either side of the midbrain–hindbrain boundary, starting at embryonic day 10.5 in the mouse [8 –10]. According to the prevailing model, the development of both 5-HT depends critically on sonic hedgehog (Shh) synthesized by the floor plate, fibroblast growth factor 8 (FGF8) generated by the midbrain–hindbrain organizer, and FGF4 produced by the primitive streak [11,12]. NPs located more rostrally, thus away from the FGF4 signal, differentiate into DA neurons.
Several transcription factors are involved in serotonergic differentiation. Mash1 and Nkx2.2 expressed in NPs of the hindbrain activate the transcription factors Gata2, Gata3, LIM homeodomain transcription factor 1b (Lmx1b), and Pet1 [13], which together define the serotonergic cell type by activating marker genes such as for tryptophan hydroxylase 2 (TPH2), aromatic amino acid decarboxylase, the serotonin transporter (SERT), and the vesicular monoamine transporter 2 [14]. Both Gata2- and Gata3-deficient mice exhibit partial loss of serotonergic neurons [13,15]. In mice lacking Pet1, 70% of serotonergic progenitors fail to differentiate, whereas in the remaining Pet1-deficient neurons diminished expression of TPH and SERT was observed [16]. Lmx1b is crucially involved in the formation of the entire serotonin system in the hindbrain, because its deletion in mice leads to the absence of 5-HT neurons in the brain [17 –19]. It is expressed in developing 5-HT neurons together with Pet1 starting around E11 in the rostral cluster of serotonergic differentiation and 1 day after in the caudal one, consistent with the delayed appearance of serotonergic cells in the latter region [18]. Lmx1b ablation does not affect expression of Nkx2.2, Gata3, and Shh, putting these factors upstream of Lmx1b [17,18]. Together with Nkx2.2, GATA3, and Pet1, Lmx1b can induce ectopically the development of serotonergic neurons in the chick neural tube, a function that Nkx2.2 and Pet1 cannot support on their own [18,20]. Lmx1b is also involved in the maintenance of the DA neurotransmitter phenotype in collaboration with the orphan nuclear receptor Nurr1 [21]. Therefore, Lmx1b plays a central role in the differentiation of 5-HT neurons, and an ancillary role in the differentiation of midbrain DA neurons.
Differentiation of mouse ES (mES) cells into serotonergic neurons is highly efficient. In a pioneer study, sequential exposure of mES cell-derived nestin+ NPs to FGF4 and then to FGF8 and Shh led to a 2.5-fold increase in the number of serotonergic neurons resulting in ∼25% of the neurons being 5-HT positive [22]. Using stromal cell cultures to induce differentiation of mES cells and adding the same combination of factors, Barberi et al. were able to define conditions in which the mouse ES cells were differentiated into 60% 5-HT expressing neurons [3]. Similar results were obtained with hES cells. Under nonoptimized differentiation conditions, <1% of the hES cell-derived neurons obtained are serotonin positive [23,24]. Addition of factors that included Shh, FGF4, FGF8, glial-derived neurotrophic factor, and brain-derived neurotrophic factor increased the number of 5-HT expressing cells to 25% of the β-III-tubulin-positive cell population. In a recent study, 40% of NPs could be converted to serotonergic neurons expressing serotonin, Tph2, and the transcription factors Mash1, Pet1, and Lmx1b [6].
Herein, we asked if overexpression of Lmx1b expression is sufficient to drive differentiation of mES cell-derived NPs to the serotonergic neuron pathway. We developed 2 experimental approaches, the first one based on lentiviral vector-based stable expression and the second one on conditional expression induced by 4′hydroxytamoxifen (4′OHT) in NPs. In both experimental systems, Lmx1b was found to promote serotonergic differentiation, either alone or in cooperation with dorsoventralizing factors.
Materials and Methods
Plasmid construction
To engineer pR4SA-EFS-CreERT2-W, the Cre-ERT2 coding sequence from pCre-ER T2 [25] was subcloned between the EcoRV and BamHI sites of plasmid pR4SA-EFS-GFP-W [26].
To engineer p2K7-HAhLmx1b, the human Lmx1b cDNA from pcDNA3.1hLmx1b (a kind gift from S. Dreyer) was first HA tagged at its N-terminus by polymerase chain reaction (PCR) amplification. It was subsequently subcloned between BamHI and EcoRV sites in plasmid pGAE-CAG-eGFP-WPRE [27]. In a second step, the CAG promoter (CMV early enhancer/chicken β-actin) from pGAE-CAG-eGFP-WPRE was cloned into pDONRP4-P1R (Invitrogen), and the HAhLmx1b cDNA from pGAE-CAG-HAhLmx1b-WPRE was cloned into pDONR221 (Invitrogen), both using Gateway BP clonase enzyme mix (Invitrogen). The resulting 2 entry vectors were then recombined into 2K7neo lentivector (kind gift of D. Suter) using Gateway LR plus clonase enzyme mix (Invitrogen) [28].
To engineer pIGTE2-R26-hPLAP, a 48-mer oligonucleotide containing a PacI site (5′-attttaattaagaagttcctattctctagaaagtataggaacttcgat-3′) and a 54-mer oligonucleotide containing an AscI site (5′-ctagagctagcgaagttcctattcttcaaatagtataggaactt cggcgcgcca-3′) were first subcloned into the SspI and AscI sites of plasmid pIGTE2-hAP [29], respectively. The resulting plasmid was digested with PacI and AscI, and the 3.8 kb fragment was subcloned between PacI and AscI in pRosa26PA [30] to generate pIGTE2-R26-hPLAP. To engineer pIGTE2-R26-hLmx1b, a 1.2 kb NheI/XbaI fragment encompassing human Lmx1b cDNA was prepared from pcDNA3.1hLmx1b (a kind gift from S. Dreyer), and subcloned in pCMV-Ires-pA to generate pCMVhLmx1b. The pCMVhLmx1b plasmid was next digested with NheI, and the resulting 2.9-kb insert was subcloned between both NheI sites into pIGTE2 to generate pIGTE2-hLmx1b. PacI and AscI sites were added as described above for pIGTE2-hAP, and the resulting PacI/AscI fragment was subcloned between PacI and AscI in pRosa26PA to generate pIGTE2-R26-hLmx1b.
mES cell culture and electroporation
mES cells (CGR8) were maintained on 0.1% gelatinized (Sigma) tissue culture dishes in Glasgow's modified Eagle's medium supplemented with 10% fetal calf serum (Biowest), 1,000 U/mL of leukemia inhibitory factor (LIF), 1 mM sodium pyruvate, 2 mM L-glutamine, 100 U/mL penicillin–100μg/mL streptomycin, and 100 μM β-mercaptoethanol (all from Invitrogen). About 5 × 106 CGR8 cells were electroporated at 0.26 kV and 960 μF with 40 μg of pIGTE2-R26-hPLAP or pIGTE2-R26-hLmx1b plasmid linearized with SwaI. Stably transfected cells were selected with 80 μg/mL hygromycine B (Roche Applied Science) for 8 days. Drug-resistant colonies were expanded before analysis and frozen.
Neuronal differentiation
Neural induction and neuronal differentiation were performed by means of coculture of mES cells with the MS5 stromal cell line as previously described [3]. In brief, mES cells were seeded at a density of 10–50 cells/cm2 on the layer of mitomycin-inactivated MS5 cells, and maintained in knockout Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 15% (vol./vol.) knockout serum replacement, 2 mM L-glutamine, and 10 μM β-mercaptoethanol (all from Invitrogen), for 8 days to allow neural induction. Neuroepithelial colonies were harvested with a fire-pulled Pasteur pipette, dissociated by gentle trypsinization, and replated on Matrigel (BD Biosciences)-coated dishes in Neurobasal medium without phenol red, supplemented with 200 mM glutamine, 5 μg/mL bovine fibronectin, N2 and B27 supplement (all from Invitrogen), and 20 ng/mL FGF2 (AbCys). FGF4 (100 ng/mL), FGF8 (100 ng/mL), and SHH (200 ng/mL) (R&D Systems) were added to the culture medium at times indicated. To induce neuronal differentiation, FGF2 was withdrawn from the medium at day 11–12, and replaced with 20 ng/mL brain-derived neurotrophic factor (R&D Systems), 200 μM ascorbic acid (Sigma), and 20 ng/mL neurotrophin 3 (R&D Systems). Final differentiation was allowed for 5–6 days. 4′OHT (Calbiochem) was added to the culture medium for the time and concentration indicated.
Neural differentiation was also induced by the formation of embryoid bodies (EBs) as previously described [5,31,32]. In brief, 6 × 106 ES cells were cultured in suspension in 10 mL of the medium (Glasgow's modified Eagle's medium supplemented with 10% vol./vol. knockout serum replacement, 2 mM L-glutamine, 1 mM sodium pyruvate, and 1 × non essential amino acids (NEAA); all from Invitrogen) in a 10-cm dish. After 8 days, EBs were dissociated and replated at a density of 2.5 × 104 cells/cm2 on poly-
Production of lentiviral vectors and infection
The method for simian immunodeficiency virus (SIV)-based vector production in 293T cells is fully described elsewhere [26,33]. In brief, 293T cells were transfected by the calcium phosphate method with a mixture of DNA containing pGRev plasmid encoding the vesicular stomatitis virus glycoprotein envelope, pSIV3+ plasmid encoding the gag, pol, tat, and rev proteins, and pR4SA-EFS-CreERT2-W. The next day, cells were refed with the fresh medium and further cultured for 48 h. The supernatant was then collected, cleared by centrifugation (3,000 rpm and 15 min), and passed through a 0.8-μM filter to remove cell and debris. For infection, 104 freshly trypsinized mES cells were resuspended into 1 mL of 293T cell supernatant producing the desired virus in the presence of 8 μg/mL sequabrene (Sigma), plated on a 2-cm-diameter dish, and cultured for 48 h before cloning by limiting dilution. The production of lentiviral particles from plasmid p2K7neo-HAhLmx1b was performed as described [28,34].
DNA and RNA extraction, Southern blot, and real-time PCR
For genomic DNA extraction, cells were lysed in 10 mM Tris-HCl (pH 8), 1 mM ethylenediaminetetraacetic acid (pH 8), 100 mM NaCl, and 0.5% sodium dodecyl sulfate. The resulting lysate was treated with 0.2 mg/mL RNase for 1 h at 37°C, followed by proteinase K treatment (0.2 mg/mL, 55°C, overnight). DNA was separated from proteins by phenol–chloroform extraction, precipitated with isopropanol, and resuspended in 1 mM Tris-HCl and 0.1 mM ethylenediaminetetraacetic acid (pH 8). Ten micrograms of EcoRV-digested genomic DNA was separated on a 0.8% agarose gel, transferred to a nylon membrane (Hybond-N+; Amersham), and hybridized with 32P-labeled (Ready-to-Go Labeling Kit; Amersham) probe. RNA was extracted using RNAeasy kit (Qiagen) with on-column DNAse (Qiagen) digestion, and reverse transcription carried out with MuMLV-RT (Promega) according to the manufacturer's recommendations. Quantitative PCR was performed using the LightCycler™ 1.5 system and the LightCycler Fast Start DNA Master SYBR Green I kit (Roche Applied Science) according to the manufacturer's instructions. Reactions were carried out in a total volume of 10 μL, comprising 0.4 μM of each primer, 0.75 μL SYBR Green, 2.5 μL of diluted cDNA, and 2–4 mM MgCl2 according to primers. Amplification and online monitoring was performed using the LightCycler 1.5 system. After 40 amplification cycles, melt-curve analyses were performed to verify that only the desired PCR product had been amplified. PCR efficiency of both the target and reference genes was calculated from the derived slopes of standard curves by LightCycler software (Roche Molecular Biochemicals; LightCycler Software, version 3.0). These PCR efficiency values were used to calculate the relative quantification values for calibrator-normalized target gene expression by LightCycler relative quantification software (version 1.0). All normalizations were carried out with β-actin. Semiquantitative PCR was performed using Euroblue Taq polymerase according to supplier (Eurobio) instructions. Sequences of primers are given in Supplementary Table S1 (available online at
Histochemical detection and immunolabeling
Histochemical analysis for hAP expression in cultured cells and quantification of hAP activity in protein lysates were carried out as described previously [29]. For immunolabeling, cells were washed in phosphate-buffered saline (PBS) and fixed in 4% paraformaldehyde in PBS at room temperature for 30 min. After the rinse with PBS, cells were permeabilized in 0.2% Triton X-100 in PBS for 15 min. Nonspecific binding was blocked with nonimmune normal goat serum (Zymed) for 30 min at room temperature. Primary antibodies were applied in 1% normal goat serum (Invitrogen) in PBS. The following primary antibodies were used: human placental alkaline phosphatase (hPLAP; Sigma), β-III-tubulin (Chemicon and Covance), PSA-NCAM (Chemicon), and 5-HT (Sigma). After incubation at 37°C for 1 h, followed by several rinses in PBS, appropriate fluorochrome-conjugated secondary antibodies were added (Molecular Probes). The cells were incubated at 37°C for 30 min and, after extensive washes in PBS, mounted on glass microscope slides using Vectashield HARD mounting medium (Vector Labs, Inc.) containing DAPI for DNA counterstaining.
Enzyme-linked immunosorbent assay
To evaluate the 5-HT level in postmitotic neuronal culture, cells were lysed in PBS containing 0.1% ascorbic acid (Sigma) by repeated freezing (−80°C) and thawing. The extracts were sonicated and separated from debris by centrifugation (30 min 14,000 rpm). The protein concentration was estimated using Coomassie blue staining. Enzymatic immunoassay for 5-HT was performed with nano-detection kit Serotonin EIA BA 10-0900 (Labor Diagnostika Nord) following manufacturer's instructions. Serotonin concentration measured with the EIA kit was normalized to total protein content.
Results
Neuronal differentiation of mES cells overexpressing Lmx1b
CGR8 mES cells were infected with p2K7-HAhLmx1b, a lentiviral vector expressing human Lmx1b cDNA driven by the ubiquitous CAG promoter. Clones expressing hLmx1b at low (L1, L2, L3, L4, L9, and L10), intermediate (L5, L6, L7, L8, and L12), and high (L11) levels were isolated (Fig. 1A). They were cultured onto MS5 stromal cells for 8 days to form neuroepithelial colonies, which were subsequently dissociated and replated on Matrigel-coated dishes in the presence of FGF2 to amplify a morphologically homogenous population of NPs, which is uniformly immunoreactive for NP-specific antigens such as nestin and PSA–NCAM [3] (hereafter called MS5 protocol). NPs were induced to differentiate into Tuj1+ neurons by withdrawal of FGF2 for 4–6 days (data not shown). We observed that hLmx1b expression dramatically influenced both the yield of neuroepithelial colonies at day 8 and the yield of NPs after colony dissociation and replating at day 12. More specifically, clone L11 (high expressor) produced significantly smaller neuroepithelial colonies in comparison either with control mES cells (56% reduction in size) or with low expressor L1 (43% reduction) (Fig. 1B, C). Of note, the number of neuroepithelial colonies did not vary significantly between clones, indicating that hLmx1b expression did not interfere with clonigenicity and survival of dissociated mES cells in these culture conditions (data not shown). We thus hypothesized that hLmx1b overexpression altered proliferation and/or survival of transient amplifying NPs. This hypothesis was confirmed after counting NPs upon dissociation of neuroepithelial colonies at day 8, and subsequent culture for 4 days. Clone L11 (high expressor) exhibited a 50% reduction in the yield of nestin+ NPs at day 12 compared with wild-type ES cells (Fig. 1D). Moreover, after withdrawal of FGF2, most L11-derived NPs failed to differentiate into mature neurons and degenerated (data not shown). In sharp contrast, clone L1 (low expressor) produced a 63% increase in nestin+ NP number at day 12 (Fig. 1D) and, at day 15 (ie, 3 days after mitogen withdrawal), a 12-fold increase in pan-neuronal marker Map2ab expression. Comparable results were obtained with the low expressor L2 (Fig. 1E).
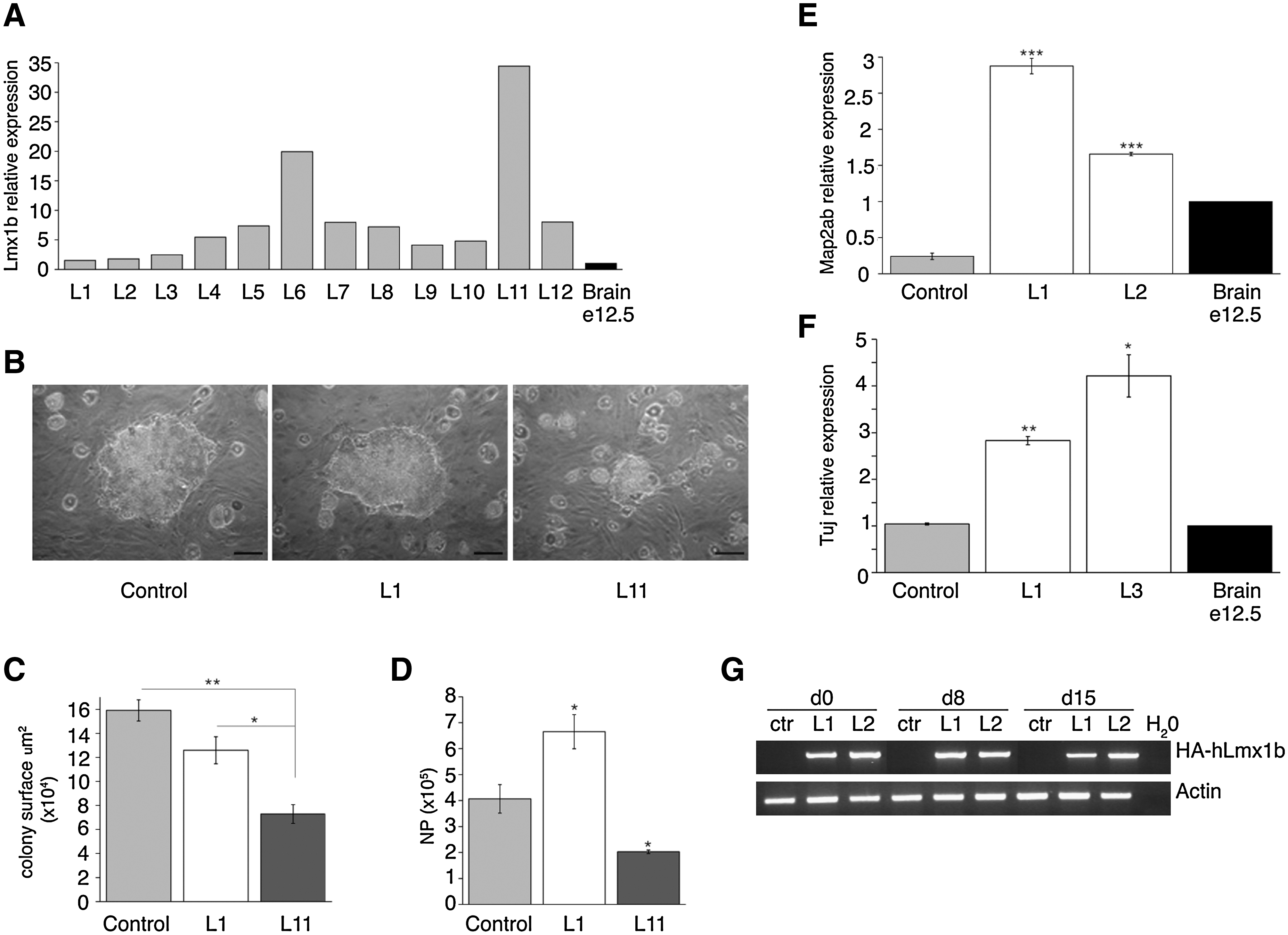
Neuronal differentiation of mouse embryonic stem (mES) cells overexpressing human LIM homeodomain transcription factor 1b (hLmx1b).
Similar data were obtained after neuronal differentiation induced by formation of EBs for 8 days, followed by replating for 2 days in the presence of FGF2, and subsequent mitogen withdrawal for 7 days (EB protocol). Compared with control cells, the low expressors L1 and L3 showed a 2.7- and 4-fold increase in pan-neuronal marker Tuj1 expression, respectively (Fig. 1F). Of note, hLmx1b was expressed at constant levels throughout differentiation (Fig. 1G). Together, these results suggest that moderate overexpression of Lmx1b significantly increases the yield of neural/neuronal differentiation of mES cells.
ES cells overexpressing Lmx1b show preferential differentiation toward serotonergic lineage in vitro
We next wanted to determine if Lmx1b overexpression influences differentiation of NPs into serotonergic, DA, and motoneurons. The low expressors L1 and L3 were used in this study as they both exhibited vastly elevated neuronal differentiation compared to high expressors. Expression of serotonergic-, DA-, and motoneuron-specific markers, after differentiation induced by EB formation, was measured by real-time PCR. Since both L1 and L3 clones showed differences in the yield of neuronal differentiation compared with control cells (see Fig. 1), the expression level of each marker was normalized to Tuj1 (in addition to β-actin) to eliminate the bias resulting from overproduction of neurons by hLmx1b-expressing clones. E12.5 dpc brain extract was used as a reference throughout these experiments. Clones L1 and L3 exhibited increased expression of serotonergic markers, 9- and 3-fold for brain-specific serotonin producing enzyme Tph2, 7.7- and 2.5-fold for serotonin transporter (SERT), and 2.2-fold for serotonergic neuron-specific transcription factor Pet1 (only L1) (Fig. 2A). Of note, a slight increase in DA markers tyrosine hydroxylase (TH) and Lmx1a was also evidenced, whereas expression of Isl1, a marker of motoneuron differentiation, was significantly decreased (Fig. 2B). The enrichment in serotonergic neurons at day 17 of differentiation in Lmx1b-overexpessing clones was confirmed by immunostaining against serotonin (Fig. 2C). Quantification of serotonin level by enzymatic immunoassay showed a 20-fold increase in 5-HT content in both L1 and L3 clones compared with control cells (Fig. 2D). These results indicate that neural precursors overexpressing hLmx1b at a low level show increased propensity for differentiation into 5-HT neurons.
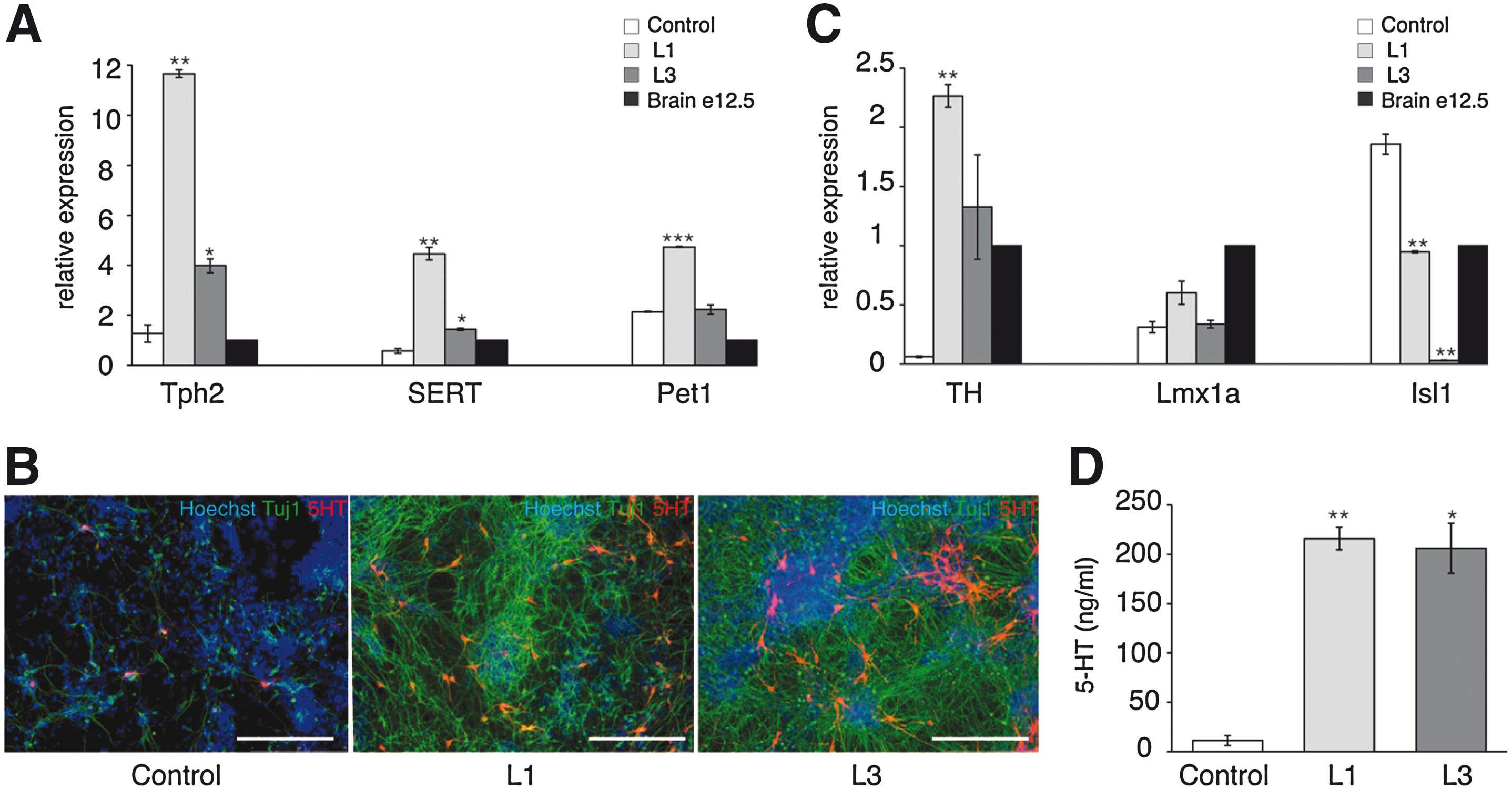
Serotonergic and dopaminergic differentiation of mES cells overexpressing hLmx1b (embryoid body protocol). Real-time PCR analysis of tryptophan hydroxylase 2 (Tph2), serotonin transporter (SERT) and Petl
Generation of an inducible expression vector and ES cells suitable for conditional gene expression in NPs and postmitotic neurons
Since Lmx1b expression appeared detrimental to neural differentiation when overexpressed at high levels in mES cells, we generated an inducible expression system to drive conditional expression of hLmx1b in the NP population. To this aim, we made use of a hormone-dependant Cre-ERT2 recombinase and of an expression vector in which transgene transcription is blocked by 3 floxed transcription termination signals (Fig. 3A). This system displays 2 significant improvements with respect to our previously published system [29]: (1) the Cre-ERT2 recombinase is expressed from a lentiviral vector in which the Cre-ERT2 coding sequence is driven by the ubiquituously active truncated version of the EF1α promoter [26]; (2) the expression vector contains 7 kb of genomic sequences that allow targeted integration into the ROSA26 locus and subsequent transgene expression under the ubiquitously active regulatory elements of ROSA26 [30]. A mES cell line expressing Cre-ERT2 was produced by infection with pR4SA-EFS-CreERT2-W. This line, hereafter called WTC15, expresses CreERT2 at high level in ES cells and in their differentiated derivatives (data not shown). Next, an inducible vector called pIGTE2-R26-hPLAP that harbors hPLAP, the gene encoding hPLAP, was introduced by electroporation into WTC15 cells. Ten individual hygromycin-resistant clones were analyzed, of which 3 displayed homologous recombination into the ROSA26 locus (Fig. 3B). All 3 clones showed a 10- to 14-fold increase in hPLAP activity upon treatment with 4′OHT (100 nM, 48 h) (Fig. 3C). One of them, hereafter called WTC-hPLAP-7, was selected for further analysis. Treatment with 4′OHT resulted in excision of the hygro-polyA cassette (Fig. 3D). Immunofluorescence staining showed hPLAP expression in most cells after treatment with 100 nM 4′OHT for 48 h (Fig. 3E). Virtually, no hPLAP-positive cells could be detected in the untreated cell population, indicating that transgene expression was repressed completely in the absence of hormone.
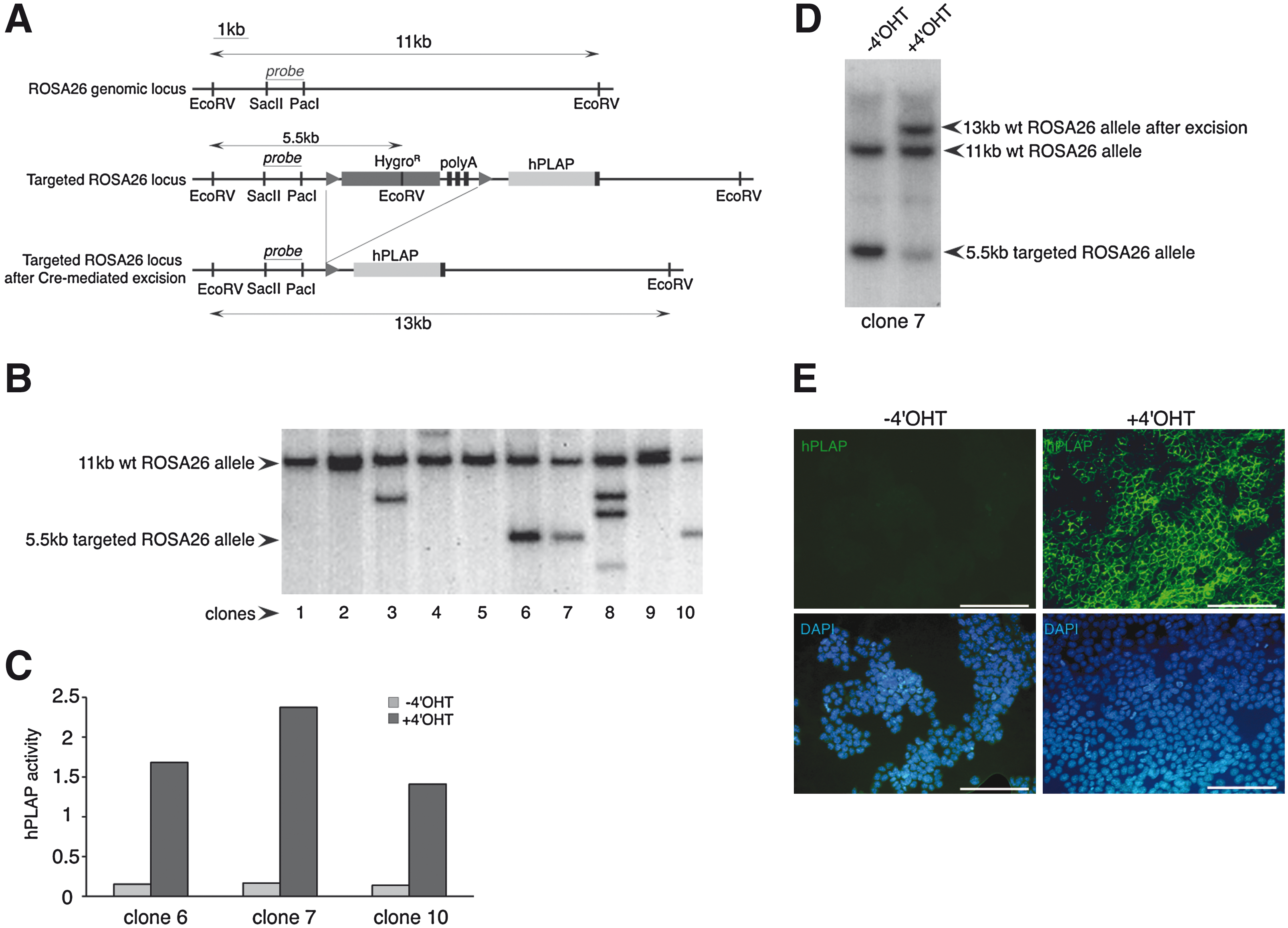
Generation of an inducible expression vector and ES cell line for conditional gene expression.
WTC-hPLAP-7 cells were induced to differentiate into neuroepithelial colonies by means of coculture with MS5 stromal cells for 8 days, followed by amplification of the NP population by FGF2 for 3 days. To assess transgene induction in the neuroepithelial colonies, day 5 cultures were treated with 100 nM 4′OHT for 48 h. Expression of hPLAP was subsequently observed in >95% of the neuroepithelial colonies (Fig. 4A, panels b, c). Day 11 NPs, derived from neuroepithelial colonies in which expression of the transgene was induced at day 5, expressed hPLAP in virtually all cells (Fig. 4A, panels e, f). Most neurons generated at day 17 also expressed it (Fig. 4A, panels h, i). Analysis of hPLAP mRNA level by real-time PCR showed a 49-fold increase in day 11 NPs and a 9-fold increase in postmitotic neurons after induction with 4′OHT at the ES cell stage (day 0). When induction was performed at the neuroepithelial colony stage (day 5), a 51-fold increase in day 11 NPs and a 22-fold increase in postmitotic neurons were observed (Fig. 4B). Together, these results show that once induced, either in the undifferentiated state or during neural differentiation, hPLAP expression level is maintained throughout neuronal differentiation.

Conditional expression of hAP in neuroepithelial colonies, NPs, and postmitotic neurons.
Inducible expression of Lmx1b in ES-derived NPs stimulates neuronal production
A mES cell line conditionally expressing hLmx1b was engineered by electroporating pIGTE2-R26-hLmx1b plasmid into WTC15 mES cells. After selection, 9 hygromycin-resistant clone in 12 displayed targeted integration into the ROSA26 locus (Fig. 5A, B). Two homologous recombinant clones were tested, showing excision of the transcription termination cassette after 4′OHT treatment (100 nM, 48 h) (Fig. 5C). Clone 6, hereafter called WTC-hLmx1b-6, was chosen for all subsequent experiments. Real-time PCR analysis of hLmx1b expression in WTC-hLmx1b-6 showed a 55-fold increase in mRNA level upon 4′OHT induction at the ES cell stage (day 0), and a 9- and 14-fold increase in postmitotic neurons after induction at day 0 and at the neuroepithelial colony stage (day 5), respectively (Fig. 5D). Of note, hLmx1b expression levels in neurons produced from WTC-hLmx1b-6 ES cells after induction at day 5 were similar to expression levels measured in hLmx1b-stably expressing clones L1 and L2 (Fig. 5E). We also confirmed that the induction of hLmx1b expression in WTC-hLmx1b-6 ES cell-derived NPs increased Map2ab expression at day 18 (Fig. 5F), as it was observed with hLmx1b-stable clones L1, L2, and L3 (see Fig. 1E, F).
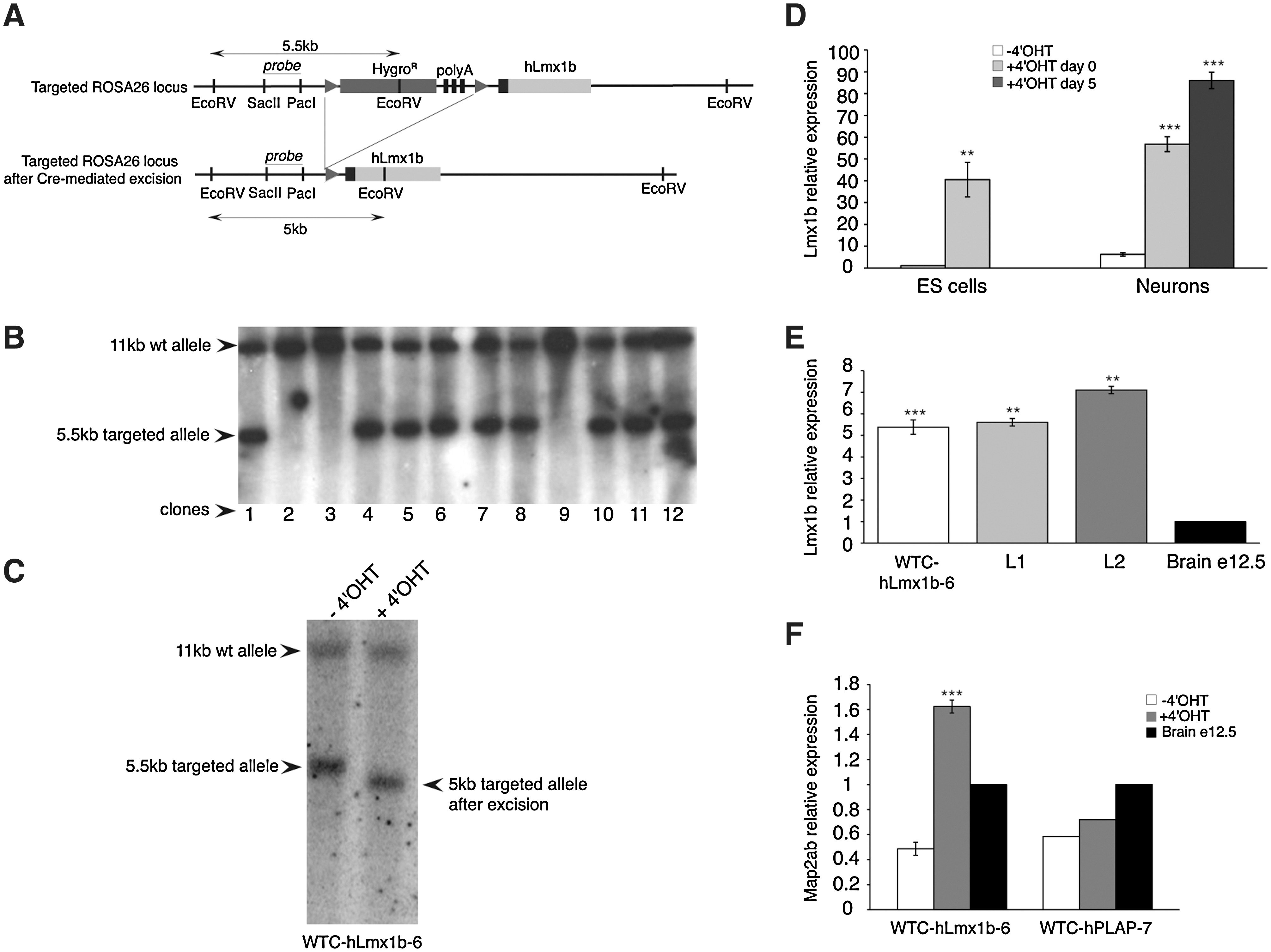
Conditional expression of hLmx1b in neurons.
Inducible expression of Lmx1b cooperates with Shh, FGF4, and FGF8 to induce serotonergic differentiation
We next asked whether the induction of hLmx1b expression in NPs promotes expression of serotonergic markers. Expression of hLmx1b was induced in the neuroepithelial colonies. After amplification in the presence of basic FGF, NPs were induced to differentiate into postmitotic neurons by mitogen withdrawal and the expression of serotonergic markers analyzed by real-time PCR. mRNA levels measured in the WTC-hLmx1b-6-derived neurons were normalized: first, to pan-neuronal Map2ab marker level to eliminate variations in the yield of neuronal differentiation; second, to mRNA levels measured in the WTC-hPLAP-7-derived neurons to eliminate nonspecific effects of 4′OHT. We then observed a 1.4-, 1.6-, and 2.5-fold increase of Pet1, Tph2, and SERT mRNA levels, respectively (Fig. 6A).
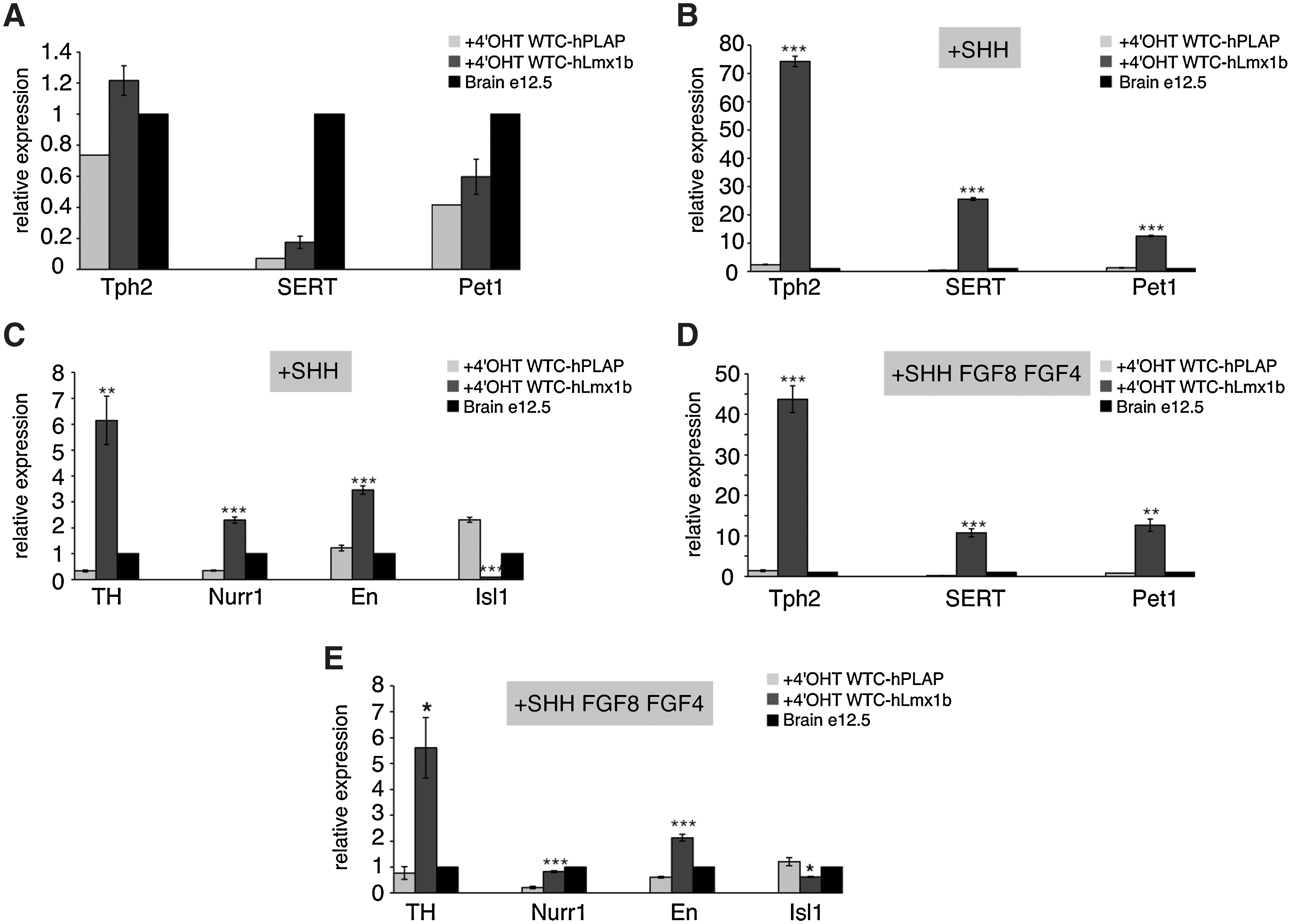
Inducible expression of hLmx1b in ES-derived NPs increases expression of serotonergic markers.
Neuroepithelial colonies and amplifying NPs derived from both WTC-hPLAP-7 and WTC-hLmx1b-6 were exposed, either to Shh alone or to combinations of Shh and FGF4 (day 5–8) followed by Shh and FGF8 (day 8–12). This latter condition was described to promote serotonergic differentiation [3,6]. Expression of hLmx1b and hPLAP transgenes was induced by 4′OHT in the developing neuroepithelial colonies at day 5. In the presence of Shh, induction of hLmx1b expression—when it was compared to induction of hPLAP—resulted in a 10-, 30-, and 55-fold increase in Pet1, Tph2, and SERT expression levels in postmitotic neurons at day 18, respectively (Fig. 6B). Treatment with Shh and FGF4, followed by Shh and FGF8, did not further increase expression of Tph2 (31-fold) and SERT (57-fold), and moderately increased expression of Pet1 (16-fold) (Fig. 6C). Treatment with Shh combined with induced expression of hLmx1b also increased expression of DA markers, En (2.8-fold), TH (18.5-fold), and Nurr1 (6.6-fold), when compared with expression levels in Shh-treated control cells (Fig. 6D). Further, addition of FGF4 and FGF8 to the differentiation cocktail reduced expression of TH and Nurr1 by 2-fold in comparison with Shh alone (Fig. 6E), which is likely to reflect the capacity of FGF4 to inhibit DA differentiation [11].
Taken together, these results show that enforced expression of Lmx1b, combined with Shh, robustly stimulates expression of serotonergic markers in the differentiating NPs derived from mES cells.
Discussion
The focus of this study was to demonstrate the role of Lmx1b in promoting differentiation of mES cell-derived NPs into serotonergic neurons. Lmx1b is essential to the development of the entire serotonergic system [17 –19], as well as to the maintenance of the DA neurotransmitter phenotype in the mouse [21]. We thus asked if overexpression of Lmx1b in mES cells is sufficient to specify serotonergic and DA phenotypes.
Forced expression of a transgene that drives differentiation often results in inability to form undifferentiated mES colonies and consequent recovery only of low expressing or deviant cells. We successfully generated mES cell lines that express hLmx1b ectopically, but only the lines expressing it at low level retained the capacity to differentiate into neural precursors harboring the growth characteristics of control cells. In contrast, mES cells expressing Lmx1b at high levels exhibited reduced capacity to form NPs, and those progenitors to differentiate into postmitotic neurons. Failure to differentiate could result either from the detrimental effect of Lmx1b when overexpressed at elevated levels, or from the selection of mutant mES cells that resisted to the differentiation-promoting effect of Lmx1b. To overcome this difficulty, we have engineered an inducible gene expression system that makes possible to overexpress differentiation-promoting genes in mES cell-derived NPs and to maintain their expression in differentiating neurons. Activation of hLmx1b in mES cell-derived NPs and its subsequent expression in postmitotic neurons, as revealed by the hPLAP reporter gene, reflect the expression pattern of Lmx1b in the mouse, starting in NPs of the hindbrain and being maintained in mature 5-HT neurons [17].
In both stably induced and 4′OHT-induced Lmx1b expressing mES cells, we observed that Lmx1b increases expression Tph2, SERT, and Pet1, the landmarks of serotonergic neurotransmitter phenotype. The effect of Lmx1b was observed using 2 different differentiation protocols, the first one based on coculture of ES cells with a stromal feeder cell line [3] and the second one based on EB formation [5], both of them including selection and amplification of nestin+ NPs. These results indicate that Lmx1b expression is rate limiting for specifying the 5-HT phenotype, in accordance with the genetic data in the mouse [17 –19]. The inductive effect of Lmx1b on serotonergic differentiation is strongly enhanced when NPs are treated with the floor plate signal Shh. This result can be explained by the capacity of Shh to activate expression of ancillary factors acting in concert with Lmx1b to promote serotonergic differentiation. Indeed, the specification of serotonergic neurons requires a Shh-regulated cascade of transcription factors to generate 5-HT neurons in vivo. Shh-activated homeodomain proteins Nkx2.2 and Nkx6.1 cooperate to induce closely related zinc-finger transcription factors GATA2 and GATA3. Preceding the induction of 5-HT neurons, GATA2 activates both Lmx1b and Pet1 consistent with the timing of their expression in vivo [17,18,20]. Ultimately, Pet1 contributes to serotonergic differentiation since mice lacking Pet1 shows a loss of 70%–80% of 5-HT neurons in the CNS [16], whereas Lmx1b knockout mice lack all central 5-HT neurons [14,16,17]. In addition, both Lmx1b and Pet1 have been shown to be necessary and sufficient to specify 5-HT transmitter phenotype when overexpressed in the chick ventral spinal cord [18]. Whether Lmx1b and Pet1 act strictly in parallel to specify 5-HT neurotransmitter phenotype [18], or whether Pet1 is also a target gene of Lmx1b [17] is still unclear. During development, expression of Lmx1b precedes that of Pet1. Moreover, Pet1 expression is lost in Lmx1b knockout mice, raising the possibility that the maintenance of Pet1 expression is dependent on Lmx1b [17,18]. Whether this reflects a loss of 5-HT neurons or gene regulation by Lmx1b remained unclear. Interestingly, the study of conditional deletion of Lmx1b at e12.5 showed that initiation of Pet1 expression is independent from Lmx1b [18], but maintenance of its expression requires Lmx1b [19]. Therefore, our observation that Pet1 expression is moderately increased in neurons overexpressing hLmx1b suggests that Pet1 is responsive to Lmx1b induction, and that Lmx1b reinforces the transcriptional activity of Pet1 [17,18].
The development of 5-HT neurons depends critically on FGF8 generated at the midbrain-hindbrain boundary and on FGF4 produced by the primitive streak. In vivo, FGF4 inhibits the development of midbrain DA neurons and promotes the development of 5-HT neurons [11]. In mES cell-derived NPs treated with 4′OHT to activate Lmx1b expression, addition of Shh, FGF4, and FGF8 has no effect on the yield of serotonergic differentiation compared to a sister culture treated with Shh alone. Thus, when Lmx1b is overexpressed, FGF8 and FGF4 become dispensable for serotonergic differentiation. This finding suggests that activation of Lmx1b expression is the main consequence of FGF8 and FGF4 induction in the developing NPs acquiring a 5-HT phenotype.
Our experiments show that overexpression of Lmx1b, either alone or in combination with Shh, has little or no effect on the yield of DA neuron differentiation. This is in accordance with a previous report [35] showing that transduction of mouse and hES cells with Lmx1b does not induce maturation to the midbrain DA neuron phenotype unless Nurr1 is cotransduced in the same cell. Noteworthy, addition of FGF8 and FGF4 slightly reduced expression of midbrain DA neuron markers in accordance with the function of FGF4 in vivo [11].
To summarize, our results underline the need for proper extrinsic signals combined with intrinsic clues when engineering and manipulating ES cells for differentiation purposes. The vectors and cell lines described might be of great value for studying the gene function in serotonergic neuron development and disease pathology and in trying to understand how drugs that affect the serotonergic system alter neurotransmitter release.
Footnotes
Acknowledgments
This work was supported by research grants from the European Union 6th Framework Program (FunGenES, contract No. LSHG-CT-2003-503494), INSERM AVENIR program, Région Rhône-Alpes (Cluster Handicap, Vieillissement, Neuroscience), Fondation Bettencourt-Schueller, European Science Fundation, FuncDyn Program, and Fondation CERA.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.