
Abstract
Destruction of the motor neurons will lead to loss of innervation of the somatic muscle, which has long been considered an illness with no remedy. The only possible treatment is to substitute the injured motor neurons by neurons differentiated from stem cells. It has been recently reported that embryonic stems cells can be induced to differentiate to motor neurons. However, the use of embryonic stem cells has innate problems. The ideal source of motor neurons should be the cells from the patients themselves, which have the potential to be induced to motor neurons. Our previous study demonstrated that mature astrocyte has the potential of being dedifferentiated to neural stem cell. The present study was aimed to investigate if the neural stem cells of astrocytic origin can be induced to motor neurons. The results demonstrated that neural stem cells of astrocytic origin could be induced to differentiate into motor neurons and their progenitor cells with rich harvest. Further, it has been reported that astrocytes can be readily obtained via biopsy from the cerebral cortex of the patient, rendering autologous transplantation possible. In conclusion, matured astrocytes can be induced to motor neurons and be autologously transplanted to patients suffering from motor neuron destruction.
Introduction
T
In this study, we confirmed that astrocytes from day 15–20 postnatal rat spinal cord, after being cultured in a defined culture system, could dedifferentiate and generate neural stem cells. More importantly, we found that the neural stem cells could be further induced by RA and Shh to motor neuron progenitors and motor neurons with high yields.
Materials and Methods
Cell culture
Postnatal (15–20 days) Sprague Dawley rats were used. The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health (NIH Publication No. 85-23, revised 1996), and the protocol was approved by the Animal Research Committee of Shanghai Jiao Tong University. A longitudinal cut was made along the dorsal root entry zone of the spinal cord and the tissue lateral to the cut was dissected out, thereby excluding the ependyma and periependymal zone, where stem cells and radial cell body reside. The spinal cord tissues were pooled into D-Hank's balanced sodium salts without Ca2+ and Mg2+ (Gibco) and the meninges and blood vessels were quickly peeled off. After the spinal cord tissue was mechanically minced and further dissociated in D-Hank's containing 0.125% trypsin (Sigma-Aldrich), 0.05% collagenase (Sigma-Aldrich), 30 U/mL papain (Sigma-Aldrich), and 0.03% dispase (Sigma-Aldrich) for 20 min, the pieces were mechanically dissociated by gentle trituration for 40–60 times through a pipette. The cell suspension was then passed through a sterile nylon gauze (80 μm pore size). These dissociated cells were plated onto 60- or 35-mm dishes coated with poly-
Immunostaining
The cells were fixed in 4% paraformaldehyde for 15 min at room temperature, rinsed with phosphate-buffered saline (PBS), and fixed in methanol for 15 min at −20°C. Nonspecific antibody binding was blocked by incubation with 10% donkey serum in PBS. The cells were then stained overnight at 4°C with the following primary antibodies for different purposes: mouse monoclonal antibodies against Nestin (1: 1,000, MAB353; Chemicon), βIII-tubulin (1:1,000, T8660; Sigma-Aldrich), Map2 (1:1,000, clone HM-2, M4403; Sigma-Aldrich), and O4 (1:80, IgM, MAB345; Chemicon); rabbit antibodies against glial fibrillary acidic protein (GFAP, 1:3,000, G9269; Sigma-Aldrich) and Olig2 (1:500, AB9610; Chemicon); and goat antibodies against HB9 (1:50, sc-22542; Santa Cruz) and choline acetyl transferase (ChAT, 1:200, AB144P; Chemicon). The cells were stained with the corresponding secondary antibodies for 50 min at room temperature: Alexa Fluor 594/488-conjugated donkey anti-mouse (1:800; Molecular Probes), Alexa Fluor 594/488-conjugated donkey anti-goat (1:800; Molecular Probes), and Alexa Fluor 594/488-conjugated donkey anti-rabbit (1:800; Molecular Probes). When necessary, Hoechst 33342 was used at a concentration of 10 μg/mL for 6 min to label the nuclei of cells. The labeled cells were observed under an Olympus confocal microscope (FV1000) or fluorescence microscope (BX-61).
Because of relatively large size of the neurospheres, they had to be sectioned. The neurospheres were collected and fixed in 4% paraformaldehyde overnight, washed with PBS, and put into 25% sucrose overnight. The neurospheres were then cut to 16 μm in a Leica CM3050 cryostat. Staining of the slices followed the procedures described earlier.
Western blot
The protocol of Jin et al. was followed [14]. Cells from the spinal cord of postnatal (15–20 days) SD rats were collected and proteins were extracted by KCl lysis buffer with a complete protease inhibitor cocktail (Roche Applied Science). Thirty micrograms of protein was loaded onto 8% sodium dodecyl sulfate–polyacrylamide gel and resolved by standard sodium dodecyl sulfate–polyacrylamide gel electrophoresis. The proteins were electrophoretically transferred onto PVDF membranes (Roche Applied Science). The membranes were blocked with 1% blocking solution in Tris buffer solution for 60 min and incubated overnight at 4°C with the primary antibodies against GFAP at 1:50,000 dilution and Nestin at 1:500 dilution. Subsequently, the membranes were incubated with POD-labeled secondary antibody (1:12,500; Roche Applied Science). HRP-labeled anti–glyceraldehyde-3-phosphate dehydrogenase at dilution 1:5,000 (Kang Chen) was used as loading control. The signals were revealed by BM Chemiluminescence Western Blotting substrate (Roche Applied Science).
Flow cytometric analysis
Cultured cells were detached with 0.125% trypsin, rinsed, and triturated to single cell suspension in PBS. The cells were then fixed in 2% paraformaldehyde for 15 min on ice, washed, and incubated in PBS–0.1% saponin with 5% donkey serum for 15 min on ice. After washing with PBS–1% FBS, the cells were incubated for 30 min with primary antibodies against rabbit GFAP (1:3,000) and mouse Nestin (1:1,000). Rabbit IgG (Sigma-Aldrich) was used as isotype control. After 2 washes, the cells were incubated for 30 min with Alexa fluor 488 anti-rabbit IgG and R-phycoerythrin–conjugated anti-mouse IgG (Jackson ImmunoResearch, West Grove, PA). The cells were then washed and analyzed using a Becton Dickinson FACSCalibur instrument and CellQuest Pro software. Photomultiplier tube voltage and spectral compensation were established using cells single-stained with Alexa Fluor 488 alone or phycoerythrin alone. Data were collected for at least 10,000 cells per sample.
Transfection
Astrocytes were cultured 2–3 days in MCM on glass-bottomed dishes and then Nestin-eGFP plasmid (pNH163, kind donation of Dr. Naihe Jing [15]) was transfected into the culture by Fugene HD (Roche Applied Science) for 24 h according to the manufacturer's protocol. Two days later, time-lapse images were picked up under a Leica DMI6000B microscope.
Reverse transcript-polymerase chain reaction assay
Total RNA was extracted from motor neuron differentiation cultures using Trizol Reagent (Invitrogen) and then treated with RNase-free DNase (Roche Applied Science). The cDNA was synthesized using the BcaBESTTM RNA PCR Kit (Takara) according to the manufacturer's instructions. PCR was performed in a final volume of 25 μL containing resulting cDNAs. The following primers were used: rat Olig2, 5′-ACGCCAGCCTGGTGTCTAGT-3′, 5′-TCGGGCTCAGTCATCTGCT T-3′, 304 bp; rat Pax6, 5′-CTTGGTGGTGTCTTTGTCA-3′, 5′-GTCCGT TCAGCATCCTTAGTTT-3′, 424 bp; rat Nkx6.1, 5′-GCGGACCAAGTGGAGAAAGA-3′, 5′-CTCCGCTGGATTTGTGCTTT-3′, 191 bp; rat glyceraldehyde-3-phosphate dehydrogenase, 5′-TCCCTCAAG ATTGTCAGCAA-3′, 5′-AGATCCACAACGGATACATT-3′, 308 bp.
Cell counts and statistical analysis
Data for the percentage of Olig2, βIII-tubulin, ChAT, Map2, and HB9/Hoechst cells were collected from a total of 3 independent experiments. Twenty fields were chosen in a uniform random fashion for quantification. Each field was scored for Hoechst-positive nuclei as the total cells. For each independent experiment, an average of >300 cells were selected. Data are presented as means ± standard error of the mean.
Results
Dedifferentiation of astrocytes in vitro
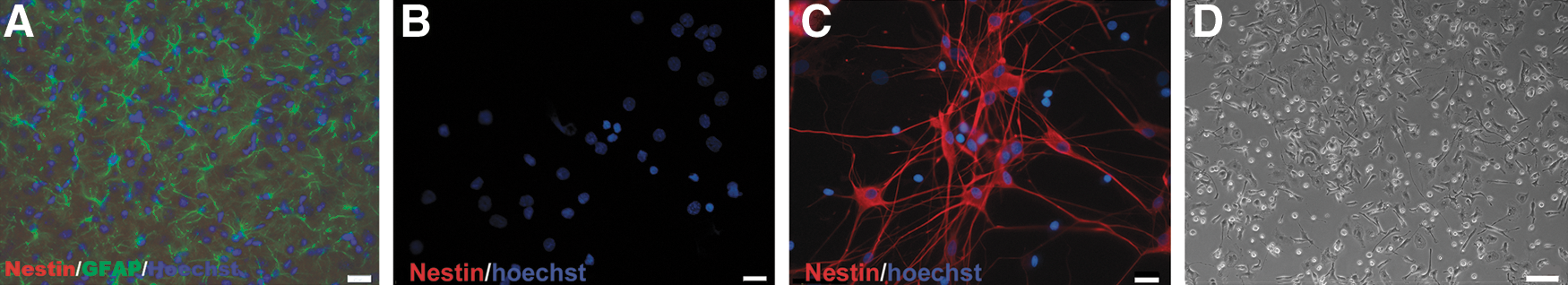
Our previous study showed that culture of primary astrocytes in MCM is optimal for astrocytes proliferation, and also it inhibits or reduces the population of fibroblasts, oligodendrocytes, neurons, and microglia [13]. In this study, the isolated cells were cultured in DMEM +20% FBS for 4 days before they were transferred into MCM. The cells were immunostained with GFAP and Nestin. Figure 1 shows the process of dedifferentiation of astrocytes from purely GFAP-immunoreactive (ir) to purely Nestin-ir with different degree of coexistence in between. At day 1, only a few double-ir cells can be identified (Fig. 1A). They increased in number at days 4 and 7, concomitant with an increase in the intensity of Nestin immunoreactivity (Fig. 1B, C). The number of double labeled cells and the intensity of GFAP immunoreactivity subsided at day 14 (Fig. 1D). The strongly Nestin-ir bipolar cells with long processes but little GFAP immunoreactivity, which first appear at day 4 (Fig. 1B), are indicative of a more advanced stage of dedifferentiation of astrocytes [16,17]. They increased in number at days 7 and 14 (Fig. 1C, D). Western blotting showed that the spinal cord tissue in postnatal (15–20 days) rats did not express Nestin; the culture cells expressed Nestin at day 3, and higher levels were demonstrated at days 7 and 14 (Fig. 1E). Fluorescence activated cell sorting analysis showed that 95.8% ± 0.8% of the cells expressed GFAP, 71.2% ± 3.8% of the cells expressed Nestin, and 69.3% ± 4.0% of the cells coexpressed Nestin and GFAP (Fig. 1F–H). A challenge to the above study is whether there was contamination of stem cells in the starting material. The frozen cross-sections of the lateral part of the spinal cord of a mature rat (15–20 days postnatal) were GFAP and Nestin immunostained. There was no Nestin-ir cell (Fig. 2A). The fresh dissociated cells were immediately plated with a cytospin device onto a glass slide and double stained with Hoechst and anti-Nestin. Again, no Nestin-ir cells could be identified (Fig. 2B, C). Further, the cells were dissociated before transferred into MCM and then cultured in DF-12 medium with 20 ng/mL EGF and 20 ng/mL bFGF. No formation of neurosphere was observed after 14 days (Fig. 2D), which provided additional evidence of the purity of the starting material.
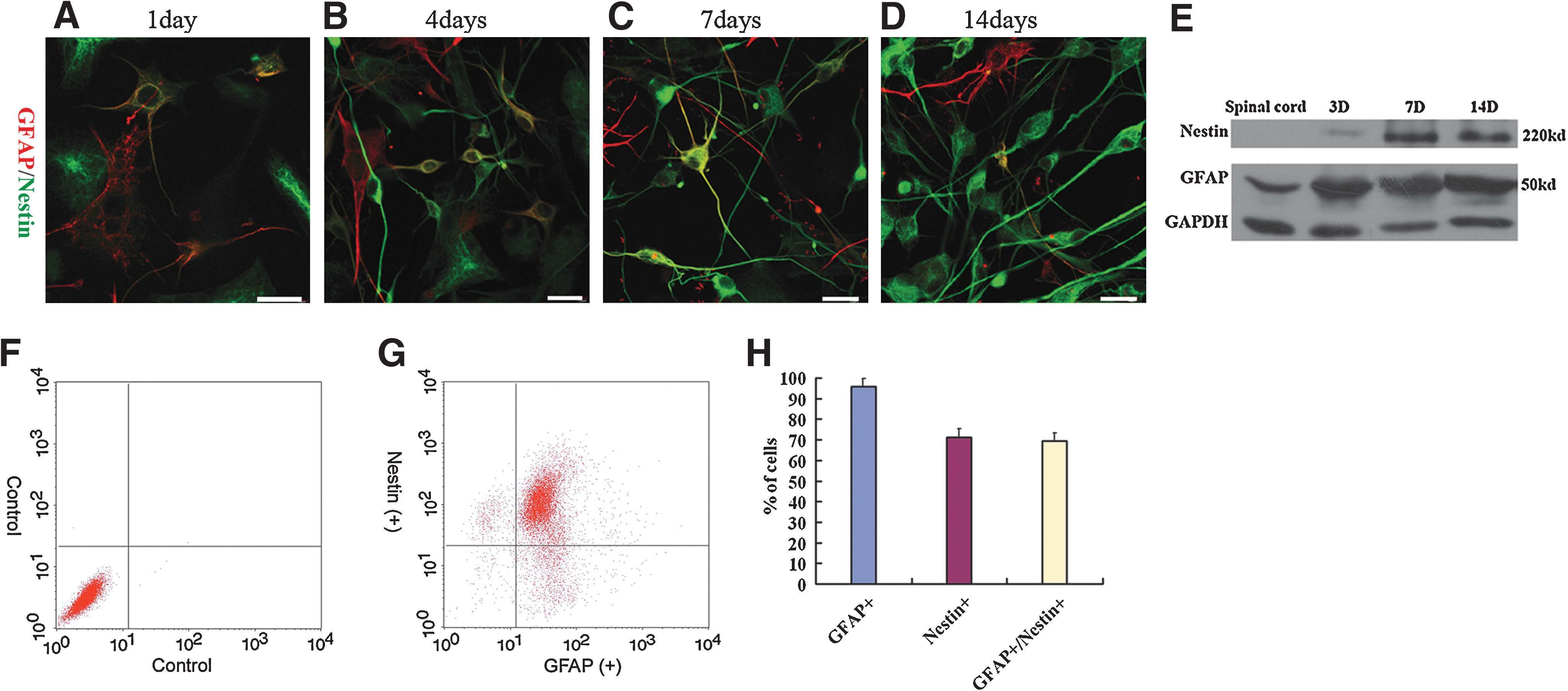
Dedifferentiation of astrocytes in vitro.
To prove the genuineness of Nestin immunoreactivity in the above experiments, the cells were transfected with Nestin-enhanced green fluorescence protein (eGFP) vector, which can specially express eGFP in Nestin-expressing cells [15]. Two days after culture, the eGFP-expressing cells could be identified (Fig. 3A). The number of the eGFP-expressing cells increased successively at days 6 and 15, and most of the cells became bipolar with long and thin processes (Fig. 3B, C). For tracing transformation of the cells, 2 days after transfection, time-lapse microphotos were taken from 0 h onward every 30 min till 25 h and 30 min. Figure 3D–F shows 3 photos highlighting a single cell that gained increasing intensity of eGFP expression and changed from polygonal to bipolar.
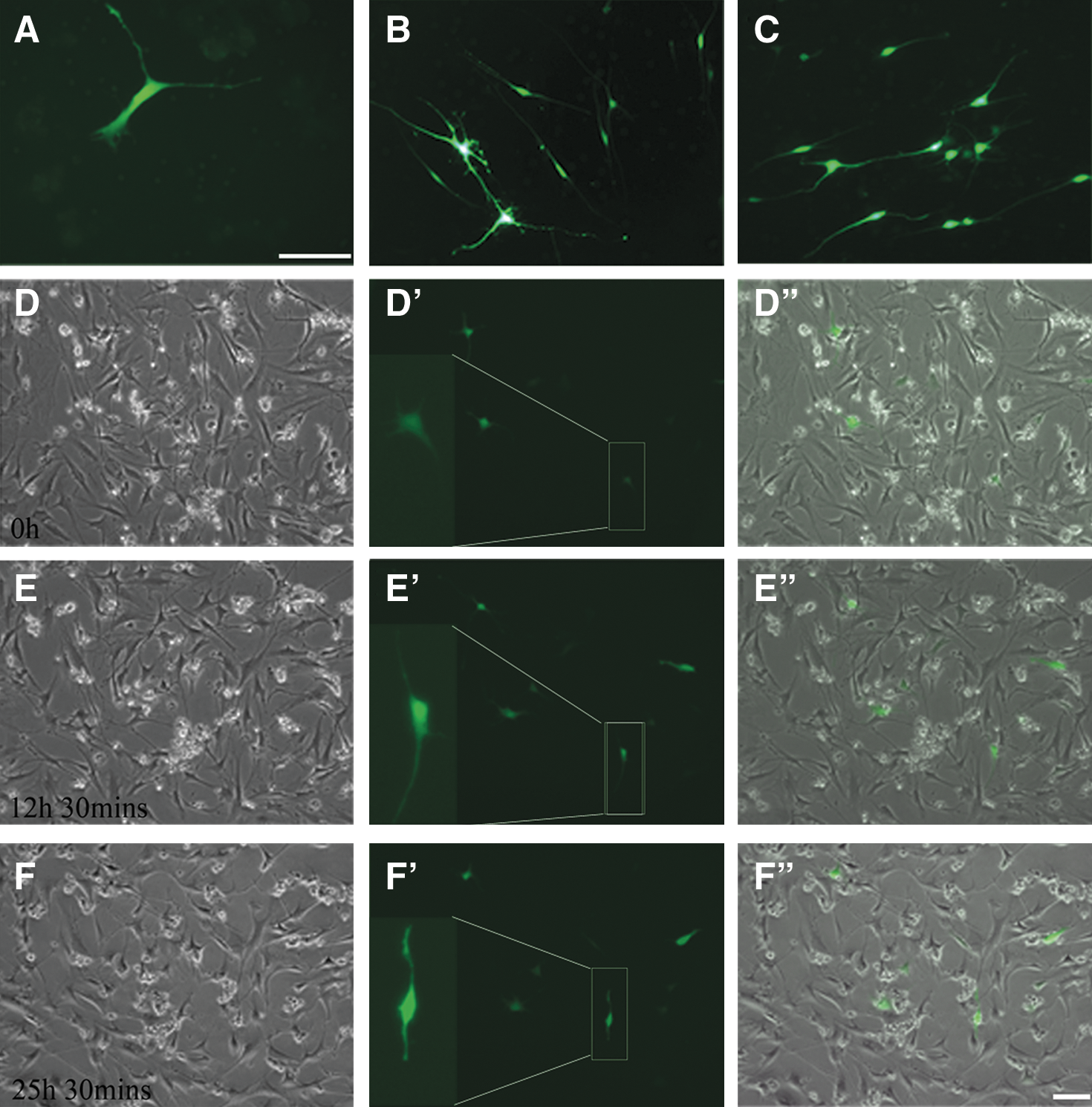
Time-lapse microscopy images showing the transformation of astrocytes transfected with Nestin-eGFP.
Formation and characterization of neurospheres
The cells cultured in MCM for 14–18 days were dissociated and cultured with 20 ng/mL EGF and 20 ng/mL bFGF. Primary neurospheres were found generated at days 9–12. The neurospheres were triturated into single cells, which were then cultured in the same medium. After 5–7 days, secondary (P2) neurospheres were generated and grew robustly (Fig. 4A). Further, the neurospheres were dissociated into single cells and plated onto a 96-well plate at a density of 10 cells/well. The stem cells in each well were checked every day thereafter. None of the stem cells aggregated, and each stem cell formed a small neurosphere by 1 week. After additional 3 weeks, the size of the neurosphere could reach >100 μm in diameter (Fig. 4B). The cells from the neurospheres were successfully passaged once per week to at least 16 passages. After the neurosphere cells were plated at a density of 100 cells/well in 48 wells of the 96-well plate, 22.7 ± 9.9 new neurospheres were formed in each well after 3 weeks, with sizes ranging from 50 to 150 μm. To test the proliferation potential of the neurosphere cells, when 1 × 106 neurosphere cells were plated, (6.6 ± 0.4) × 106 (n = 3) cells were obtained in 7 days. A similar number of cells were generated when neurospheres from passages 2 to 16 were examined. The neurospheres were frozen sectioned for immunostaining. They were full of Nestin-ir cells (Fig. 4C). To examine the potential of differentiation of the neurosphere cells, the P2–P6 neurospheres were triturated into single cells, then seeded onto poly-
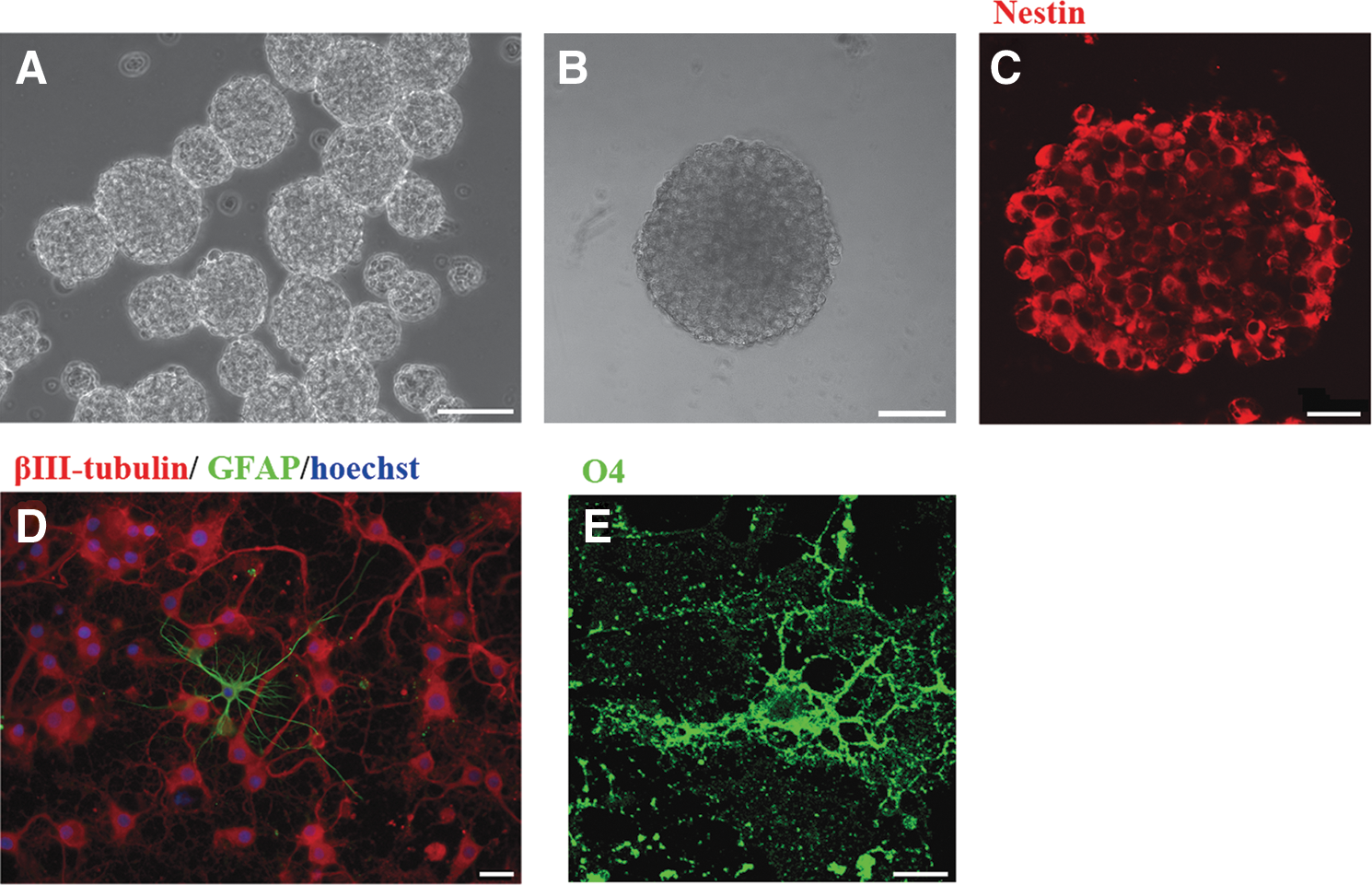
Dedifferentiation of astrocytes to neural stem cells.
Differentiation of the neural stem cells into motor neuron progenitors and motor neurons
It has been reported that spinal motor neurons can be successfully induced from ES cells by RA and Shh [2,3,18]. We followed the method of Wichterle et al. [2] with slight modification as described in the Materials and Methods section. The expression of the basic helix–loop–helix transcription factors Olig2, a specific marker for motor neuron progenitor [19], and βIII-tubulin was examined. After 5 days of induction, as many as 97.7% ± 0.5% of the nuclei of the cells were Olig2-ir, and 97.6% ± 0.5% cells were βIII-tubulin-ir (Fig. 5A, B). The merged figure (Fig. 5C) clearly demonstrated that almost all of the Olig2-ir cells were βIII-tubulin-ir. These data showed that these astrocyte-derived neural stem cells can be highly and efficiently induced into Olig2/βIII-tubulin–coexpressing motor neuron progenitors.
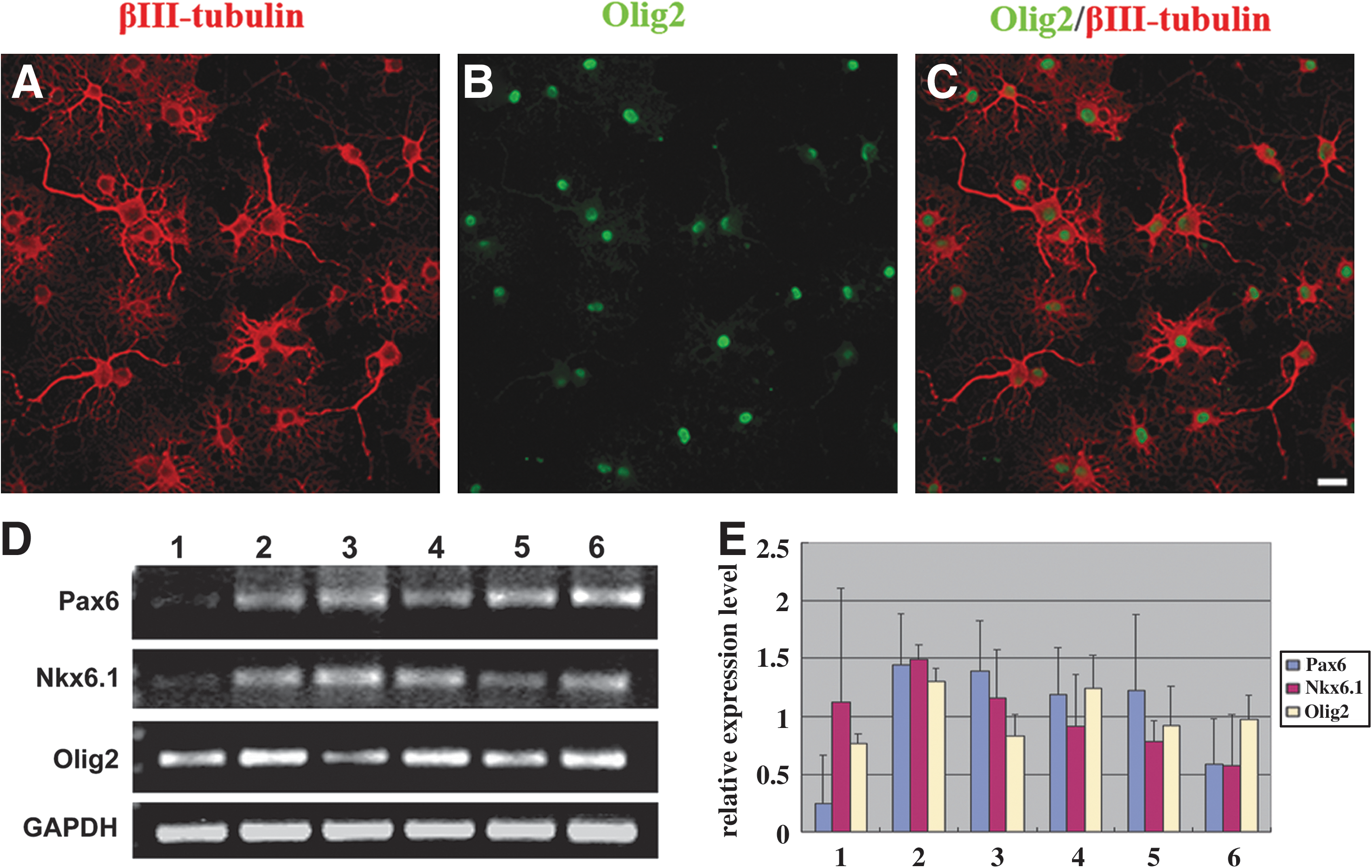
Differentiation of neural stem cells to motor neuron progenitors.
RT-PCR analysis of Olig2 and the homeodomain transcription factors Pax6 and Nkx6.1, which are involved in the specification of motor neurons [20,21], showed that, after treating RA and Shh for 5 days, the Pax6, NKx6.1, and Olig2 expressions were all upregulated, and from 12 to 19 days the expressions decreased successively (Fig. 5D, E). This proves that the neural stem cells are responsive to RA and Shh factors to regulate the Pax6, NKx6.1, and Olig2 expressions, which are essential for motor neuron differentiation.
For differentiation of motor neurons, the neurospheres were first cultured for 5 days in the presence of RA and Shh, and then, for mature differentiation, BDNF, GDNF, CNTF, NT-3, 2% FBS, and Shh reduced to 50 ng/mL were added and cultured for additional 14 days. Most of the cells coexpressed βIII-tubulin and ChAT (Fig. 6A–A”). Immunostaining with a specific motor neuron marker, HB9, which is an homeodomain transcription factor that participates in motor neuron specification and is expressed in postmitotic motor neurons [22,23], demonstrated that all of them coexpressed βIII-tubulin immunoreactivity (Fig. 6B–B”). Figure 6C–C” shows colocalization of ChAT and Map2, a marker for dendrites. Statistical analysis of immunostaining revealed that 93.3% ± 1.7% of the cells were βIII-tubulin-ir, 87.2% ± 1.4% of the cells were ChAT-ir, 27.6% ± 4.6% cells were MAP2-ir, and 24.4% ± 1.1% cells were HB9-ir at 2 weeks (Fig. 6D). The cells without RA and Shh treatment generated significantly fewer HB9-ir motor neurons (4.0% ± 0.4%; P < 0.001; Mann–Whitney test). These data demonstrated that RA and Shh can significantly stimulate the neural stem cells to produce motor neurons. Also, if differentiation time is increased, the yield of motor neuron could be further increased (34.1% ± 2.9% at 4 weeks). Statistical analysis demonstrated that the percentage of ChAT-ir cells is more than that of HB9-ir cells (Fig. 6D), indicating that the ChAT-ir neurons involves populations other than motor neurons.
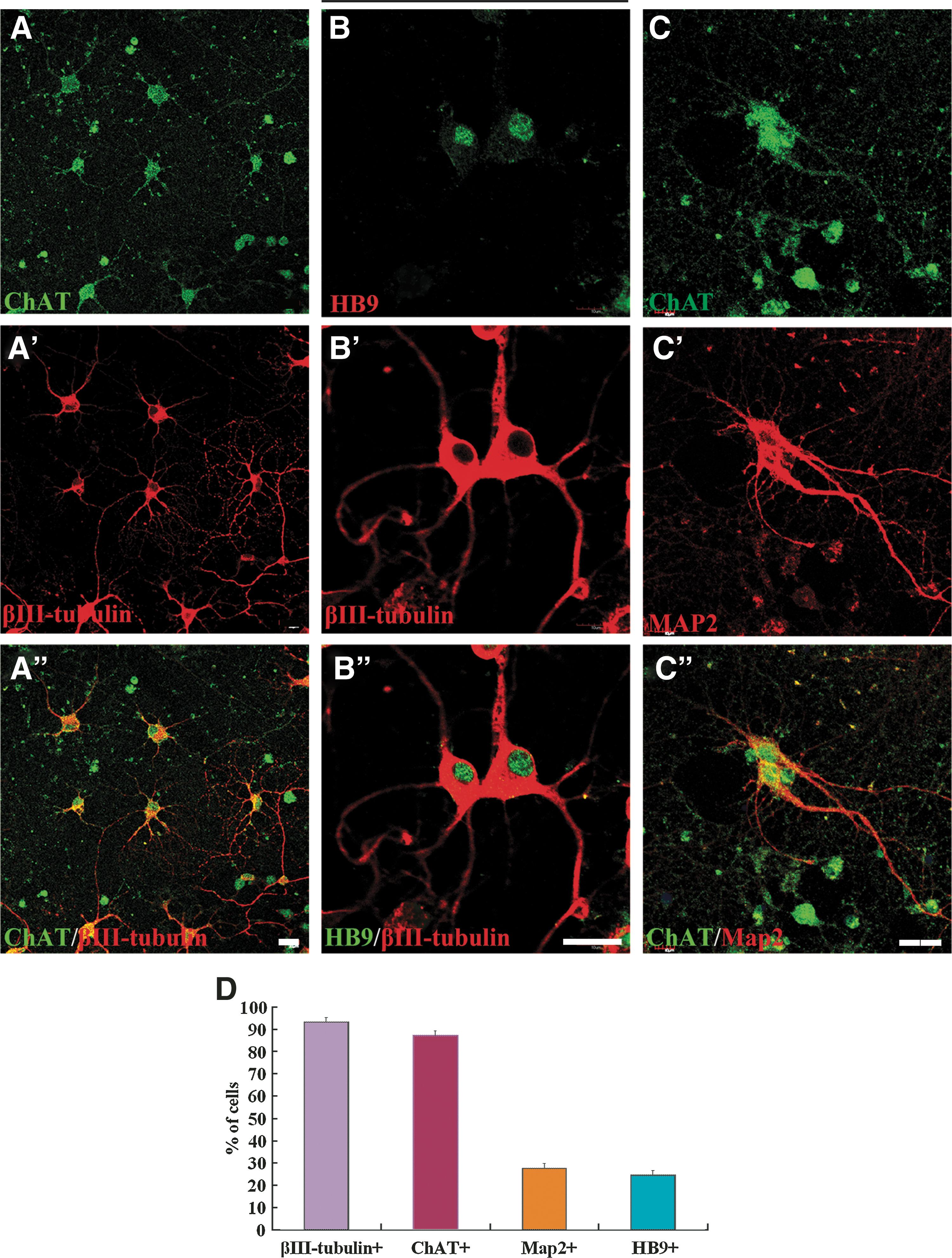
Differentiation of neural stem cells to motor neurons. After being treated with RA and Shh for 5 days, the cells were cultured for additional 14 days with Shh plus brain-derived neurotrophic factor, glial-derived neurotrophic factor, ciliary neurotrophic factor, neurotrophin-3, 2% fetal bovine serum, and 50 ng/mL Shh to promote maturation of motor neurons.
Discussion
The motor neurons are the final common path of the somatic nervous system. Destruction of the motor neurons will lead to loss of innervation of the somatic muscle, which has long been considered an illness with no remedy. The advance of stem cell study ignites a hope to substitute the injured motor neurons by neurons differentiated from stem cells. It has been recently reported that ES cells of mouse [2] or human [8] can be induced to differentiate to motor neurons. However, there are drawbacks in using ES cell and heterologous transplantation. An ideal solution could be to dedifferentiate certain type of mature neural cells from the patients themselves to neural stem cells, which can then be induced to differentiate to motor neurons and transplanted autologously. Our previous study has demonstrated that astrocytes from mature rats have the potential to be dedifferentiated into neural stem cells [11]. The present study further proved that astrocytes for mature rats can be successfully induced to motor neurons and progenitor cells, and astrocytes can be readily obtained via biopsy from the cerebral cortex of the patient [10]. There are different cell sources for autologous transplantation. Dimos et al. demonstrated that induced pluripotent stem (iPS) cells generated from patients with amyotrophic lateral sclerosis could differentiate into motor neurons [6]. Takahashi et al. successfully generated iPS cells from adult human dermal fibroblasts with Oct3/4, Sox2, Klf4, and c-Myc [24]. However, the low efficacy of reprogramming primary human cells and the high risk of tumor formation limit its therapeutic application [25]. Huangfu et al. reprogrammed the primary human fibroblasts with only 2 factors, Oct4 and Sox2, omitting the oncogenes c-Myc or Klf4 to reduce the risk of oncogenesis [25]. Olfactory neuroepithelium may be another source of stem cells to differentiate into motor neurons for autologous transplantation in spinal cord injury. Zhang et al. found that simultaneous transfection of Ngn2 and HB9 cDNA into neurosphere-forming cells from cultures of human adult olfactory neuroepithelium generated motor neurons, when the cells were treated with RA, forskolin, and Shh [5]. However, both iPS and olfactory neuroepithelium cells aforementioned as sources of autologous cells all face the safety problem. Takahashi et al. [24] and Huangfu et al. [25] applied gene engineering with retrovirus infection. Zhang et al. [5] introduced cDNA with plasmid. The foreign genes or proteins introduced and the unpredictable changes that may happen in the genome will arouse hesitation in accepting this therapy. But none of these procedures is needed for generating motor neurons from astrocytes in our studies.
All the so far reported transplantation of stem cell-derived motor neurons [26 –28] actually had transplanted a mixture of cells, including at least progenitor cells. Thus, strictly speaking, the final outcome should not be due to motor neuron alone. It is not known what role the progenitor cells may play when transplanted, good or bad by themselves or their interactions with the motor neurons. Also, can the transplanted progenitor cells differentiate into motor neurons? Can the transplanted motor neuron survive in their new environment? Further studies are needed to clarify these issues. Moreover, the motor neurons are generated with many manipulations in vitro. The possibility of changes in phenotype should not be overlooked and should be screened and evaluated before transplantation into the spinal cord can be applied. In summary, this study confirmed our previous studies that mature astrocytes can dedifferentiate to neural stem cells and, for the first time, demonstrated that they can be induced to differentiate to 97.7% of motor neuron progenitors and at least 30% of motor neurons. It offers a valuable alternative for substitution of the lost motor neurons in spinal cord injuries.
Footnotes
Acknowledgments
This study was supported by the National Basic Research Program of China (2003CB515301). Advices from Drs. S.T. Li, W.L. Jin, and L.Z. Gao are highly appreciated.
Author Disclosure Statement
The authors declare no competing financial or commercial interests.