
Abstract
Pluripotent human embryonic stem cells (hESCs) provide appropriate systems for developmental studies and prospective donor cell sources for regenerative medicine. Identification of surface markers specific to hESCs is a prerequisite for studying hESC biology and can be used to generate clinical-level donor cell preparations that are free from tumorigenic undifferentiated hESCs. We previously reported the generation of monoclonal antibodies that specifically recognize hESC surface antigens using a decoy immunization strategy. In this study, we show that monoclonal antibody 57-C11 recognizes a phosphorylated form of adenovirus early region 1B-associated protein 5 (E1B-AP5). E1B-AP5 is a nuclear RNA-binding protein, but we report that 57-C11-reactive E1B-AP5 is expressed on the surface of undifferentiated hESCs. In undifferentiated hESCs, 57-C11-reactive E1B-AP5 is localized to SSEA3-, SSEA4-, TRA-1-60-, TRA-1-81-, OCT4-, SOX2-, and NANOG-positive hESCs. In mixtures of undifferentiated hESCs and hESC-derived neurons, 57-C11 exclusively recognizes undifferentiated hESCs but not hESC-derived neuronal cells. Further, the expression of 57-C11-reactive E1B-AP5 decreases upon differentiation. Our results demonstrate that 57-C11-reactive E1B-AP5 is a novel surface molecule that is involved in the undifferentiated state of hESCs. As far as we know, this is the first report demonstrating that heterogeneous nuclear RNA-binding protein is expressed on the surface of undifferentiated hESCs.
Introduction
H
Adenovirus early region 1B-associated protein 5 (E1B-AP5) is a member of the heterogeneous nuclear ribonucleoprotein (hnRNP) family. It has significant sequence homology to the hnRNP-U/scaffold attachment factor A and appears to be responsible for selective accumulation of late viral mRNA [13]. E1B-AP5 can bind to mRNA and single-stranded DNA and functions in RNA transport and processing [13]. E1B-AP5 represses basic transcription from several viral and cellular promoters [14]. When associated with BRD7, it activates transcription of a glucocorticoid-responsive promoter in the absence of ligand stimulation [14]. E1B-AP5 also inhibits p53 transcriptional activity [15] and acts as a key component of the ataxia telangiectacia, mutated and Rad 3-related (ATR) signaling pathway during adenovirus infection [16]. Thus, E1B-AP5 is a multifunctional nuclear RNA-binding protein. In addition, recent microarray and proteome studies generated expression profiles and predicted the function of E1B-AP5 in hESCs [17 –21].
We previously used a decoy immunization strategy to generate a panel of monoclonal antibodies (MAbs) specific to undifferentiated hESCs [22]. In this study, we reveal that a phosphorylated form of E1B-AP5 is the target protein for 57-C11, one of the MAbs specific to undifferentiated hESCs. The phosphorylated E1B-AP5 is specifically expressed on the surface of undifferentiated hESCs and downregulated upon differentiation.
Materials and Methods
Cell cultures
H1, H7, and H9 cells were cultured on mouse embryonic fibroblast (MEF) feeder cells as described previously [22]. CHA-hES4 was kindly provided by Dr. Hyung Min Chung (Pocheon CHA University, Korea). HSF6 cells were obtained from the University of California-San Francisco and maintained as previously described [23]. Differentiation of H9 cells was induced by incorporating all-trans-retinoic acid (RA, 10−5 M; Sigma-Aldrich, Seoul, Korea) into the medium before culturing for 14 days. Human embryoid bodies (EBs) were prepared from H9 cell colonies as previously described [24]. H9 cells were also induced to differentiate to neuronal cells by sequential procedures of coculturing on MS5 stromal cell feeders, neural precursor cell (NPC) proliferation, and terminal differentiation of NPCs into neuronal cells, as previously described [25]. Mouse ESCs (mESCs), J1 [26], R1 [American Type Culture Collection (ATCC), Manassas, VA], and E14Tg2a.4 (Mutant Mouse Regional Resource Center, University of California-Davis, Davis, CA), TC-1 (University of Connecticut Health Center, Farmington, CT) were cultured as previously described [22]. The human embryonal carcinoma cell lines NT-2/D1 and NCCIT were cultured according to the instructions provided by the ATCC. Human peripheral blood mononuclear cells were isolated by the Ficoll-Paque Plus method (GE Healthcare, Seoul, Korea). Human dermal fibroblast cells were purchased from MCTT (Seoul, Korea). Cancer cell lines and MRC-5 were obtained from the ATCC and maintained according to the protocol provided by the supplier.
Antibody purification and biotinylation
MAb was purified from the culture supernatant of hybridoma by Protein G-Sepharose column chromatography as previously described [22]. Biotinylation of MAbs was performed according to the supplier's protocol with EZ-Link Sulfo-NHS-LC-Biotin (Pierce Biotechnology, Rockford, IL). Briefly, 1.63 mg of NHS-LC-biotin was dissolved in 1 mL phosphate-buffered saline (PBS, pH 8.0), and 50 μL (l46.4 nmol) of this solution was used to biotinylate 1 mg of MAb. After 1 h incubation at room temperature (RT), the reaction mixture was dialyzed at 4°C against 0.1 M NaHCO3 (pH 8.0) for 48 h.
Flow cytometry
Undifferentiated hESCs, RA-treated hESCs, and mESCs were treated with collagenase IV for 10 min in normal growth medium, treated with cell dissociation buffer (Invitrogen, Carlsbad, CA) for 10 min in a 37°C incubator, and filtered through a 40-μm cell strainer as previously described [24]. The dissociated cells were immediately resuspended at approximately 2 × 105 cells per mL in PBA (1% bovine serum albumin, 0.02% NaN3 in PBS) and incubated with various primary antibodies for 30 min at 4°C. Then, the cells were further incubated with fluorescein isothiocyanate (FITC)-conjugated anti-rat immunoglobulin (Ig) M, anti-mouse IgM, or anti-mouse IgG (BD Biosciences, Seoul, Korea), depending on the isotype of the primary antibody. After washing, propidium iodide (PI)-negative cells were analyzed for the antibody binding using FACSCalibur (BD Biosciences) and Cell Quest software (BD Biosciences). Primary antibodies used were anti-SSEA3, anti-SSEA4 (R&D Systems, Minneapolis, MN), anti-TRA-1-60, anti-TRA-1-81 (Millipore, Billerica, MA), mouse MAb 57-C11, rat MAb 4A11 [27], or rabbit polyclonal anti-E1B-AP5 antibodies (40586 [Novus Biologicals, Littleton, CO]; SC-101976 [Santa Cruz Biotechnology, Santa Cruz, CA]). Multicolor flow cytometric analyses were performed using biotin-conjugated 57-C11 and hESC-specific antibodies as previously described [22]. Briefly, H9 cells were incubated with appropriate primary antibodies for 30 min at 4°C. The primary antibodies used were anti-SSEA3, anti-SSEA4, anti-TRA-1-60, and anti-TRA-1-81. The cells were then further incubated with FITC-conjugated anti-rat IgM, anti-mouse IgG, or anti-mouse IgM (BD Biosciences). After washing with PBA, the cells were incubated with biotin-conjugated 57-C11, followed by streptavidin–phycoerythrin (BD Biosciences).
For intracellular staining, cells were fixed in 2% paraformaldehyde (PFA) in PBS (pH 7.4) for 10 min at RT and then washed with PBA. The cells were then incubated with 57-C11 and rabbit anti-NANOG (Santa Cruz Biotechnology), rabbit anti-OCT4 (Millipore), or rabbit anti-SOX2 (Santa Cruz Biotechnology) diluted in PBA containing 0.5% saponin (Sigma-Aldrich) for 30 min at RT, subsequently incubated with FITC-conjugated rabbit IgG (Vector Laboratories, Burlingame, CA) and PE-Cy5.5-conjugated anti-mouse IgG1 (Invitrogen) in PBA containing 0.5% saponin for 30 min at room temperature (RT), and analyzed on a Becton-Dickinson FACSCalibur.
Cell surface biotinylation, immunoprecipitation, and western blotting
Cell surface biotinylation, immunoprecipitation, and western blotting were performed as previously described [22]. Biotin-labeled H9 or NT-2/D1 cells were treated with lysis buffer (25 mM Tris-HCl, pH 7.5, 250 mM NaCl, 5 mM ethylenediaminetetraacetic acid, 1% Nonidet P-40, 2 μg/mL aprotinin, 100 μg/mL phenylmethylsulfonyl fluoride (PMSF), and 5 μg/mL leupeptin) at 4°C for 20 min. Nuclei were removed by centrifugation, and the cell lysates were stored at −70°C prior to use. To remove the cellular proteins that nonspecifically bind to Protein G plus-Sepharose (Santa Cruz Biotechnology), the cell lysate from approximately 1 × 107 cells was incubated with 20 μL of Protein G plus-Sepharose at 4°C for 2 h, and the beads were then recovered and extensively washed with lysis buffer for use as a negative control for immunoprecipitation experiments. To immunoprecipitate the antigens recognized by antibodies, the precleared lysates were incubated with approximately 1 μg of monoclonal or polyclonal antibodies at 4°C overnight and were further incubated with Protein G plus-Sepharose as described above. The beads were extensively washed with lysis buffer, and the bound proteins were eluted from the beads by heating at 100°C for 5 min. The precleared lysate and eluted proteins were fractionated by sodium dodecyl sulfate (SDS)–polyacrylamide gel electrophoresis on a 10% polyacrylamide gel under denaturing conditions. Proteins were then transferred to a nitrocellulose membrane for western blotting. The membrane was blocked in 5% skim milk in PBST (PBS containing 0.1% Tween 20) at RT for 1 h. After 2 rinses with PBST, the membrane was incubated with streptavidin–horse radish peroxidase (SA-HRP, 1:4,000; GE Healthcare) at RT for 1 h. After extensive washing, the biotinylated proteins were visualized using enhanced chemiluminescence (ECL) detection reagent (GE Healthcare).
For western blot analysis, proteins on polyacrylamide gels were transferred to polyvinylidene fluoride membranes and incubated with anti-E1B-AP5 antibodies followed by HRP-conjugated anti-rabbit Ig (Santa Cruz Biotechnology) for 1 h at RT, or directly incubated with SA-HRP for 1 h at RT. The membranes used were stripped at 50°C for 30 min in stripping buffer (62.5 mM Tris-Cl [pH 6.7], 100 mM 2-mecaptoethanol, 2% SDS). The membranes were then extensively washed with PBST and subjected to western blotting with SA-HRP for 1 h at RT. The immunoblots were visualized using ECL detection reagent as described earlier.
Mass spectrometry
The protein of interest was enzymatically digested in gel in a manner similar to that previously described [28]. Gel pieces were washed with 50% acetonitrile to remove SDS, salt, and stain, dried to remove solvent, rehydrated with trypsin (8–10 ng/μL; Promega, Seoul, Korea), and incubated for 8–10 h at 37°C. The proteolytic reaction was terminated by the addition of 5 μL of 0.5% trifluoroacetic acid. Tryptic peptides were recovered by combining the aqueous phase from several extractions of gel pieces with 50% aqueous acetonitrile. After concentration, the peptide mixture was desalted using C18ZipTips (Millipore), and peptides were eluted in 1–5 μL of acetonitrile. An aliquot of this solution was mixed with an equal volume of a saturated solution of α-cyano-4-hydroxycinnamic acid in 50% aqueous acetonitrile, and 1 μL of the mixture was spotted onto a target plate. Protein analysis was performed using an Ettan MALDI-TOF spectrometer (GE Healthcare). Peptides were evaporated with an N2 laser at 337 nm, using a delayed extraction approach, and were accelerated with a 20 kV injection pulse for time-of-flight analysis. Each spectrum represents the cumulative average of 300 laser shots. The search program ProFound, developed by The Rockefeller University (
Transfection
HEK293T cells were transfected with pHA-E1B-AP5wt [27] or pcDNA3 using ExGen 500 (Fermentas, Burlington, Ontario, Canada) according to the supplier's protocol. E1B-AP5 protein was transiently expressed for 48 h, and cell pellet was subjected to 3 freeze–thaw cycles. The resulting cell lysate was centrifuged and collected, and 20 μg was used for western blotting with 40586 and 700 μg was used for immunoprecipitation with 57-C11.
Calf intestine phosphatase treatment
H9 cells were biotinylated at 4°C for 30 min and extracted with lysis buffer with phosphatase inhibitors (1 mM NaF and 1 mM NaVO3) at 4°C for 30 min. Cell extracts were immunoprecipitated with 57-C11, 40586, or 4A11, and immunoprecipitates were left untreated or incubated with calf intestine alkaline phosphatase (CIP; Roche, Mannheim, Germany) at 37°C for 30 min. Both types of sample were subjected to a second immunoprecipitation with 57-C11 or 40586, and immunoprecipitates were analyzed with SA-HRP or 40586.
Immunocytochemistry
Cell surface antigens were stained as previously described [22]. H9 cells were cultured for 3 days on a glass cover-slip coated with 0.5% gelatin and MEF in a 12-well culture dish and were then fixed in 3.7% PFA for 10 min at RT. To stain cell surface molecules, the cells were directly incubated with 57-C11 and anti-TRA-1-81 at RT for 1 h before fixation and then further incubated with Alexa 660-conjugated anti-mouse IgG and/or FITC-conjugated anti-IgM (Vector Laboratories) at RT in the dark for 1 h. For double immunofluorescence staining for 57-C11 and NANOG, 57-C11 and SOX2, or 57-C11 and TuJ1, H9 cells were incubated with 57-C11 before fixation and permeabilized with 0.1% Triton X-100. The cells were blocked with blocking solution (10% goat serum and 0.1% bovine serum albumin in Ca2+- and Mg2+-PBS) for 30 min and were then incubated with anti-NANOG, anti-SOX2, or TuJ1 (Covance, Princeton, NJ) for 1 h at RT followed by Alexa 660-conjugated anti-mouse IgG, FITC-conjugated anti-mouse IgG, FITC-conjugated anti-rabbit IgG (Vector Laboratories), and/or Cy3-conjugated anti-rabbit IgG at RT in the dark for 1 h, depending on the isotype of the primary antibodies. Between each step, cells were washed with Ca2+- and Mg2+-PBS. Nuclei were stained with 4′,6-diamidino-2-phenylindole. Fluorescence signals were detected with a fluorescence microscope or a Leica TCS SP5 confocal microscope.
Karyotype analysis
H9 cells (passages 68) were incubated with 20 μg/mL colcemid (Invitrogen) at 37°C in a 5% CO2 incubator for 4 h, and the cells were harvested and incubated with 5 mL of 0.075 M KCl at 37°C for 10 min. The cells were then added with 500 μL of Carnoy's fixative (methanol:acetic acid = 3:1) and centrifuged at 1,000 rpm for 10 min. The harvested cells were fixed with 3 mL of Carnoy's fixative for 20 min, and the fixing step was repeated twice. The resultant cells were dropped on a glass slide and baked at 60°C for 30 min. The slide was then treated with 50% H2O2 for 3 min and baked at 60°C for 30 min again. GTG banding was performed by incubating the glass slide in a 0.05% trypsin solution (Invitrogen), followed by rinsing in PBS and staining with a 5% Giemsa stain solution (Sigma-Aldrich) for 5 min. The slide was then rinsed with water and air-dried to detect chromosomal abnormalities. The slide of the chromosomes was examined using ChIPS-Karyo program (GenDix, Seoul, Korea).
Results
Specificity of MAb 57-C11 for pluripotent human stem cells
To study cell surface molecules on pluripotent and undifferentiated hESCs, we previously generated 70 MAbs against hESCs using a modified decoy immunization strategy [22]. Mice were immunized in the right hind footpads with RA-differentiated H9 cells as a decoy immunogen and in the left hind footpads with undifferentiated H9 cells. The left popliteal lymph node cells were fused with myeloma cells, resulting in 70 hybridomas secreting MAbs that bound to the undifferentiated H9 cells, but not to the differentiated H9 cells [22]. One of the MAbs, 57-C11 (IgG1, κ), attracted our most interest because of prominent specificity of the MAb in detecting human pluripotent stem cells. By flow cytometric analysis, 57-C11 bound to hESC lines H1, H9, and CHA-hES4, but not to peripheral blood mononuclear cells, MEFs, mESCs (J1, R1, E14Tg2a.4, and TC-1), MRC-5, and human dermal fibroblasts (Fig. 1 and Table 1). Immunocytochemical analysis showed that 57-C11 binds to hESCs HSF6 and H7 (Supplementary Fig. S1; Supplementary Data are available online at
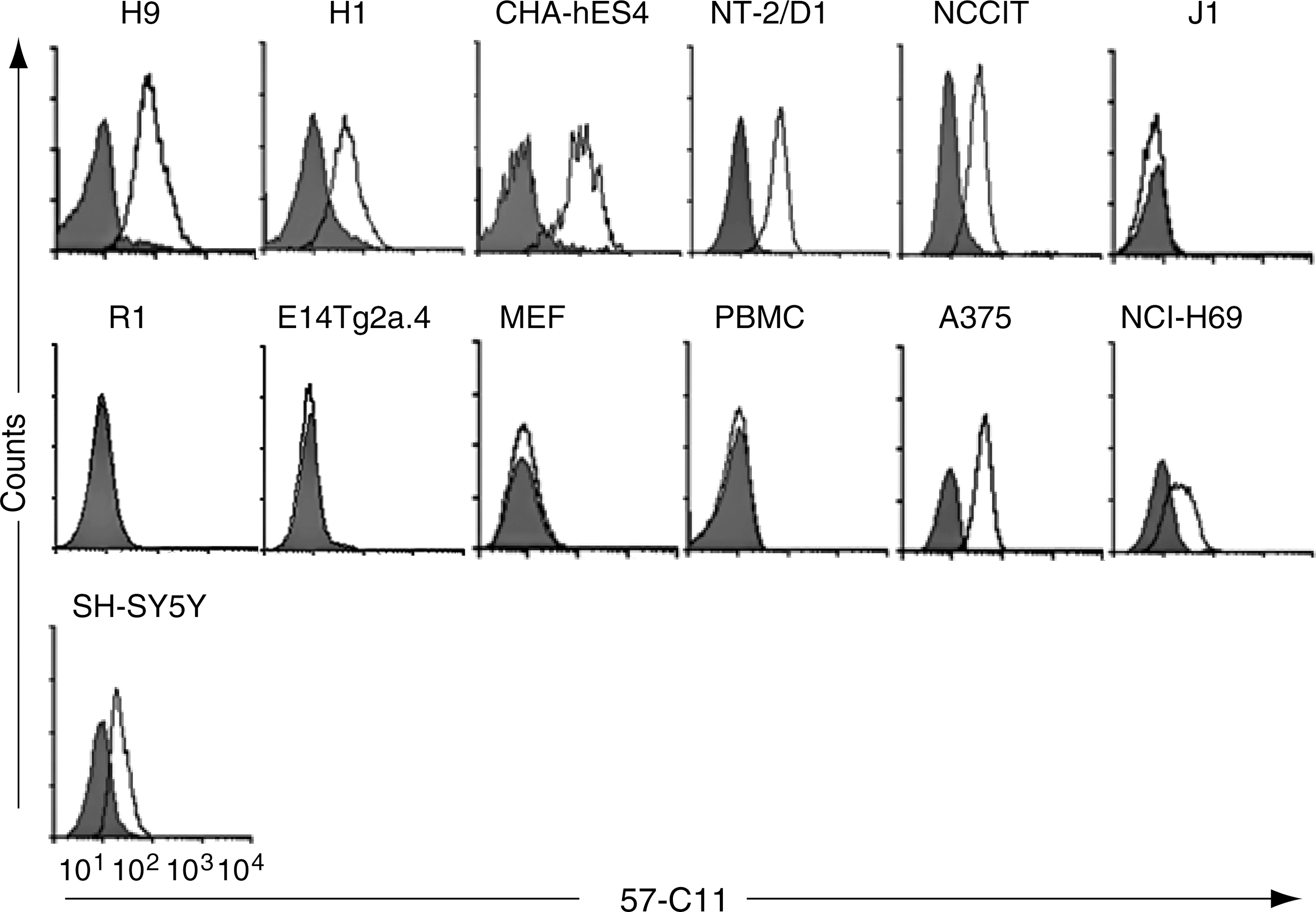
57-C11 antigen expression is examined by flow cytometry in hESCs (H1, H9, and CHA-hES4), malignant pluripotent stem cell lines (NT-2/D1 and NCCIT), mouse ESCs, (J1, R1, E14Tg2a.4), MEFs, PBMCs, and cancer cells A375, NCI-H69, and SH-SY5Y. Cells were stained with 57-C11 followed by FITC-conjugated anti-mouse IgG. Nonviable cells identified with PI were excluded. The unfilled population indicates 57-C11 staining; filled population indicates FITC-conjugated secondary antibody staining as a control. FITC, fluorescein isothiocyanate; hESC, human embryonic stem cell; Ig, immunoglobulin; MEF, mouse embryonic fibroblast; PBMC, peripheral blood mononuclear cell.
hESC, human embryonic stem cell; mESC, mouse ESC; +++, strong binding; ++, medium binding; +, weak binding; −, no binding, *, by immunocytochemistry.
We further examined the 57-C11 specificity with H9 cells using multicolor flow cytometric analysis. Approximately 85%, 96%, 84%, and 81% of 57-C11–positive H9 cells were also positive for the expression of the hESC-specific surface molecules SSEA3, SSEA4, TRA-1-60, and TRA-1-81, respectively (Fig. 2A), indicating that most 57-C11–positive cells also express hESC-specific surface markers. When H9 cells were permeabilized, 57-C11 immunoreactivity was also well localized with the intracellular pluripotency markers NANOG, SOX2, and OCT4. Approximately 100%, 91%, and 100% of 57-C11–positive H9 cells were positive for the expression of NANOG, SOX2, and OCT4, respectively (Fig. 2B). Immunocytochemical analyses further confirmed the colocalization of 57-C11 immunoreactivity with TRA-1-81 on H9 surface (Fig. 3, upper panels), NANOG, and SOX2 (Fig. 3, middle and lower panels).
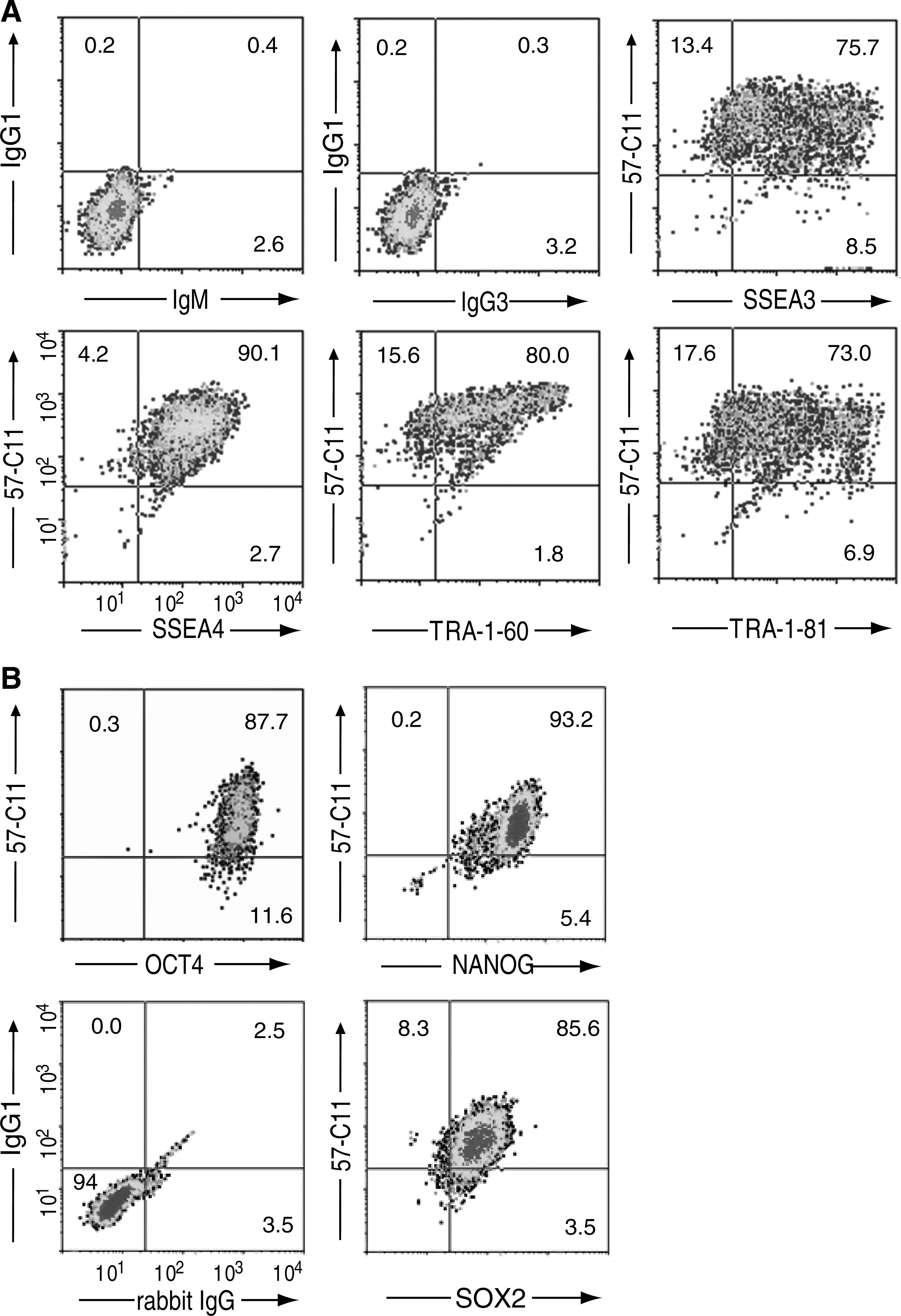
57-C11 antigen is localized to undifferentiated and pluripotent hESCs.

57-C11 antigen is localized to the cell surface of undifferentiated and pluripotent hESCs. 57-C11 antigen and pluripotent stem cell markers were analyzed by immunocytochemistry. H9 cells were incubated with 57-C11 and TRA-1-81, fixed with 3.7% PFA, and incubated with Alexa 660-conjugated anti-mouse IgG and FITC-conjugated anti-mouse IgM. Antibody staining is shown in green or red and nuclear DAPI staining in blue (first row of panels). For double immunofluorescence staining for 57-C11 and NANOG, or 57-C11 and SOX2, H9 cells were incubated with 57-C11 before fixation, permeabilized, and incubated with rabbit anti-NANOG or anti-SOX2 antibodies. The cells were incubated with Alexa 660-conjugated anti-mouse IgG and FITC-conjugated anti-rabbit IgG. Antibody staining is shown in red for 57-C11 and in green for NANOG and SOX2 (second and third rows of panels). Scale bars are 60, 30, and 40 μm in the first, second, and third rows of panels, respectively. DAPI, 4′,6-diamidino-2-phenylindole. Color images available online at
57-C11 exclusively recognizes undifferentiated state of hESCs
To investigate whether 57-C11–mediated detection of hESCs is altered depending on the differentiation stages of these cells, we induced hESC differentiation by 2 different ways—RA treatment and EB formation. Regardless of the differentiation protocols, 57-C11 binding efficiencies to the hESCs, estimated by flow cytometry (Fig. 4A), immunoprecipitation, and western blot analyses (Fig. 4B), were drastically decreased upon cell differentiation. The similar cell differentiation-dependent reduction of antibody binding was also shown with other undifferentiated markers SSEA3, TRA-1-60, and TRA-1-81.
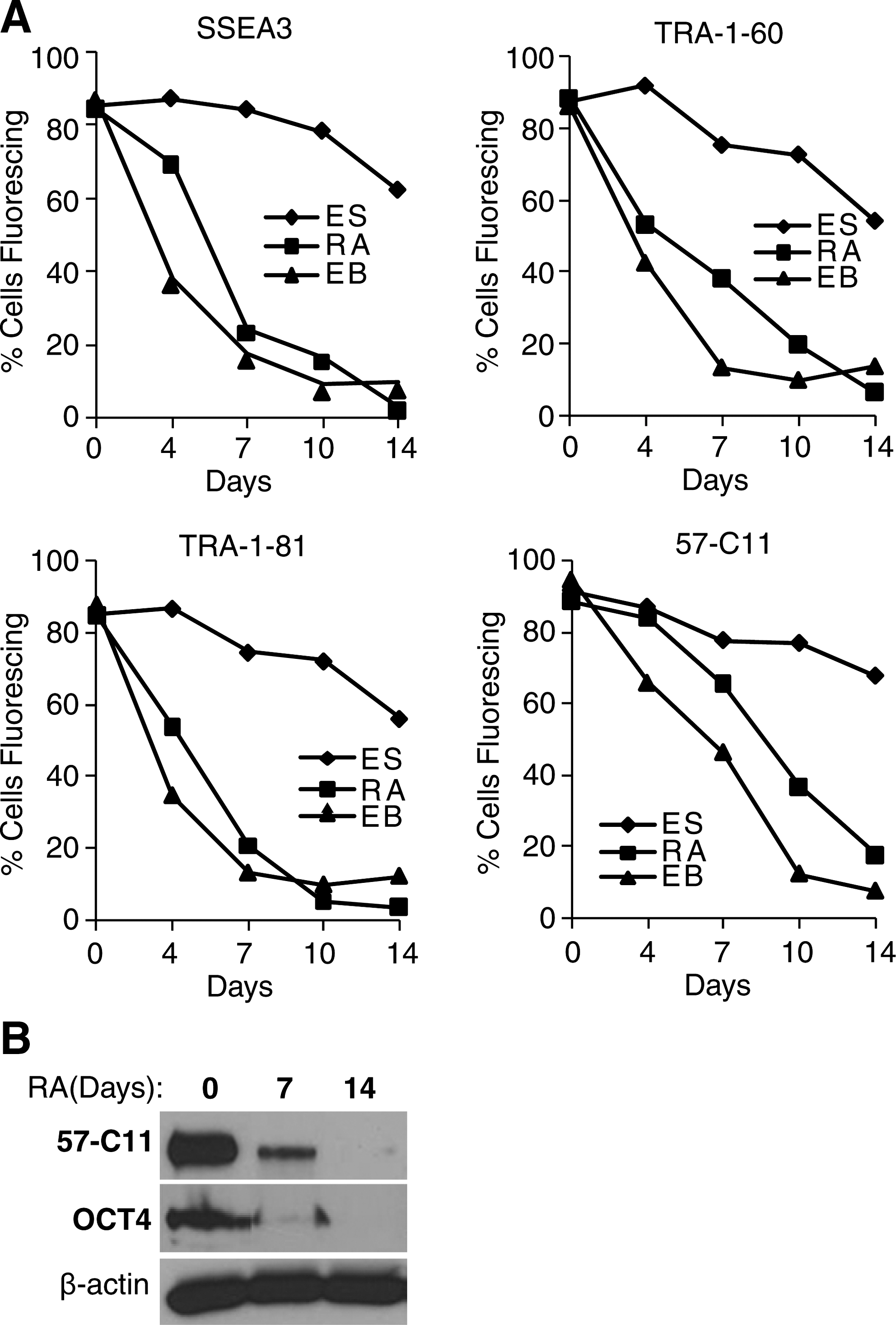
57-C11 antigen is downregulated upon differentiation.
We previously reported an efficient neural differentiation of hESCs [25]. In terminally differentiated cultures, a majority (>60%) of cells are neuronal cells positive for the neuron-specific marker TuJ1. The hESC-derived neuron-enriched cultures could be a prospective cell source for cell therapeutic approaches in neurodegenerative disorders. Cell sorting with hESC-specific surface antibodies is the most practical strategy for obtaining undifferentiated hESC-free donor cells. To examine the potential use of 57-C11 in donor cell preparation, we arbitrarily mixed undifferentiated H9 cells and differentiated neurons from H9 cells and used double immunocytochemistry to examine the specificity of 57-C11 for recognizing undifferentiated hESCs. 57-C11 immunoreactivity was consistently localized at the surfaces of cells positive for undifferentiated hESC markers NANOG and SOX2 (Fig. 5, left and middle panels), but was completely excluded in TuJ1-positive neuronal cells (Fig. 5, right panel), confirming the highly specific and selective detection of undifferentiated hESCs by 57-C11.

57-C11 antigen is exclusively localized to NANOG- and SOX2-positive hESCs but not to TuJ1-positive differentiated hESCs. Undifferentiated H9 cells and neuronal cells differentiated from H9 cells (7:3) were mixed and fixed with 4% PFA, permeabilized with Triton X-100, and incubated with 57-C11 and rabbit anti-NANOG, -SOX2, and -TuJ1 antibodies before incubation with FITC-conjugated anti-mouse IgG and Cy3-conjugated anti-rabbit IgG. Scale bars are 40 μm. Color images available online at
Identification of a 57-C11 target protein
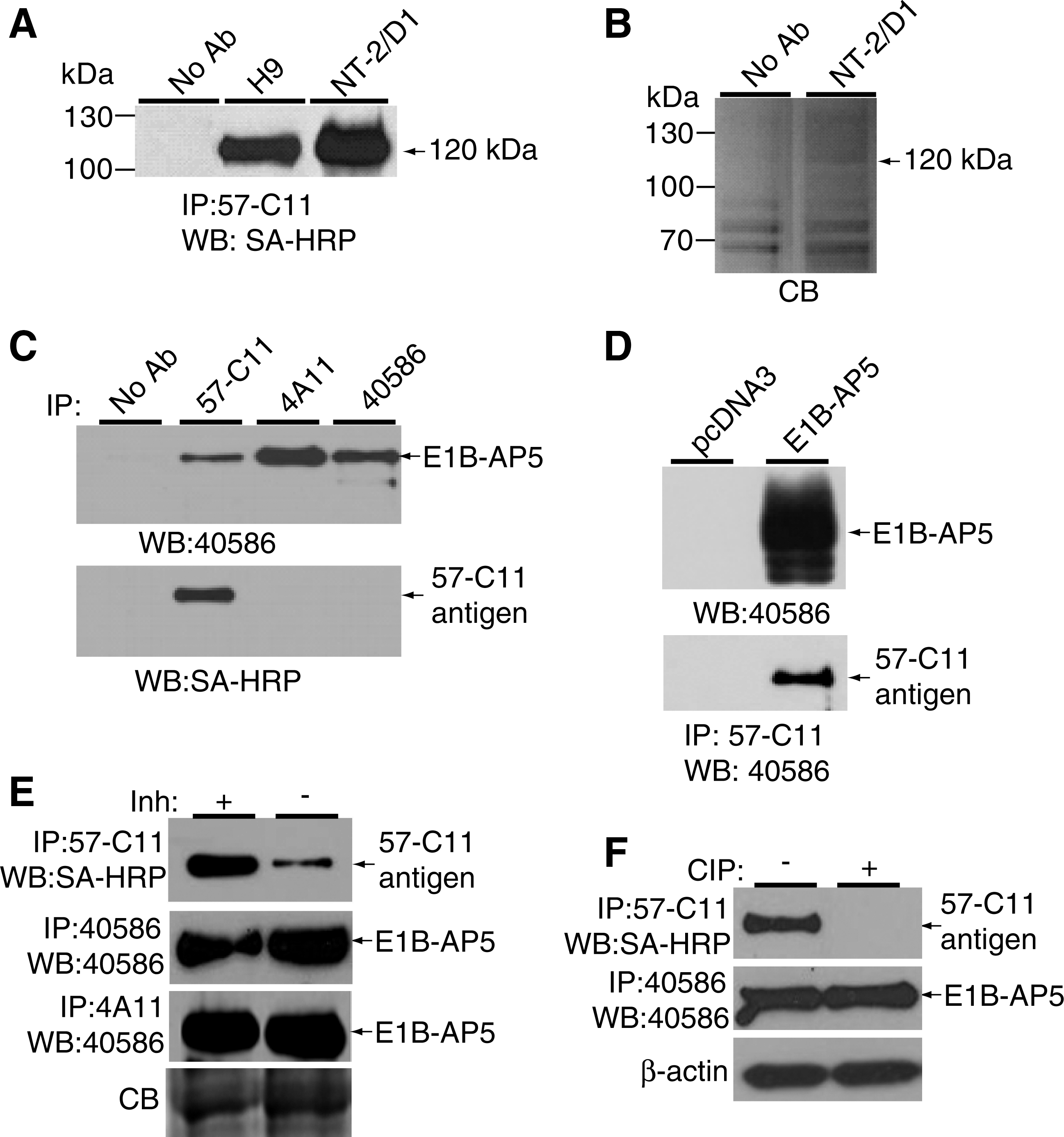
To identify the cell surface antigens recognized by 57-C11, the surface proteins of H9 and NT-2/D1 cells were biotinylated, and cell lysates were immunoprecipitated with 57-C11. Approximately 120 kDa was immunoprecipitated from H9 and NT2/D1 cell lysates (Fig. 6A). The 120-kDa protein on a Coomassie G250-stained gel was removed from the NT-2/D1 lane (Fig. 6B) and subjected to mass spectrometry. From a protein database search with the peptide obtained, the 120-kDa protein was identified as E1B-AP5 (Fig. 7) [13].
57-C11 recognizes phosphorylated E1B-AP5 on the surface of undifferentiated hESCs.
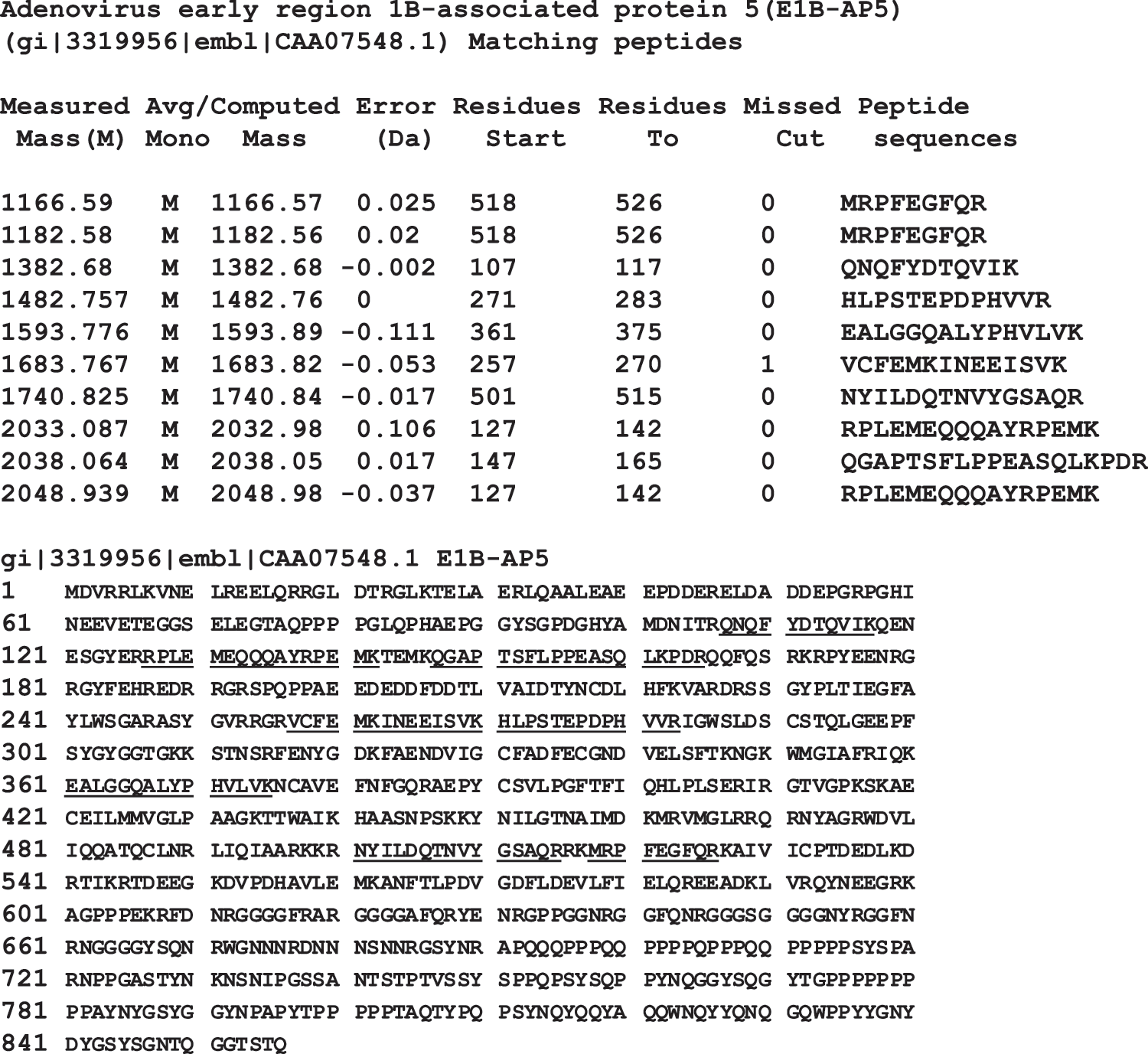
Mass spectrometric identification of the 120-kDa protein after immunoprecipitation with 57-C11. The 120 kDa band in SDS-PAGE (Fig. 6B) was excised and treated with trypsin, and resulting peptides were analyzed by MALDI peptide mass fingerprinting. Peaks were searched against the NCBInr database. Eight tryptic peptides (underlined) originating from NT-2/D1 cells matched E1B-AP5.
To confirm that 57-C11 recognizes E1B-AP5 on the cell surface, H9 cell surface proteins were biotinylated, and cell lysates were immunoprecipitated with 57-C11 and 2 available anti-E1B-AP5 antibodies, 4A11 and 40586. The immunoprecipitates were analyzed by western blot with 40586 or 57-C11. We found that 40586 detected the same 120-kDa protein in lanes 57-C11, 4A11, and 40586 (Fig. 6C, upper panel), indicating that 57-C11 antigen is indeed E1B-AP5. However, the 120-kDa protein was not detected in western blotting with 57-C11 (data not shown), suggesting that 57-C11 recognizes E1B-AP5 in a conformation-dependent manner. To confirm whether the 120-kDa protein is a cell surface protein, we stripped the western blot membrane and attempted to detect the protein using SA-HRP. Only the 57-C11 lane showed the 120-kDa protein again (Fig. 6C, lower panel), indicating that 57-C11 specifically recognizes cell surface-expressed E1B-AP5.
To further demonstrate that 57-C11 recognizes E1B-AP5, the E1B-AP5 expression plasmid pHA-E1B-AP5wt [27] was transfected into HEK293T cells, and cell lysates were subjected to western blotting with 40586, which detected E1B-AP5 protein in transfected cells (Fig. 6D, upper panel). The same lysate was subjected to immunoprecipitation with 57-C11, followed by western blotting with 40586. The 120-kDa protein was detected from cells transfected with pHA-E1B-APwt, but not from cells transfected with the control plasmid pcDNA3 (Fig. 6D, lower panel), confirming that 57-C11 antigen is E1B-AP5. However, cell surface expression of 57-C11 antigen was not detected in the transfected cells by flow cytometry (data not shown), indicating that 57-C11–reactive E1B-AP5 is not expressed on the surface of HEK293T cells.
57-C11 recognizes phosphorylated E1B-AP5 at the cell surface of undifferentiated hESCs
E1B-AP5 is a nuclear RNA-binding protein [13,27]. Therefore, we used other E1B-AP5 antibodies to examine the expression of E1B-AP5 on the undifferentiated H9 surface. Of the antibodies examined, 40586 recognized surface-expressed E1B-AP5 by flow cytometry (Fig. 8A), supporting that E1B-AP5 is really expressed at the cell surface. To compare the subcellular locations of 57-C11–reactive and 40586-reactive E1B-AP5s by immunocytochemistry, H9 cells were permeabilized and incubated with both 57-C11 and 40586. 57-C11–reactive E1B-AP5 was strongly detected at the cell surface and a low level of 57-C11–reactive E1B-AP5 was detected in the cytoplasm and nucleus (Fig. 8B). In contrast, 40586-reactive E1B-AP5 was not clearly detected on the surface (Fig. 8B). Instead, 40586-reactive E1B-AP5 was mainly detected in the nucleus, consistent with previous studies [13,27]. Therefore, we hypothesized that 57-C11–reactive E1B-AP5 is a posttranslationally modified form of E1B-AP5 that is expressed at the cell surface. We found that immunoprecipitation of 57-C11–reactive E1B-AP5 was markedly decreased in the absence of phosphatase inhibitors, whereas immunoprecipitation of 40586- and 4A11-reactive E1B-AP5 was not affected (Fig. 6E). When 57-C11–reactive and 40586-reactive E1B-AP5s were left untreated or treated with CIP, before a second immunoprecipitation with 57-C11 or 40586, 40586 was able to immunoprecipitate E1B-AP5 irrespective of CIP treatment, but 57-C11 was not able to immunoprecipitate CIP-treated E1B-AP5 (Fig. 6F). Taken together, these results indicate that 57-C11 specifically detects the phosphorylated form of E1B-AP5 proteins at the cell surface.
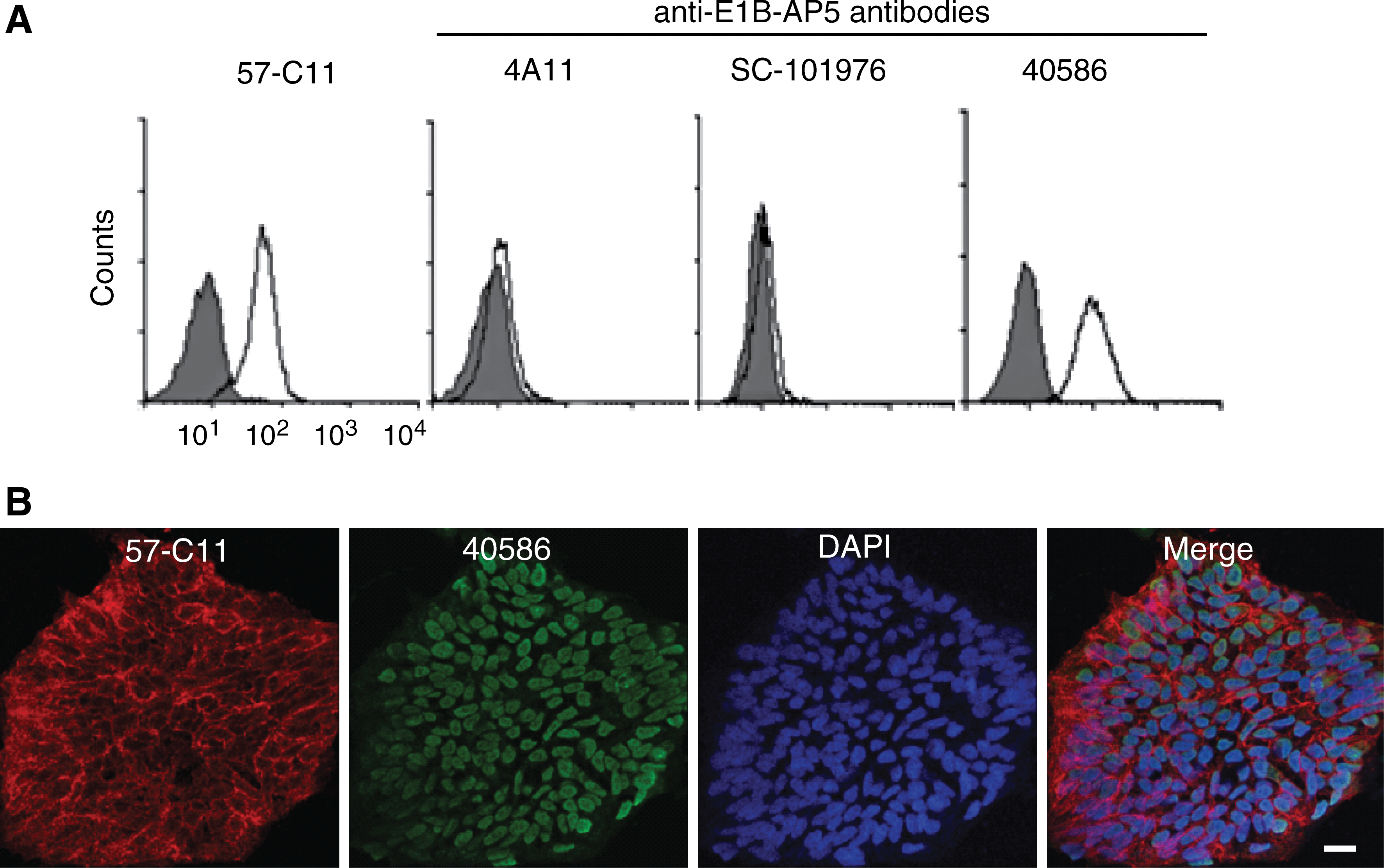
57-C11–reactive E1B-AP5 is expressed at the cell surface.
Discussion
Identification and characterization of cell surface molecules on pluripotent and undifferentiated hESCs are useful for understanding the extrinsic signaling pathways involved in hESC pluripotency and self-renewal. MAbs specific to functional proteins on intact hESCs can identify and isolate functional proteins with posttranslational modifications. Therefore, we and others generated a panel of MAbs against hESCs by simple intraperitoneal injection or a modification of the decoy immunization strategy [22,33 –35]. In the previous study, we generated hybridoma 57-C11, which secretes MAb that binds to undifferentiated but not RA-differentiated H9 cells [22]. In this study, we found that 57-C11 recognizes E1B-AP5 on the surface of undifferentiated hESCs (Figs. 1 –3, 6, and 8). E1B-AP5 is a nuclear RNA-binding protein and is localized in the nucleus and to a lesser extent in the cytoplasm [13,27]. Therefore, we analyzed the subcellular location of E1B-AP5 by flow cytometery and immunocytochemistry with various available E1B-AP5 antibodies. We compared results from several E1B-AP5 antibodies and found that the majority of E1B-AP5 recognized by 57-C11 was located at the cell surface, whereas the majority of E1B-AP5 recognized by other E1B-AP5 antibodies was located in the nucleus (Figs. 3 and 8). Although the recognition of E1B-AP5 by several available E1B-AP5 antibodies was not affected by dephosphorylation, the recognition of surface-expressed E1B-AP5 by 57-C11 was inhibited by alkaline phosphatase treatment (Fig. 6E, F), indicating that 57-C11 recognizes phosphorylated E1B-AP5. Therefore, we report for the first time that the phosphorylated E1B-AP5 is expressed at the cell surface. Interestingly, the surface expression of 57-C11–reactive E1B-AP5 was limited to human pluripotent stem cells, with rare exceptions (Fig. 1 and Table 1). Further, the surface-expressed E1B-AP5 was downregulated upon differentiation (Fig. 4). These results indicate that 57-C11–reactive E1B-AP5 is a novel hESC cell surface marker. This is unexpected, but microarray and proteome studies also support our findings. E1B-AP5 is expressed in all hESC lines examined by microarray [17,18] and is downregulated in EB cells, hepatocyte-like cells, and neuroectoderm-like cells derived from hESCs [19]. Recent membrane proteomic analyses suggest that E1B-AP5 is expressed in the membrane fraction of undifferentiated hESCs [36]. Further, recent phosphoproteomic analyses of hESCs show that phosphorylated E1B-AP5 is detectable in undifferentiated but not differentiated hESCs [20,21], suggesting that the phosphorylated E1B-AP5 may be related to the undifferentiated state of hESCs. Tyrosine 717 and serine 718 are suggested to be the E1B-AP5 phosphorylation sites in undifferentiated hESCs [20], along with serine 512 [21] and serine 194 in HEK293 cells [37]. Regardless of the exact site(s), the phosphorylated E1B-AP5 was localized at the surface of hESCs and downregulated upon differentiation, indicating that phosphorylated E1B-AP5 is a novel surface marker for undifferentiated hESCs.
As far as we know, 57-C11–reactive E1B-AP5 is the first RNA-binding protein ever found on the hESC surface. However, 57-C11–reactive E1B-AP5 is not the first RNA-binding protein to be identified as a cell surface molecule. The Ewing sarcoma (EWS) protein is reported to be expressed on the cell surface [38], and the hnRNP M4 is a cell surface receptor for a carcinoembryonic antigen [39]. Nucleolin also acts as a cell surface receptor, which, by shuttling between the surface and the nucleus, may provide a mechanism for extracellular regulation of cell growth or virus infection [40 –43]. Interestingly, nucleolin, a multifunctional nucleolar RNA-binding protein, is expressed in a phosphorylated form on the cell surface and implicated in tumor cell proliferation and angiogenesis [40,44 –49]. In addition, a recent study shows that nucleolin stability is essential for maintaining self-renewal in mouse ESCs [50], which suggests the functional role of 57-C11–reactive E1B-AP5 in undifferentiated hESCs. As 57-C11–reactive E1B-AP5 is found on the surface of hESCs (Figs. 1 –3), it is conceivable that 57-C11–reactive E1B-AP5, like nucleolin, might act as a cell surface receptor for unknown ligands and participate in the regulation of hESC self-renewal in response to external stimuli.
Our current findings raise questions about how nuclear E1B-AP5 is translocated to the plasma membrane. One possible mechanism that may regulate E1B-AP5 translocation to the cell surface is protein methylation. E1B-AP5 has repeated RGG motifs that are methylated in vivo [27]. Previous work showed that surface expression of EWS, an RNA-binding protein, is accelerated by RGG methylation, and treatment with the methylation inhibitor adenosine dialdehyde (AdOx) results in decreased EWS expression on the cell surface [38,51]. We attempted to examine whether surface-expressed E1B-AP5 is regulated by methylation by treating H9 cells with AdOx, but treatment did not change the surface expression of E1B-AP5 and did not affect the recognition of surface-expressed E1B-AP5 by 57-C11 (data not shown). These results suggest that methylation does not affect E1B-AP5 translocation. How nuclear E1B-AP5 is translocated to the plasma membrane in undifferentiated hESCs is an interesting question for future studies.
Footnotes
Acknowledgments
The authors thank Professor Chang-Hwan Park (Hanyang University, Seoul, Korea) for the H7 analysis and Professor Goo Taeg Oh (Ewha Women's University, Seoul, Korea) for the cell line E14Tg2a.4. This study was supported by the National Research Foundation of Korea (2009-0084052, 2009-0053111, and 2010-0015752).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.