
Abstract
Recently, regenerative medicine with bone marrow stromal cells (BMSCs) has gained significant attention for the treatment of central nervous system diseases. Here, we investigated the activity of BMSCs under simulated microgravity conditions. Mouse BMSCs (mBMSCs) were isolated from C57BL/6 mice and harvested in 1G condition. Subjects were divided into 4 groups: cultured under simulated microgravity and 1G condition in growth medium and neural differentiation medium. After 7 days of culture, the mBMSCs were used for morphological analysis, reverse transcription (RT)–polymerase chain reaction, immunostaining analysis, and grafting. Neural-induced mBMSCs cultured under 1G conditions exhibited neural differentiation, whereas those cultured under simulated microgravity did not. Moreover, under simulated microgravity conditions, mBMSCs could be cultured in an undifferentiated state. Next, we intravenously injected cells into a mouse model of cerebral contusion. Graft mBMSCs cultured under simulated microgravity exhibited greater survival in the damaged region, and the motor function of the grafted mice improved significantly. mBMSCs cultured under simulated microgravity expressed CXCR4 on their cell membrane. Our study indicates that culturing cells under simulated microgravity enhances their survival rate by maintaining an undifferentiated state of cells, making this a potentially attractive method for culturing donor cells to be used in grafting.
Introduction
U
One disadvantage of BMSCs is their scarcity; thus, to increase the number of stem cells within BMSCs to a sufficient population for grafting, the gene transfer method is conventionally used to propagate undifferentiated cells in culture [6,7]. However, because viral vectors used in gene transfer are commonly associated with a risk of activating cancerous genes as observed in induced pluripotent stem cells, it is essential to develop safer methods to produce adequate number of cells for clinical applications. In addition, the possible side effects due to the large amount of cytokines often used while culturing these cells and the potential malignancy of hyperdifferentiated grafted cells raise further concerns regarding this method. Therefore, to increase their safety for clinical applications, it is necessary to obtain higher-quality stem cells with an improved capacity for survival and self-renewal.
We are currently using a novel approach for culturing cells under simulated microgravity, with special focus on their cellular responses to physical stimulation [8 –10]. Previous studies using human mesenchymal stem cells (MSCs) and human hematopoietic progenitor cells have indicated that simulated microgravity suppresses cellular differentiation while favoring cell proliferation [10,11]. Therefore, simulated microgravity seems to be a useful and effective way to propagate and maintain the “stemness” (undifferentiated state) of stem cells. Here, we investigated (1) whether neural differentiation of BMSCs is suppressed when cultured under simulated microgravity conditions; (2) using a mouse model of cerebral contusion created cryogenically, we determined the efficacy and functional integration of mouse BMSCs (mBMSCs) cultured under simulated microgravity and grafted into the brain of these mice.
Materials and Methods
3D-clinostat
The 3D-clinostat, a multidirectional G force generator, was produced by Mitsubishi Heavy Industries, Ltd. (Kobe, Japan). By controlled simultaneous rotation of 2 axes, the 3D-clinostat cancels the cumulative gravity vector at the center around the device, producing an environment with an average of 10−3 G over time. This is accomplished by rotation of a chamber at the center of the device to disperse the gravity vector uniformly within a spherical volume, at a constant angular velocity [patented: Undifferentiated pluripotent stem cell proliferation/differentiation regulation method and system, Japanese Patent, Publication Number P2001-197182A, date of filing June 28, 2001, P2003-9852A, date of filing January 14, 2003, and overseas patents, WO2004/061092 A1 PCT (United States, Canada, China, and Korea), P/E (Italy, United Kingdom, Sweden, Germany, and France), 2004].
Preparation of mBMSCs
We used 5–8-week-old C57BL/6 mice. Donor cells for grafting were obtained from green fluorescent protein (GFP) transgenic mice (a gift from Dr. M. Okabe, Research Institute for Microbial Diseases, Osaka University). All animal procedures were conducted in accordance with human animal care standards outlined in the institutional animal care and use committees Guide for the Care and Use of Experimental Animals and were approved by the Hiroshima University Animal Testing Committee.
Mouse bone marrow cells were obtained from the femoral and tibial bones, and 1.0 × 108 cells were plated on 90-mm-diameter culture dishes (Thermo Fisher Scientific Nunc A/S brand, Roskilde, Denmark). The medium was exchanged to eliminate floating cells after 48 h. mBMSCs adhered to the bottom of the culture dish were used as culture cells. Dulbecco's modified Eagle's medium (Sigma-Aldrich Co., St. Louis, MO) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific HyClone brand, South Logan, UT), 100 units/mL penicillin, and 100 μg/mL streptomycin (both from Sigma-Aldrich Co.) was used as the culture medium.
To obtain a sufficient population of cells for the experiment, 2 subcultures of mBMSCs were proliferated and plated using OptiCell™ (Thermo Fisher Scientific Nunc brand, Rochester, NY) with the density of 2.0 × 104 cells/cm2. At 70% confluence, the cells were divided and cultured under 2 different conditions: normal 1G (group 1G) and simulated microgravity using the 3D-clinostat (group CL) (day 0). Each group was further divided into 2 groups and cultured in a growth medium (group GM) or a neural induction medium (group ND). Thus, the study included the following 4 groups: 1G-GM, CL-GM, 1G-ND, and CL-ND. Dulbecco's modified Eagle's medium/F-12 (Invitrogen Co., Carlsbad, CA) supplemented with 20 ng/mL human basic fibroblast growth factor, 10 ng/mL human β neural growth factor, 10 ng/mL human brain-derived neurotrophic factor (all from PeproTech, Inc., Rocky Hill, NJ), B27 supplement (Invitrogen Co.), 100 units/mL penicillin, and 100 μg/mL streptomycin was used as the neural induction medium.
Morphological changes
Morphological changes were examined using an inverted phase-contrast microscope (Eclipse TE 300; Nikon Co., Tokyo, Japan) and by taking random images.
Reverse transcription–polymerase chain reaction
Samples were collected using ISOGEN (Nippon Gene Co., Ltd., Toyama, Japan), and RNA was isolated according to the supplied protocol. Reverse transcription was performed with SuperScript™ II (Invitrogen Co.). Using cDNA as the template, PCR was performed using BD Advantage™ 2 PCR Kits (BD Biosciences Clontech, Palo Alto, CA). We used Oct-4 as the pluripotency marker and neurofilament heavy chain (NF-H) as the neural differentiation marker. β-actin was used as a housekeeping gene. The sequence of the prepared primer as well as PCR conditions are shown in Supplementary Table S1 (Supplementary Data are available online at
Immunostaining of neural differentiation markers
Fixed mBMSCs were stained using an immunostaining method and examined using a multifunctional microscope (BZ-9000; Keyence Co., Osaka, Japan). For the neural differentiation markers, NF-H and microtubule-associated protein 2 (MAP2), monoclonal anti-neurofilament 200 and monoclonal anti-MAP2 (both from Sigma-Aldrich Co.) were used as primary antibodies, respectively. Alexa Fluor® 488 goat anti-mouse IgG (H + L) (Invitrogen Co.) was used as the secondary antibody. For nuclear staining, 4′,6-diamidine-2-phenylindole dihydrochloride (DAPI) (Kirkegaard & Perry Laboratories, Inc., Gaithersburg, MD) was used. Images were stored on a computer, and the number of positive cells was divided by the total number of cells to calculate the rate of positivity.
Construction of a mouse model of cerebral contusion and cell grafting
We used 8-week-old C57BL/6 mice (8 mice/group) for constructing a cerebral contusion model using a cryogenic method [12 –14]. Mice were anesthetized and a 15-mm incision was made on the scalp along the craniocaudal axis. Then, a metal probe chilled with liquid nitrogen was inserted between the coronal suture and bregma and 3.0 mm lateral to the left interparietal suture for four 30-s periods, with intervals of 30 s to create a 4.0-mm-diameter contusion.
Cells were grafted at 7 days after inducing damage by injecting 3.0 × 105 cultured cells diluted in 100 μL phosphate-buffered saline into the retro-orbital venous plexus. Group C received no cellular grafting. Group S, a control group, underwent a placebo operation consisting only of a scalpel incision.
Evaluation of motor function
Beam-walking and rotarod tests were conducted to test motor function (8 mice/group).
In the beam-walking test, mice walked on a wooden square prism (6 mm in width, 120 mm in length), and the number of slips due to the paralyzed right hind leg in 50 steps was noted. All mice were trained to walk with <5 slips in 3 days prior to causing cryogenic damage.
In the rotarod test, mice ran on a rod rotating at 20 rpm using rotarod system (KN-75; Natsume Seisakusho Co., Ltd., Tokyo, Japan). The duration before falling from the rod was measured. All mice were trained to walk for at least 100 s on a rod rotating at 10 rpm in 3 days prior to causing the cryogenic damage.
Histological analysis
Mice were first anesthetized, and after perfusion, cerebral tissue was excised at 28 days after the damage was induced. The cerebral tissue was subsequently embedded in Tissue-Tek® O.C.T™ compound (Sakura Finetechnical Co., Ltd., Tokyo, Japan), frozen with liquid nitrogen, and sliced into 10-μm segments using cryostat (Leica Microsystems GmbH, Wetzlar, Germany). The segments were then used for hematoxylin and eosin staining, neural differentiation marker staining for NF-H and MAP2, and astrocyte marker glial fibrillary acidic protein (GFAP) staining. Primary antibodies for NF-H and MAP2 have been described previously, and for GFAP, monoclonal GFAP (Abcam plc, Cambridge, United Kingdom) was used. Alexa Fluor® 594 goat anti-mouse IgG (H + L; Invitrogen Co.) was used as the secondary antibody. Stained segments were examined under a multifunctional microscope (BZ-9000; Keyence Co.).
Immunostaining of chemokine receptors
The staining protocol was the same as that for the neural differentiation markers described above. A 100-fold dilution of anti-CXCR4 [fusin (H-118); Santa Cruz Biotechnology, Inc., Santa Cruz, CA] was used as the primary antibody for the chemokine receptor CXCR4.
Statistical analysis
The results are shown as mean ± standard deviation from 2-way analysis of variance using JSTAT for Windows. When 2-way analysis of variance showed a significant difference, the significance between the groups was calculated with the Bonferroni test for multiple comparison. A probability value of <0.01 was considered statistically significant.
Results
Effects of simulated microgravity on cellular differentiation: cell morphology, reverse transcription–polymerase chain reaction, and immunocytochemical analysis
On day 0, cells from groups 1G-GM and 1G-ND showed a spindle and oval shape (Fig. 1) and cells in 1G-GM did not show significant changes over time. 1G-ND cell cultures were characterized by numerous bipolar cells that formed a network with their long cell processes. In contrast, cells from both groups cultured under simulated microgravity (groups CL) showed no significant morphological changes during the following 7 days, regardless of whether they were cultured in GM or ND. Cultures from the CL-ND group had numerous small cells, and despite the presence of neural induction factors, cells did not develop any cell process. When these cultures were placed back in 1G-ND conditions for 7 days, numerous cells developed long neurite-like cell processes and developed contacts with neighboring cells (see Supplementary Fig. S1).
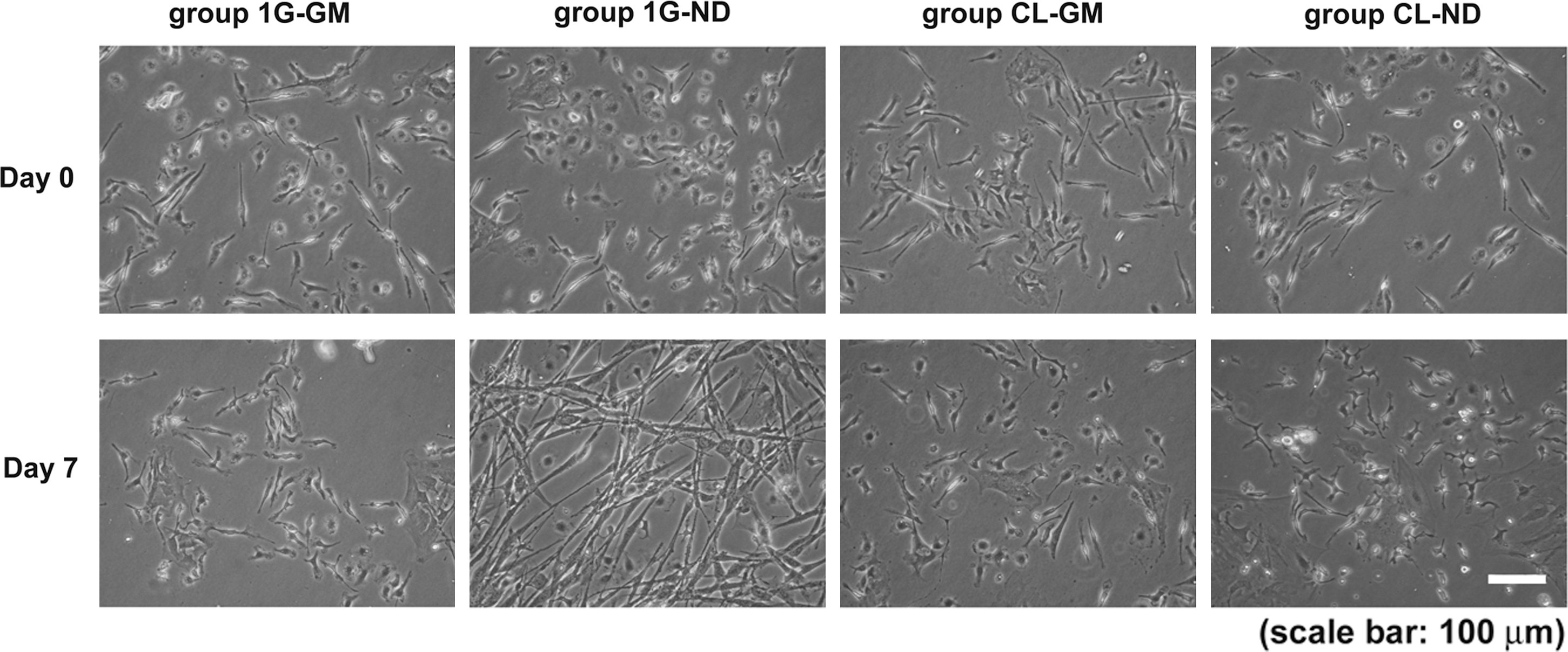
Effect of simulated microgravity on cellular morphology. Phase-contrast microscopic images of the cells from each group on days 0 and 7 are shown. Many cells with long processes were observed in the neural-induced group cultured under 1G conditions (group 1G-ND). However, under simulated microgravity, despite the presence of neural induction factors, no cells with neuron-like characteristics were observed. The scale bar indicates 100 μm.
On day 0, the expression of Oct-4 mRNA but not NF-H mRNA was detected (Fig. 2A). Oct-4 mRNA expression was observed in cells under 1G-GM conditions where only a few cells expressed NF-H or MAP2 protein (Fig. 2B). Cells maintained in 1G-ND showed downregulation of Oct-4 mRNA with a concomitant upregulation of NF-H mRNA, indicating neural differentiation. No difference was seen in the levels of expression of the NF-H and MAP2 proteins. In contrast, cells from groups CL-GM and 1G-GM and the neural-induced group CL-ND failed to express the differentiation markers but did express Oct-4 and retained an undifferentiated morphology. Cells maintained in CL-ND for 7 days were then shifted to 1G conditions and cultured in the induction medium for 7 days, and these showed decreased expression of Oct-4 mRNA, with the concomitant upregulation of NF-H and MAP2 (Supplementary Fig. S2).

Effect of simulated microgravity on pluripotency and the expression of differentiation markers. The figure shows
Effects of grafted cells cultured under simulated microgravity: recovery of motor function and neurogenesis
Immediately after the procedure, mice began to show paralysis on the right side of the body. To avoid interference from inflammatory cells, mBMSCs were grafted 7 days after the induction of the head injury. Damage to the left cortical surface was confirmed by hematoxylin and eosin staining at 28 days after injury (Supplementary Fig. S3).
Mice grafted with CL-ND cells had the highest scores and showed the greatest improvement compared with all other groups. Eleven days after injury they showed more significant improvement in the beam-walking test than group C (which received intravenous injections of phosphate-buffered saline), and by day 14 they performed significantly better than group 1G-ND (P < 0.01; Fig. 3A). Groups 1G-GM, CL-GM, and 1G-ND showed significant improvement compared with group C after day 14. The improvement of group 1G-ND was slow, whereas that of groups CL-GM and 1G-GM was about the same. In the rotarod test, mice grafted with CL-ND cells showed significant improvement compared with group C on day 14, group 1G-ND on day 21, and groups CL-GM and 1G-GM on day 28 (P < 0.01; Fig. 3B).
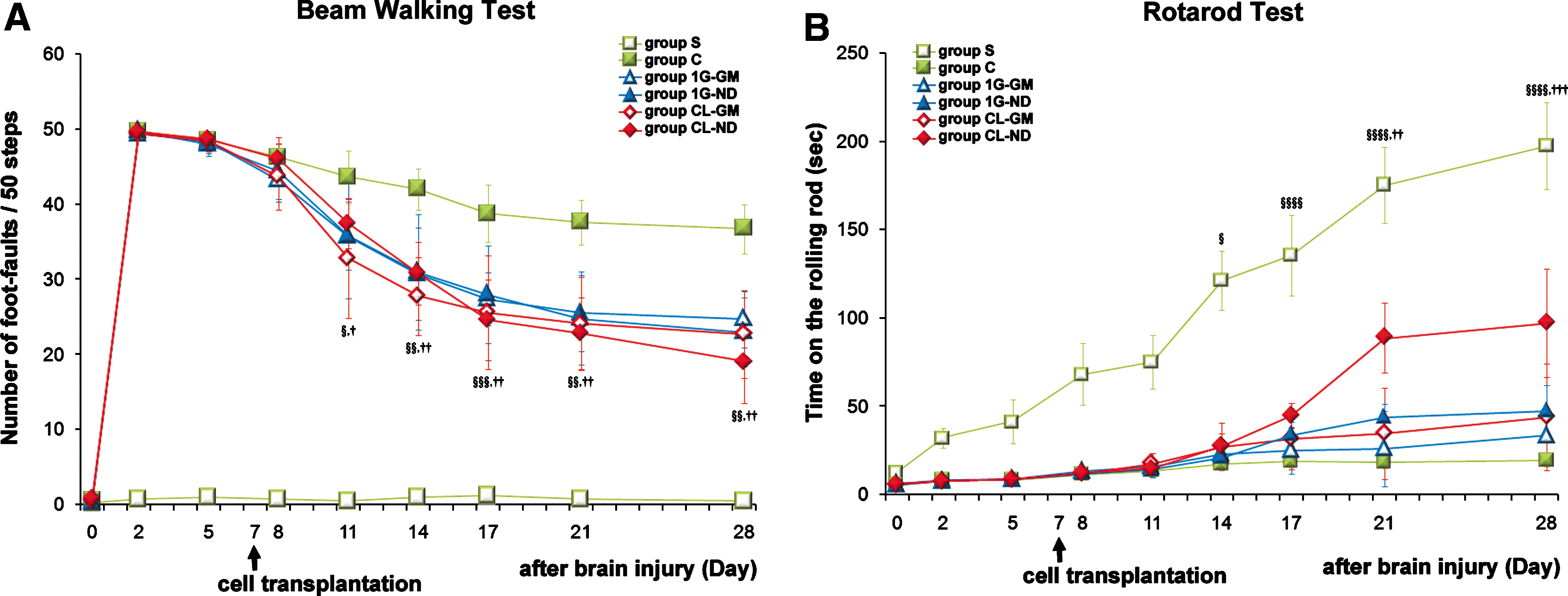
Effect of grafting cells cultured under simulated microgravity on motor function improvement. The results of motor function tests are indicated from before inducing damage (day 0) to 28 days later.
On day 28 (21 days after grafting), grafted cells were detected in host brains by the GFP label. These cells were mainly observed at the contusion level in all mice that received cell grafts: 1G-GM, CL-GM, 1G-ND, and CL-ND (Fig. 4). However, mice grafted with cells cultured under simulated microgravity had more GFP-expressing cells at the lesion and many more of these grafted cells expressed the neuronal markers NF-H and MAP2 compared with mice grafted with cells cultured under 1G conditions (group 1G-GM compared with group CL-GM, or group 1G-ND compared with group CL-ND) (Fig. 4A, B). Further, comparing with group CL-GM, more grafted cells expressed NF-H and MAP2 in group CL-ND. Grafted cells that had differentiated into GFAP-positive cells were also observed, albeit in low numbers when compared with cells expressing NF-H or MAP2, and numerous GFAP-expressing cells were observed in mice grafted with 1G-GM cells (Fig. 4C).
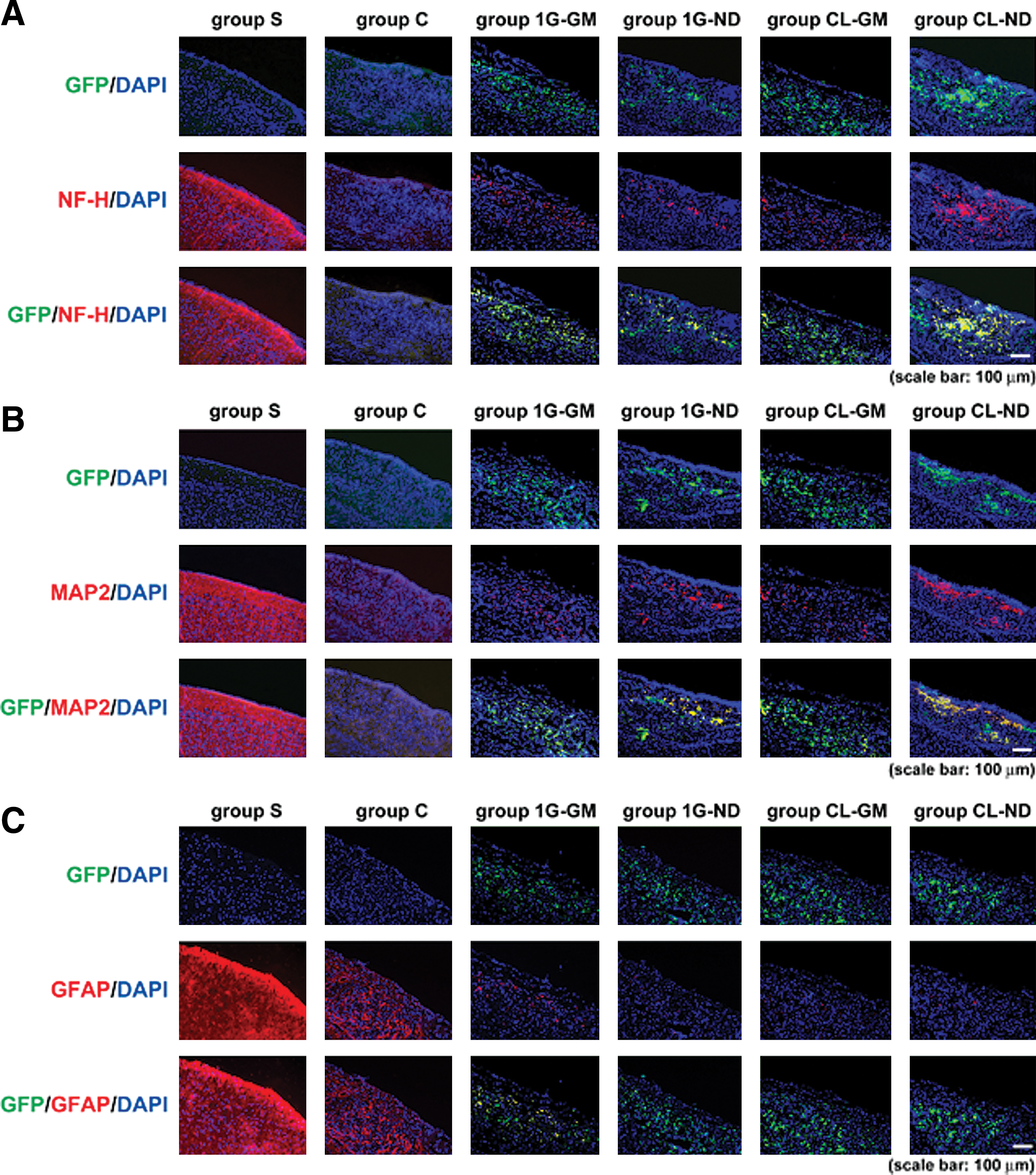
NF-H, MAP2, and GFAP expression in grafting cells cultured under simulated microgravity. Immunostained images of each group at 28 days after inducing damage are shown. The images of
CXCR4 expression in grafted cells cultured under simulated microgravity
Groups 1G-GM and CL-GM displayed similar levels of CXCR4. Mice grafted with 1G-ND cells had a smaller number of cells expressing CXCR4, whereas hosts grafted with CL-ND had a significantly larger number of cells expressing CXCR4 (Fig. 5).

CXCR4 expression in grafting cells cultured under simulated microgravity. Compared with the growth medium–cultured groups 1G-GM and CL-GM, the neural-induced group 1G-ND had smaller number of CXCR4-expressing cells. On the other hand, numerous group CL-ND cells expressed CXCR4. Percentage values indicate the positivity ratios calculated by dividing the number of positive cells by the total number of cells. The scale bar indicates 25 μm. Color images available online at
Discussion
In this study, we investigated the impact of simulated microgravity on the self-renewal and neurogenic potential of mBMSCs cultured in either growth or neural induction medium. We chose these cells because of the advantages they offer, which include their mode of administration. The transcription factor Oct-4 is involved in the self-renewal of embryonic stem cells as well as the maintenance of pluripotency. Recently, MSCs were also found to express Oct-4 [15,16], making it an ideal pluripotency marker for our study. When cultured in neural induction agents and standard 1G conditions, mBMSCs differentiated into neural cells. In contrast, under simulated microgravity, cells exhibited long cell processes and expressed neural differentiation markers in conjunction with Oct-4, indicating that simulated microgravity leads to preservation of the uncommitted stage of mBMSCs. When cells that were cultured in CL-ND were placed from simulated microgravity to 1G and sustained in differentiation induction medium for 7 days, mBMSCs developed cell processes and began to express NF-H and MAP2. These results indicate that simulated microgravity impacts mBMSCs by overriding the effects of neural induction medium. Nonetheless, this suppression of self-renewal is transient and can be reversed by placing mBMSCs grown under simulated microgravity back into standard conditions.
Our previous studies indicated that one of the mechanisms contributing to the maintenance of the undifferentiated state is the specific inhibition of p38 phosphorylation in the mitogen-activated protein kinase (MAPK) cascade [9], but the process is not fully understood. Our present study showed that the cellular size of mBMSCs cultured under simulated microgravity was small (Supplementary Fig. S4). The expression of cytoskeletal proteins has been reported to decrease under simulated microgravity, whereas the structure becomes fragile and unorganized [17,18]. This study suggests that the cytoskeletal change is one of the factors causing the mBMSCs to become smaller under simulated microgravity. As the cytoskeleton is thought to be involved in intracellular signaling, it may also contribute to the mechanism responsible for retaining pluripotency in mBMSCs. In addition, mBMSCs cultured under simulated microgravity showed decreased mitochondrial activity (Supplementary Fig. S5). Our study involving brain tumor cells cultured in simulated microgravity showed decreased mitochondrial activity in the cancerous cells, which indicates that the suppression of cancerous cell proliferation may not occur via enhanced apoptosis under simulated microgravity, but rather through slowing down of the cell cycle [19]. This would seem to suggest that a decrease in mitochondrial activity induces a hibernation-like state because of the lowering of cellular metabolism, resulting in the maintenance of an undifferentiated state under simulated microgravity.
To test future clinical applications of cells grown in simulated microgravity, we examined the ability of these cells to survive, migrate, and functionally integrate upon grafting into the contused mouse brain. In our study, grafting was achieved by injecting the cells intravenously. Intravenously injected cells have been shown to cross the blood brain barrier to reach cerebral tissues, and this phenomenon has been reported to burst after ischemia [20 –22]. We found GFP-labeled grafted cells in the lesion at 21 days after grafting. mBMSCs cultured under simulated microgravity survived in the damaged region in larger numbers and the motor function of the grafted mice improved significantly. The group receiving CL-ND mBMSCs showed the greatest recovery of motor function as well as the highest survival rate of graft cells compared with the other groups. These results provide insight on the use of this approach as a solid basis for clinical applications.
The focus of our cell therapy studies involving grafting of BMSCs is to repair lost tissues and regenerate function. It is absolutely necessary for graft cells to survive for a long time in the damaged region as well as to promote neuroregeneration and/or restructuring of effective neural circuits. Previous studies involving BMSCs grafted into ischemic animals confirmed the neural differentiation of BMSCs in vivo [1,2,5,23 –26]. In the present study, we believe that more pluripotent cells from the mBMSCs of group CL-ND migrated to the damaged region, contributing to neuroregeneration through neural differentiation in the damaged region and to improvement in motor function. On the other hand, recently, the effects of grafted BMSCs have been attributed to the nutritional effect caused by the secretion of neuro-nutritional factors. BMSCs grafted after ischemia are believed to release neural nutritious factors and are thought to contribute to the survival of neural cells in the damaged region as well as the differentiation and growth of endogenous neural stem cells and neural progenitor cells [1,27 –29]. Numerous cells differentiated into astrocytes in mice grafted with group 1G-GM mBMSCs, and the protective effect by grafted cells may have contributed to motor function improvement. Accumulation of astrocytes in the damaged region is called a glia scar, and it is known to interfere with the elongation of axons during neuroregeneration. Group C, which received no cellular grafting, had mostly astrocytes and few neural cells on day 28. Conversely, the grafted group had significantly fewer astrocytes and more NF-H- and MAP2-expressing cells than GFAP-expressing cells. With the motor function evaluation, the cell-grafted group showed a greater improvement than the nongrafted group. In the study by Li et al. [5] with BMSCs injected through tail vein of ischemic rats at 7 days after brain damage, the results were similar to our study. The efficacy of grafting BMSCs was evidenced not only by the cells differentiating into neuronal cells but also by how the factors released by grafted cells suppressed the formation of glial scar and enhanced the elongation of the axons.
Previous studies have shown that neural cells are more suitable for grafting directly to ischemic animals than pluripotent cells [30]. However, many studies report that with intravenous grafting, better results are obtained with pluripotent cells, such as MSCs [1 –3,23,24,31]. Culturing under simulated microgravity enables cells to maintain pluripotency and express CXCR4 and can, therefore, be a useful culturing method for donor cells for transplant treatments. In addition, the characteristic small size of mBMSCs cultured under simulated microgravity is believed to contribute to easier migration to the periphery after transplantation. Future investigation regarding the mechanism for the increase in CXCR4 expression in the membrane and the preservation of pluripotency when differentiation is induced under simulated microgravity is necessary.
We propose that 3D-clinostat provides a novel way to expand stem cell populations successfully in vitro for use in cell therapy and developmental biology, particularly for cell and tissue transplantation.
Footnotes
Acknowledgments
The authors thank Dr. Masaru Okabe at Osaka University for kindly providing GFP mice; Mr. Ryuji Hirose, Mr. Toshimasa Ochiai, and Mr. Eiji Matsubara, Mitsubishi Heavy Industries, Ltd., for providing information and suggestions; and Dr. Araceli Espinosa-Jeffrey and Prof. Jean de Vellis at Department of Neurobiology, Semel Institute for Neuroscience, UCLA, for their comments during data analysis and preparation of the manuscript. This study received the award from the 23rd Annual Meeting of the American Society for Gravitational and Space Biology at NASA Ames Research Center, San Francisco, October 27, 2007.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.