
Abstract
Unrestricted somatic stem cells (USSCs) represent an intrinsically multipotent CD45-negative fetal population from human cord blood. They show differentiation into neuronal cells of a dopaminergic phenotype, which express neuronal markers such as synaptophysin, neuronal-specific nuclear protein, and neurofilament and release the neurotransmitter dopamine accompanied by expression of dopaminergic key factors tyrosine hydroxylase and Nurr1 (NR4A2). MicroRNA expression analysis highlighted their importance in neural development but their specific functions remain poorly understood. Here, downregulation of a set of 18 microRNAs during neuronal lineage differentiation of unrestricted somatic stem cells, including members of the miR-17-92 family and additional microRNAs such as miR-130a, -138, -218, and -335 as well as their target genes, is described. In silico target gene predictions for this microRNA group uncovered a large set of proteins involved in neuronal differentiation and having a strong impact on differentiation-related pathways such as axon guidance and TGFβ, WNT, and MAPK signaling. Experimental target validations confirmed approximately 35% of predictions tested and revealed a group of proteins with specific impact on neuronal differentiation and function including neurobeachin, neurogenic differentiation 1, cysteine-rich motor neuron protein 1, neuropentraxin 1, and others. These proteins are combined targets for several subgroups from the set of 18 downregulated microRNAs. This finding was further supported by the observed upregulation of a significant amount of predicted and validated target genes based on Illumina Beadstudio microarray data. Confirming the functional relationship of a limited panel of microRNAs and predicted target proteins reveals a clear network-like impact of the group of 18 downregulated microRNAs on proteins involved in neuronal development and function.
Introduction
M
Demonstrating stage- and tissue-specific expression patterns during development [8,9], microRNAs appear to not only act as key regulatory molecules in a variety of cellular processes including apoptosis [10,11], cancer [12], proliferation [13], development [14], and differentiation [15] but also play important roles in stemness and pluripotency of embryonic stem cells [16]. Several expression profiling approaches revealed enrichment of groups of microRNAs in brain [17] and certain microRNAs appear to play important roles in nervous system development [9,18], neurite outgrowth, and synaptic plasticity [19]. Most importantly, brain-specific miR-124 promotes neural transcription by targeting the splicing regulator PTBP1 [20] and miR-9 is involved in differentiation of neural progenitor cells [21].
Unrestricted somatic stem cells (USSCs) constitute a rare CD45-negative population from human cord blood [22]. Adherently growing in vitro USSCs can be induced to cells from all three germinal layers [22]. Using a specific cocktail of growth and differentiation factors containing all-trans-retinoic acid as a major component (XXL medium), differentiation of USSCs into neural cells (XXL-USSC), which express neurofilament (NEF) and sodium channel proteins as previously described [23]. In addition, XXL-USSCs display various neurotransmitter phenotypes including expression of GABA [22] and tyrosine hydroxylase, the key enzyme of the dopaminergic pathway [23]. This neuronal lineage differentiation of USSCs, however, seems to be limited because patch-clamp analysis failed to detect voltage-activated fast inactivating Na+ current [22,23], indicating that neuronal lineage-differentiated USSCs have not developed a fully functional neuronal phenotype as yet.
To understand microRNA-guided neuronal lineage differentiation of USSCs, we analyzed changes in the microRNA expression profile following XXL-mediated differentiation of USSCs. Remarkably, downregulation of a group of 18 microRNAs including members of the miR-17-92 family was observed. In silico target gene predictions for this microRNA population revealed a large set of proteins important for neuronal differentiation and function. Experimental target validations for 10 microRNAs and 11 predicted target genes demonstrated crosswise overlapping of microRNA downregulation and upregulation of target gene expression in XXL-differentiated USSCs.
Materials and Methods
Neuronal lineage differentiation of USSCs
For expansion, USSCs were incubated with Dulbecco's modified Eagle's medium (Lonza, Cologne, Germany) supplemented with 30% heat-inactivated fetal bovine serum (Lonza) and penicillin/streptomycin (100 U/mL; Gibco, Invitrogen, Karlsruhe, Germany). Neural differentiation was performed as previously described [23]. Briefly, USSCs were seeded on laminin-precoated glass cover slips and incubated with differentiation medium XXL containing Dulbecco's modified Eagle's medium GlutaMAX™ (Gibco), 15% fetal bovine serum, 100 U/mL penicillin/streptomycin, 50 ng/mL beta-nerve growth factor, 20 ng/mL basic fibroblast growth factor (both from Tebu, Offenbach, Germany), 1 mM dibutyryl-cAMP, 0.5 mM 3-isobutyl-methylxanthine, and 10 μM all-trans-retinoic acid (all from Sigma-Aldrich, Taufkirchen, Germany) for up to 14 days.
Immunocytochemical analysis
For immunocytochemical analysis, cells were fixed with 4% formaldehyde (Merck, Darmstadt, Germany) for 10 min, rinsed three times with phosphate-buffered saline, and incubated with blocking solution including 10% normal goat serum and 0.03% Triton X-100 for 1 h. USSCs were incubated with the primary antibody directed against NEF protein (recognizing NEFL, NEFM, and NEFH; 1:1000; BioTrend, Cologne, Germany) and the anti-rabbit secondary antibody conjugated with Alexa 594 (1:1000; Invitrogen), both diluted in phosphate-buffered saline. Cell nuclei were labeled with 4,6′-diamidino-2-phenylindoline (Roche, Mannheim, Germany). As negative controls and to ensure specificity, first antibody was omitted.
MicroRNA expression analysis
Small and large RNA fractions from USSCs and XXL-USSCs were prepared using the Ambion mirVana microRNA Isolation kit (Applied Biosystems, Darmstadt, Germany) according to the manufacturer's instructions, with the sole exception of direct lysis of adherent USSCs, thereby omitting trypsinization.
Small RNA fractions were used for microRNA expression analysis using the TaqMan microRNA Megaplex array (pool A; Applied Biosystems) [24] according to the manufacturer's instructions. In brief, 10 ng small-fraction RNA was reverse transcribed and preamplified for 12 polymerase chain reaction (PCR) cycles, followed by TaqMan probe-based array amplification for 40 additional PCR cycles. Raw Ct values were normalized to U6 RNA data, and ddCt as well as 2−(ddCt) data were calculated.
Computational target gene predictions
Target gene predictions were performed using miRGen compilation of prediction algorithms found on
Experimental validation of target gene predictions
PCR products of full-length 3′-UTRs or fragments of 3′-UTRs spanning the predicted microRNA-binding sites on the target gene of interest were cloned at the 3′ end of firefly luciferase ORF in dual reporter (Firefly and Renilla luciferases) vector pmirGLO (GenBank accession number: FJ376737; Promega, Mannheim, Germany) using restriction enzyme pairs SacI/XbaI or XhoI/SalI. pmirGLO vector (100 ng) and equimolar amounts of pmirGLO/3′-UTR were transfected into 5 × 104 HEK293T cells using 0.5 μL Lipofectamine 2000 (Invitrogen) to normalize for effects of endogenous microRNAs of HEK293T on the given 3′-UTR. Pairwise cotransfections of 100 ng empty pmirGLO with the 2.5 pmol microRNA mimic (Dharmacon, Bonn, Germany) of interest and pmirGLO/3′-UTR with the appropriate microRNA mimic were performed and Firefly and Renilla activities were measured 24 h posttransfection using Beetlejuice and Renillajuice reagents (PJK, Kleinblittersdorf, Germany). Percental relations of Renilla-normalized Firefly activities of each pmirGLO-microRNA-mimic and pmirGLO/3′-UTR-microRNA-mimic cotransfections with normalized firefly activities of pmirGLO and pmirGLO/3′-UTR were established. Percental reductions of ≥20% were denoted positive. Transfections were performed in at least two independent biological experiments with triple or quadruple transfections each.
Transfection of USSCs
To test the effects of endogenous USSC microRNAs on the predicted 3′-UTRs during XXL differentiation, 200 ng of pmirGLO and equimolar amounts of pmirGLO/3′-UTR were transfected in 2 × 104 USSCs using Lipofectamine 2000. Twenty-four hours after transfection, neuronal lineage differentiation was induced for 5 days using XXL medium. Renilla-normalized Firefly activities were measured 24 h and 6 days after transfection as described in the preceding section. Transfections were performed in four independent experiments with triple or quadruple transfections each.
Gene expression analysis
For transcriptome profiling, 500 ng of DNA-free large-fraction RNA was used as input for a linear cRNA synthesis (Illumina TotalPrep RNA Amplification Kit; Ambion, Darmstadt, Germany) following the manufacturer's instructions (in-vitro transcription [IVT]: 14 h). Quality-checked biotin-labeled cRNA samples (750 ng) were hybridized as biological or technical duplicates for 18 h onto HumanRef-8 v3 expression BeadChips (Illumina, San Diego, CA), washed, stained, and scanned following the guidelines of the manufacturer. Bead intensities were mapped to gene information by applying BeadStudio 3.2 software (Illumina). Raw data were background subtracted and finally normalized using the rank-invariant algorithm. Expression values below the detection limit (<50) were set to the level of threshold detection in order to avoid nonsense values for expression ratios. Differences in gene expression were considered significant at a fold change of at least 50%, with a P value of ≤0.01.
Results
Comparative microRNA expression profiling of USSCs undergoing neuronal lineage differentiation
Two independent USSC lines, namely USSC SA5/03 and USSC SA5/73, were differentiated in XXL medium for 14 days as previously described [23]. Figure 1 shows expression of neuronal marker NEF in native USSCs and neuronal lineage differentiated USSCs (XXL-USSC). In contrast to USSCs, the XXL-USSCs showed an increased expression of NEF as well as a typical neuronal morphology. Elevated expression of NEF (all chains) and βIII-Tubulin was also confirmed by quantitative PCR assays (data not shown).
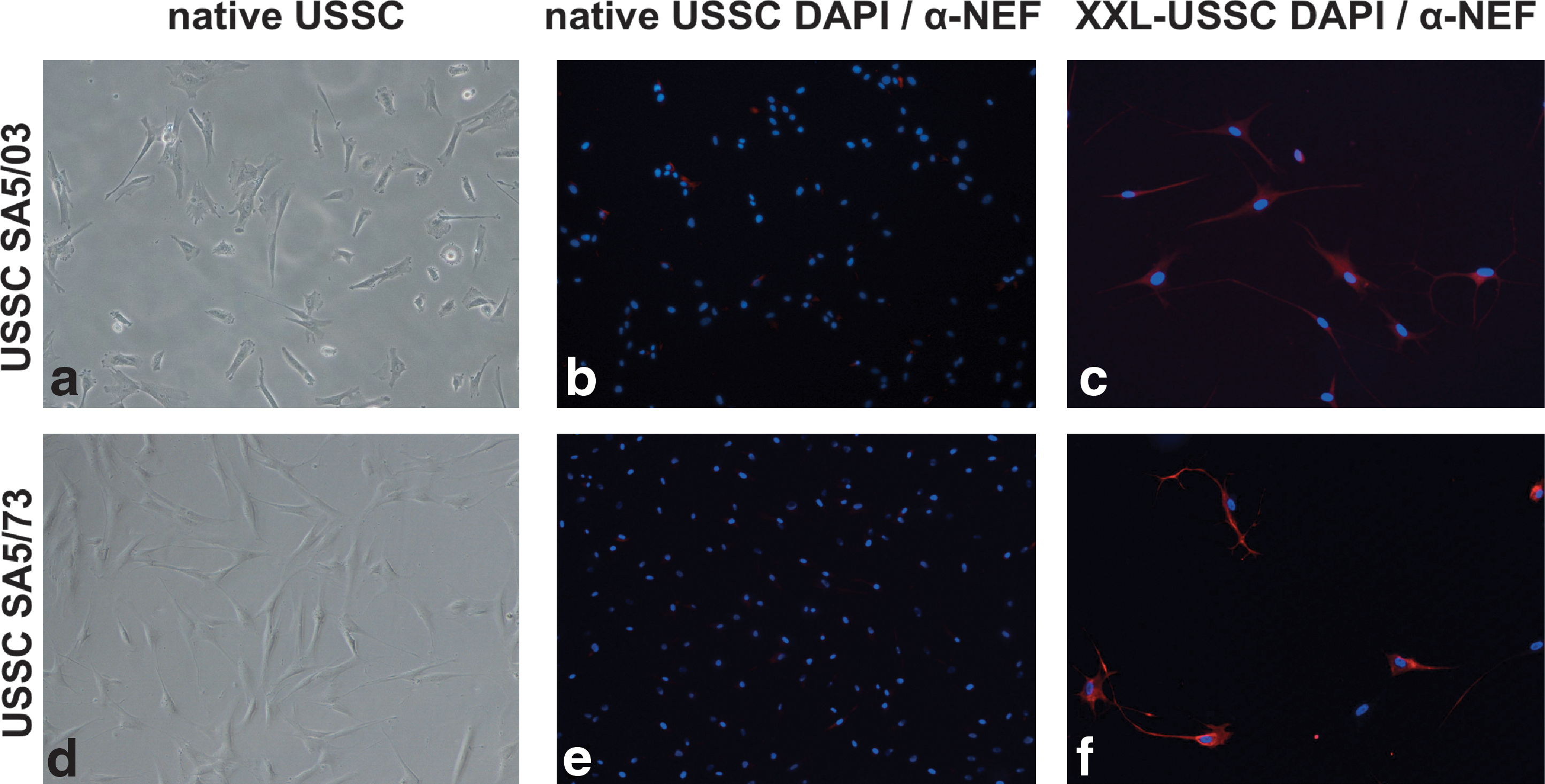
Neuronal lineage differentiation of USSC SA5/03
To determine the microRNA expression profiles in both native USSCs and XXL-USSCs, we used the ABI microRNA Megaplex TaqMan assay (pool A) covering 377 human microRNAs [24]. TaqMan results were confirmed by northern hybridization for all three microRNAs (miR-16, -22, and -222) tested (data not shown). Interestingly, in both USSC lines, SA5/03 and SA5/73, a common set of 18 microRNAs was significantly downregulated upon XXL differentiation after 14 days. Figure 2 shows the heat map of the 18 differentially expressed microRNAs from both USSC lines in both native and neuronal lineage-differentiated stages and summarizes fold changes for each member of this microRNA set. MiR-138, -335, -18a, -20a, and -130a were among the most strongly downregulated microRNAs. Interestingly, no microRNAs were found significantly upregulated during XXL treatment, and we were also unable to detect the neuronal-specific miR-124 and miR-9, neither in native USSCs nor in XXL-USSCs. The brain-specific miR-138 was found highly expressed in native USSCs and was the most strongly downregulated microRNA among the set of 18 microRNAs (Fig. 2). Further, miR-125b, which was shown to be involved in neurite outgrowth [28], was found among the most abundant microRNAs in native USSCs and XXL-USSCs. Expression of the embryonic stem cell-specific miR-302 family could not be detected in any USSC. In addition, miR-145, which has been shown to repress pluripotency factors OCT4, SOX2, and KLF4 [16], was among the most strongly expressed microRNAs in both native USSCs and XXL-USSCs.

Differential expression of microRNAs upon XXL-mediated neuronal lineage differentiation of USSC lines SA5/03 and SA5/73. The heat map shows common downregulation of 18 microRNAs in both cell lines as measured by quantitative real-time (RT)–polymerase chain reaction, together with the fold changes (2–ddCt) of microRNA expression in XXL-USSC lines SA5/03 and SA5/73 compared with the respective native USSC lines SA5/03 and SA5/73. Color images available online at
In silico target gene predictions
To evaluate the biological impact of the set of 18 microRNAs downregulated during neuronal lineage differentiation of USSCs, we performed target gene predictions using primarily the web-based miRGen compilation of prediction algorithms. The UNION prediction mode used herein lists all predictions for a certain microRNA generated by the algorithms miRanda, PicTar, TargetScan, and DIANA-microT. In the UNION mode, lists of predicted target proteins were generated independently for all 18 microRNAs found downregulated in USSCs, resulting in a total of 9053 proteins. In the DAVID database (2008), GO terms were subsequently applied to filter the lists for neuronally relevant proteins. Using the GO search terms development, differentiation, and morphology (but skipping nonneuronal terms such as muscle development) together with nerv, neur, axon, brain, and synap (8-GO condition) revealed a total of 2122 proteins predicted for the group of 18 microRNAs. More stringent filtering by using the search phrases nerv, neur, axon, brain, and synap only (5-GO condition) revealed a total of 711 proteins predicted for the complete set of 18 downregulated microRNAs. Because of target gene redundancy, many of the predicted proteins contain putative target sites for more than one microRNA out of this group. Certain proteins such as neurogenic differentiation 1 (NEUROD1), neurobeachin (NBEA), or neuropentraxin 1 (NPTX1) contain putative target sites for ≥10 microRNAs out of the 18 microRNAs group (Supplementary Fig. S1; Supplementary Data are available online at
To further underscore the putative impact of the set of 18 downregulated microRNAs on neuronal differentiation of USSCs, the pathway analysis function, implemented in the DAVID database (2008), assigned the listed proteins to biological pathways that are relevant for neuronal differentiation and found within the KEGG database. As listed in Table 1, predicted targets for 12 out of 18 microRNAs could be assigned to members of distinct KEGG pathways related to neuronal differentiation and development. Most prominently, a group of 10 microRNAs putatively target 45 out of 79 depicted genes in the axon guidance pathway, followed by seven microRNAs potentially influencing 27 out of 54 genes from the TGFβ signaling pathway, and six microRNAs may recognize 53 out of 126 genes found within the MAPK signaling pathway. Graphical maps of the pathways axon guidance, TGFβ signaling, WNT signaling, MAPK signaling, and long-term potentiation together with the corresponding predictions are shown in Supplementary Fig. S2. The putative targets of only six microRNAs (miR-18a, -27a, -140-5p, -143, -362-5p, and -431) were not related to any of these five pathways.
The number of matches (including redundancies) for each microRNA is given. The last row presents the number of putative microRNA targets (including redundancies) as well as the total number of protein members within the pathway.
Experimental validation of selected putative microRNA targets
For experimental target gene validation, we selected 11 proteins [cysteine-rich motor neuron protein 1 (CRIM1), ephrin receptor (EPHA4), methyl-CpG binding protein 2 (MECP2), NBEA, NEUROD1, neurogenin (NEUROG1), NPTX1, neuropilin 1 (NRP1), netrin 4 (NTN4), pleiotrophin (PTN), and signal transducer and activator of transcription 3 (STAT3)] from the predictions list with regard to protein function and amount of predicted target sites. In addition, the 3′-UTR of the housekeeping gene GAPDH was tested. This set of 3′-UTRs was analyzed for interactions with 10 distinct microRNAs (miR-17-5p, -18a, -19a, -20a, -106b, -130a, -137, -138, -218, and -35) covering 71 predicted 3′-UTR-microRNA interactions (Supplementary Fig. S1). As described in detail in Materials and Methods, full-length 3′-UTRs (or 3′-UTR fragments covering the predicted microRNA-binding sites) were cloned at the 3′ end of Firefly luciferase in dual luciferase reporter vector pmirGLO (Promega). In case of MECP2, the 8554-bp 3′-UTR was split into two fragments (MECP2-1 and MECP2-2), cloned, and analyzed separately. Pairs of pmirGLO and pmirGLO/3′-UTR were cotransfected into HEK293T cells with the appropriate microRNA-mimic (Dharmacon) to test for specific influence of the microRNA on the given 3′-UTR. For example, Fig. 3A depicts Firefly activities measured from cotransfections of each mimic miR-130a, -137, and -335 with pmirGLO compared with cotransfections of these mimics with pmirGLO/NRP1-3′-UTR. Figure 3B summarizes the validation results for all 3′-UTRs tested, which were normalized to (i) effects caused by endogenous HEK293T microRNA population, (ii) unspecific effects of certain microRNA-mimics on Firefly and Renilla activity per se (possibly caused by influence of mimics on luciferase ORFs [29 –31]), and (iii) transfection efficiency variations. Overall, we were able to validate at least one microRNA–target gene interaction in each 3′-UTR tested. Seven out of nine microRNAs tested were shown to target the NBEA 3′-UTR, although with different efficiencies in firefly downregulation. In summary, 32% of all individual predictions could be experimentally verified (Fig. 3B), and it remains noteworthy that measured effects of distinct microRNAs on certain 3′-UTRs ranged from strong (i.e., NEUROD1 and miR-138) to rather weak but still significant (i.e., NPTX1 and miR-130a). Detailed data for all tested 3′-UTRs are given in Supplementary Fig. S3.
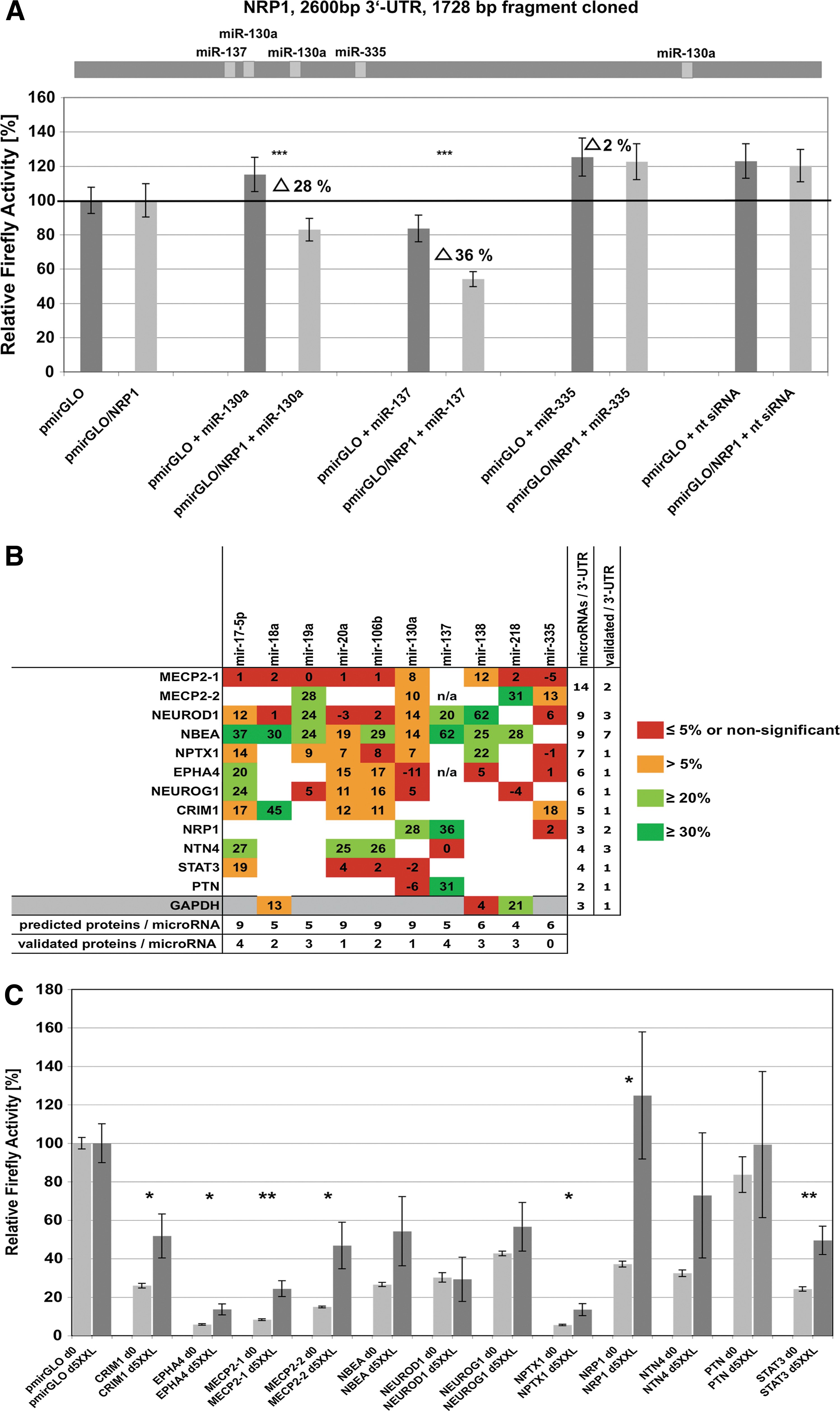
Experimental validation of target gene predictions.
Effects of endogenous microRNAs in USSCs on predicted target genes during XXL differentiation
We next aimed to analyze how changes in endogenous microRNA expression of USSCs during neuronal lineage differentiation influence expression of validated target genes by means of their 3′-UTRs. To this end, USSCs were transfected with pmirGLO and pmirGLO-full-length 3′-UTR constructs (except for NPTX1, which was represented by the 2194-bp 3′-UTR used for target validations) followed by induction with XXL for 5 days. Relative firefly activities were measured at day 0 prior to and at day 5 after XXL induction. As shown in Fig. 3C, significant upregulation of normalized firefly activity was found for constructs containing the 3′-UTRs of CRIM1, EPHA4, MECP2-1, MECP2-2, NPTX1, NRP1, and STAT3 compared with the empty pirGLO-vector. No significant changes or only nonsignificant upregulations were observed for NBEA, NEUROD1, NEUROG1, NTN4, and PTN. These findings demonstrate that, even after only 5 days of XXL treatment, changes in endogenous microRNA expression patterns diminish microRNA-mediated regulatory constraints regarding the validated targets analyzed here.
Comparative global gene expression analysis during neuronal lineage differentiation of USSCs
Then the question was addressed whether the regulatory impact of the set of 18 microRNAs downregulated upon neuronal lineage differentiation of USSCs is also reflected in endogeneous transcriptional upregulation of predicted genes in XXL-USSCs compared with native USSCs. Illumina array data generated from USSC SA5/73 and XXL-USSC SA5/73 revealed approximately 17,000 genes expressed in the native USSCs and 3726 genes being significantly upregulated upon neuronal lineage differentiation. Cross-match analyses of upregulated genes and targets predicted for the set of downregulated microRNAs revealed 2095 (23.1%) of the 9053 predicted target genes being upregulated in XXL-USSC SA5/73 (Fig. 4A), which corresponds to 56% of the 3726 upregulated genes. From the 8-GO term prediction subset (2122 genes), 481 (22.7%) were found upregulated (Fig. 4A). The 711 genes of the 5-GO prediction subset represent 42% of the 1698 genes present on the Illumina array that match this GO criterion and 125 (17.6%) of these predicted genes were found upregulated in XXL-USSC SA5/73 (Fig. 4B). All overlaps between predicted and upregulated genes represent statistically significant enrichments by applying a hypergeometric distribution model (P values: UNION ≤0.001, 8-GO terms ≤0.001, and 5-GO terms ≤0.05, respectively). Among the 12 predicted proteins validated for microRNA–target gene interaction, we detected moderate to strong upregulations of NRP1, NTN4, NPTX1, PTN, and STAT3 (Fig. 4B), but no significant differential expression of CRIM1, EPHA4, MECP2, NBEA, NEUROD1, and NEUROG1 in XXL-USSCs was seen on the transcriptional level. As expected, GAPDH was only slightly upregulated in XXL-USSCs (data not shown). A recent study by Beveridge and coworkers [32] demonstrated downregulation of the miR-17-92 family during neuronal differentiation of SH-SY5Y neuroblastoma cells accompanied by an upregulation of 2945 genes as measured with the Illumina array, compared with the 3726 upregulated genes in XXL-USSCs. As illustrated in Fig. 4C, cross-match analyses revealed an overlap of 931 genes upregulated in parallel in both XXL-USSCs and neuronally differentiated SH-SY5Y cells. Moreover, 609 (65%) of these commonly upregulated genes were included in the UNION predictions for our group of 18 microRNAs.
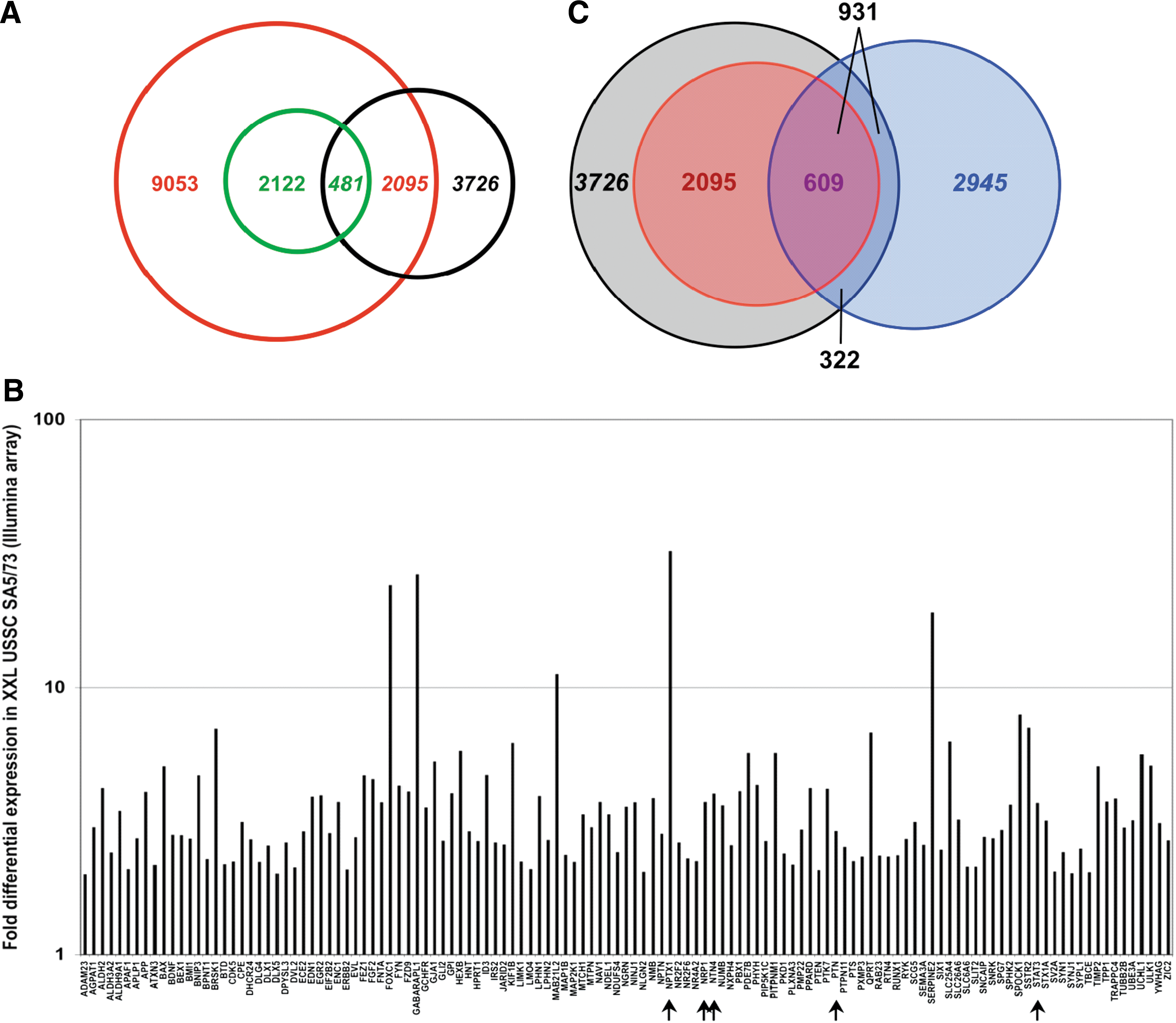
Gene expression analysis of USSC SA5/73 and XXL-USSC SA5/73 based on Illumina arrays.
Surprisingly, only a small set of 115 genes was detected as significantly downregulated in XXL-USSCs by the Illumina array (Fig. 5). This set was analyzed for functional properties (GO and assignment to KEGG pathways) using the DAVID database. Interestingly, more than 37% of the downregulated proteins were assigned to cell cycle- and cell proliferation-related GO terms, and cell cycle was the most prominent KEGG pathway the DAVID database revealed for this protein set. Members of the cell division cycle and cyclin protein families were found among the most strongly downregulated genes (Fig. 5). This finding is in line with the observed proliferation arrest of XXL-USSCs.

Subset of genes downregulated in XXL-USSC SA5/73. Fold changes in Illumina expression data are given for this group of genes.
Discussion
Given the limited neuronal functionality of XXL-USSCs, we evaluated the biological impact of the set of 18 microRNAs downregulated in XXL-USSCs with regard to neuronal differentiation pathways. The finding of several hundred proteins specific for neuronal differentiation and function, predicted in a largely overlapping manner as putative targets for these microRNAs, illustrates their network-like impact on neuronal lineage differentiation of USSCs. This was further substantiated by the high impact of these microRNAs on members of several KEGG pathways relevant for neuronal differentiation (summarized in Table 1 and Supplementary Fig. S2A). In particular, the predicted impact of 10 microRNAs on a majority of members of the axon guidance pathway clearly demonstrates the potential regulatory influence of these microRNAs on neuronal differentiation and function.
Interestingly, microRNA expression analysis by Beveridge and coworkers [32] in human SH-SY5Y neuroblasts, a cell type also capable of retinoic acid-induced neuronal differentiation and widely used for studying neuronal properties in vitro [33,34], revealed downregulation of the entire miR-17-92 cluster (miR-17-3p, -17-5p, -18, -19a, -19b, -20a, and -92, and paralogs miR-106a, -19b, -363, -92, -106b, -93, and -25). This finding strongly supports our results in XXL-USSCs, because, with the sole exception of miR-363, the miR-17-92 family-associated microRNAs were indeed also found downregulated in XXL-USSCs. This downregulation of overlapping sets of microRNAs during retinoic acid-induced neuronal lineage differentiation of both USSCs and SH-SY5Y neuroblasts strongly suggests that both neuronal specifications for a major part follow the same molecular pathways. The finding that miR-125b is highly abundant in USSCs and XXL-USSCs is of additional relevance, because it is found upregulated during neuronal differentiation of both SH-SY5Y and human neural progenitor ReNcell VM cells and has been proposed to repress antagonists of neuronal differentiation pathways [28].
As illustrated in Table 1 and Supplementary Fig. S1, many of the proteins predicted for the set of 18 microRNAs downregulated are putatively targeted by more than one microRNA from this set. This further implies that a synchronized downregulation of entire groups of microRNAs with functionally related target proteins might be necessary for coordinated expression changes of proteins during differentiation processes. To experimentally validate target predictions, we selected a panel of 10 microRNAs most abundant in predictions and 11 proteins. Among the selected proteins, we analyzed key players in neuronal development and differentiation, such as helix loop helix transcription factors NEUROD1 [35], bearing predictions for 11 out of 18 downregulated microRNAs, and NEUROG1 (involved in neuronal differentiation and cell-type specification [36]; 6 microRNAs predicted). Further relevant proteins include NBEA (required for formation and functioning of central synapses [37]; 10 microRNAs predicted), NPTX1 (also termed NP1, involved in synaptogenesis [38]; 10 microRNAs predicted), EPHA4 (critical in axon guidance and growth cone collapse [39,40]; 10 microRNAs predicted), and MECP2 (related to the neurodevelopmental disorder Rett syndrome [41]; 13 microRNAs predicted). In addition, miR-25 putatively targets all three chains of NEF (not included in experimental validations).
Further, we included the CRIM1 (implicated in central nervous system (CNS) development [42]), NRP1 (essential for neuronal development [43]), NTN4 (directing axon growth and cell migration during neuronal development [44]), PTN (increases the production of tyrosine hydroxylase-positive dopaminergic neurons from embryonic stem cells (ES) cell-derived nestin-positive cells [45]), and STAT3 (involved in retinoic acid-induced neuronal differentiation [46]). We also added housekeeping gene GAPDH, which may be expected to escape microRNA regulation [47], but yet contains predicted sites for miR-18a, -138, and -218 on its 196-bp 3′-UTR. This panel contains overall 71 predicted microRNA–3′-UTR interactions corresponding to 115 predicted individual microRNA target sites (Supplementary Fig. S1). As transfections of USSCs result in dramatically weaker efficiencies compared with HEK293T cells, particularly in plasmid/microRNA-mimics cotransfections, all validations had to be performed in HEK293T cells. Usage of human cell lines such as HEK293T or HeLa cells as host cells for microRNA target gene validations is widely accepted in the scientific literature [48 –50].
Taken together we were able to identify 23 correct microRNA target gene interactions, and with the sole exception of STAT3, at least one microRNA target site was positively validated for each protein tested (Fig. 3B). Interestingly, we found a correlation neither between the number of validated microRNA–target interactions and the length of the corresponding 3′-UTRs nor between the number of predicted target proteins for a given microRNA and the corresponding positive verifications (Fig. 3B). The most intensively targeted protein in our assay was NBEA, a protein not yet described as a microRNA target. Sequenced by targeting efficiency, mir-137, -17, -18a, -106b, -218, -138, and -19a were found to strongly target NBEA (Fig. 3B). Further, CRIM1, EPHA4, MECP2, NEUROD1, NEUROG1, and NTN4 were shown to be targets of miR-17-92 family members. In addition, MECP2 was found to be targeted by miR-218. MiR-137 is further related to neurogenesis by also affecting NEUROD1, NRP1, and PTN. NRP1 was in addition targeted by miR-130a. PTN enhances the expression of NR4A2 (also termed Nurr1), a protein important for dopaminergic phenotype [45], and putatively inhibited by miR-17-92 family members as well as by miR-130a and miR-137. Downregulation of miR-130a and miR-137 together with miR-17-92 family members might thus contribute to the dopaminergic phenotype of XXL-USSC, which is further supported by the observed upregulation of PTN and NR4A2 in XXL-USSCs based on the Illumina-derived data. MiR-137 target NEUROD1 was additionally identified as strongly inhibited by brain-specific miR-138, which is compatible with the observation that NEUROD1 expression is highest during differentiation in the cerebral cortex and decreases in maturing neurons [51,52]. Thus, downregulation of miR-137 and -138 during XXL differentiation of USSCs will allow for initial translation of NEUROD1 transcripts, together with other validated miR-138 targets NBEA and NPTX1. In addition, NEUROD1 is transcriptionally induced by miR-17 target NEUROG1 in Xenopus laevis [53]. Synaptogenesis-related NPTX1 acts together with NPTX2, also named NARP [38], a putative target for miR-22 and -27a among our set of 18 microRNAs. NPTX1 was found the most strongly upregulated gene in XXL-USSCs, as shown by the Illumina-derived array data, and expression of NPTX2 was also increased. Interestingly, the housekeeping gene GAPDH was regulated by miR-218.
Although we could only analyze the functional relationship of a limited panel of microRNAs and predicted target proteins, the results demonstrate a clear network-like impact of the group of 18 downregulated microRNAs on proteins with functions related to neuronal development and function. This is further supported by the distribution of predicted microRNAs on 125 genes upregulated in XXL-USSCs (Supplementary Fig. S1), which suggests that microRNAs and target genes are functionally connected in a network woven from overlapping interactions of a limited number of downregulated microRNAs and a large number of putative neuronally relevant target proteins. Indeed, the analysis of the posttranscriptional influence of 3′-UTRs on protein expression by endogenous microRNAs during XXL differentiation (Fig. 3C) clearly indicated microRNA-mediated expression inhibition of at least half of the validated target genes as early as 5 days after XXL induction. Because of losses of transfected vectors, considerable apoptosis occurring during XXL treatment and the observation that XXL-USSCs are not transfectable to sufficient efficiencies, this experiment could not be extended to 14 days after XXL induction, the time point where microRNA expression profiles had been analyzed and the full regulatory impact of the 18 microRNAs is to be expected. Nevertheless, even under these cell culture-based limitations, changes in endogenous microRNA expression early in XXL-mediated differentiation clearly point to a diminishing inhibitory potential of endogenous USSC-microRNAs with regard to the analyzed mRNAs (represented by their 3′-UTRs).
The statistically significant enrichment of putative targets within the fraction of upregulated genes in XXL-USSCs, which was not found in the population of genes upregulated during osteogenic differentiation of USSCs (data not shown), further supports the specificity for neuronal determination of the target gene population. It is noteworthy that upregulation of putative target proteins on the transcriptional level (Fig. 4B) is not directly caused by downregulation of targeting microRNAs but most likely the result of coordinated parallel regulatory triggers.
In addition, our observation that 23.1% of predicted targets were indeed found upregulated in XXL-USSCs (Fig. 4A) is in good agreement with the recent observation that neuronal differentiation of the SH-SY5Y neuroblastoma line mentioned above led to upregulation of 26% of genes predicted as targets for the miR-17-92 family [32]. Interestingly, cross-match analyses of proteins upregulated in both XXL-USSCs and neuronally differentiated SH-SY5Y [32] revealed an overlap of 931 proteins. Given that neuronal developments are observed in these different cell types, this finding indicates that differentiation of both cell types at least in part proceeds with parallel biochemical pathways. This is further substantiated by the observation that >65% of the proteins upregulated in both XXL-USSCs and neuronally differentiated SH-SY5Y cells are putative targets for the group of 18 microRNAs downregulated in XXL-USSCs.
In view of the hitherto limited neuronal functionality of XXL-USSCs, our results should be understood in the context of early stages toward neuronal differentiation. Further, the parallel regulatory events of microRNAs and mRNAs found during the neuronal differentiations of independent cell types will not only allow further insight into mechanisms of neuronal differentiation but also help to validate the molecular differentiation pathways by comparing microRNA expression as guiding molecules to identify proteins of critical functional impact also in other lineage differentiation pathways.
Footnotes
Acknowledgments
The authors express their gratitude to Marion Hendicks for technical assistance and to Jürgen Enczmann, Johannes Fischer, and Fabian Kruse for statistical analyses. This work was supported by grants from the Deutsche Forschungsgemeinschaft (DFG FOR717: WE 505/2-1, MU630/10-1).
Author Disclosure Statement
No conflicts of interest exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.