
Abstract
Induced pluripotent stem (iPS) cells have great potential for regenerative medicine and gene therapy. Thus far, iPS cells have typically been generated using integrating viral vectors expressing various reprogramming transcription factors; nonintegrating methods have been less effective and efficient. Because there is a significant risk of malignant transformation and cancer involved with the use of iPS cells, careful evaluation of transplanted iPS cells will be necessary in small and large animal studies before clinical application. Here, we have generated and characterized nonhuman primate iPS cells with the goal of evaluating iPS cell transplantation in a clinically relevant large animal model. We developed stable Phoenix-RD114-based packaging cell lines that produce OCT4, SOX2, c-MYC, and KLF4 (OSCK) expressing gammaretroviral vectors. Using these vectors in combination with small molecules, we were able to efficiently and reproducibly generate nonhuman primate iPS cells from pigtailed macaques (Macaca nemestrina). The established nonhuman primate iPS cells exhibited pluripotency and extensive self-renewal capacity. The facile and reproducible generation of nonhuman primate iPS cells using defined producer cells as a source of individual reprogramming factors should provide an important resource to optimize and evaluate iPS cell technology for studies involving stem cell biology and regenerative medicine.
Introduction
R
Humanized mouse systems are usually used for studies of iPS cells [34]. However, transgenic murine models do not faithfully mirror human physiology due to the dramatic differences in anatomy, tissue composition, and physiology between these 2 species. For example, there is a >10-fold difference in the heart rates, and a 50-fold difference in age and, thus, a substantial difference in the demand on tissue regeneration between humans and mice [35,36]. In contrast, the genomes of nonhuman primates are highly similar to humans [37]. Given the differences between mice and humans, and the limited long-term follow-up possible with xenogeneic human iPS cell studies in mice, nonhuman primate studies will provide important preclinical safety and efficacy data [38].
Pigtailed macaques (Macaca nemestrina) are commonly used animal models to address preclinical questions [39 –41], and thus could be a good model to study iPS cell-derived therapy against HIV or to address other transplantation-related issues. Therefore, we wished to develop a method to efficiently and reproducibly generate iPS cells from M. nemestrina.
Methods
Reprogramming of human and nonhuman primate cells by transient plasmid/RNA transfection
A lentiviral vector expressing green fluorescent protein (GFP) from a phosphoglycerate kinase promoter (PGK-GFP) was used as sample DNA, and lentiviruses harboring the same construct were used as positive control. The toxicity and efficiency of transfection using reagents including calcium phosphate, polyethylenimine, LipoLTX (Invitrogen), Polyfect and Nanofect (Qiagen), TransIT LT1 and TransIT mRNA (Mirus), on human IMR90 cells (ATCC) were evaluated according to the individual manufacturers' protocols. In addition, the pHAGE-STEMCCA vector (gift from Dr. Gustavo Mostoslavsky) was transfected by Fugene (Roche) into human and M. nemestrina fibroblasts every 2 days for a total of 4 transfections. 5′ capped and 3′ poly A-tailed RNAs that encode OCT4, SOX2, KLF4, and c-MYC were obtained by Illumina TotalPrep RNA Amplification Kit (Ambion). Total mRNAs of M. nemestrina ES (MnES) cells were purified by RNeasy Mini Kit (Qiagen). 50K M. nemestrina CD34+ cells were repetitively transfected with either 8 μg stabilized RNAs (2 μg for each gene) or 8 μg MnES mRNAs twice every 3 days by the human CD34+ Cell 96-well Nucleofector Kit (Lonza). All DNA or RNA-transfected cells were placed on irradiated (4,500 rad) mouse embryonic fibroblast (iMEF) for reprogramming in the MEF-conditioned medium (MEF-CM).
Generation of stable packaging cell lines for production of reprogramming factors
Retroviral vectors, pMIG-Oct4, pMIG-Sox2, pMSCV-c-Myc-IRES-GFP, pMIG-Klf4, pBABE-puro-SV40LT, pBABE-puro-hTERT (all from Addgene), and pMSCV-lin28-IRES-YFP were transfected into Phoenix-GALV and Phoenix-RD114 cells using lipofectamine 2000 (Invitrogen). Virus-containing medium was harvested and used to cross-infect Phoenix-RD114 and Phoenix-GALV cell lines at 6 multiplicities of infection (MOIs), respectively. Cells with 71% transduction efficiency were sorted and single producer clones were expanded. Clones that repeatedly produced high viral titer were selected and used for virus production.
Generation of control human iPS cells by Phoenix-RD114 retroviral vectors
About 1 × 105 IMR90 cells (ATCC) were transduced by Phoenix-RD114-generated retroviruses (OCT4 MO1 = 10, SOX2 MO1 = 10, c-MYC MO1 = 10, KLF4 MO1 = 10, with and without LIN28 MO1 = 2, SV40LT MO1 = 1, hTERT MO1 = 1) twice in the original cell culture. Transduced cells were transferred to ES cell cultures on iMEF feeders 4 days after the second transduction. Cells were fed daily with mTeSR1 medium (StemCell Technologies) plus 2 mM 5′-azaC (Sigma A2385) and 2 mM valproic acid (VPA) (Sigma P6273). Candidate colonies with classic ES cell morphology were continuously expanded at a 1:2 ratio every 7 days on Matrigel with and without iMEF or irradiated (3,000 rad) human fetal fibroblast (iHFF) feeders.
Optimization of reprogramming conditions
Established IMR90-derived human iPS cells maintained in mTeSR1 medium were used to test the capacity of small molecules to prevent spontaneous differentiation. A combination of mitogen-activated protein kinase kinase (MEK) inhibitor 0.5 μM (PD0325901; Stemgent), an inhibitor of the type 1 TGFβ receptor 0.5 μM (ALK5, A8301; Tocris) and 3 μM GSK3β inhibitor (CHIR99021; Stemgent) were tested on human iPS cells without feeders. After ∼2 weeks of chemical treatment, iPS cell morphology was observed by microscopy.
A cocktail of 3 μM CHIR99021, 0.5 μM PD0325901, 2 mM VPA, and 10 μM Rock inhibitor (Y-27632; Calbiochem) that was used to supplement the gammaretrovirus-mediated reprogramming efficiency of nonhuman primate iPS cells was evaluated using oral fibroblasts. The capacity of 3 μM CHIR99021 and 0.5 μM PD0325901 to prevent spontaneous differentiation were tested individually during daily maintenance of M. nemestrina oral fibroblast-derived iPS (MnOFiPS) cells in MEF-CM supplemented with 16 ng/mL human basic fibroblast growth factor (hbFGF).
Generation of M. nemestrina iPS cells
1 × 105 oral fibroblasts from 3 separate M. nemestrina subjects (T09033 female, R08004 male, L08002 male) were transduced with Phoenix-RD114-pseudotyped vectors expressing OCT4, SOX2, KLF4, and c-MYC at an MOI of 5 daily for 2 days. Transduced cells with retroviral vectors were transferred to Matrigel-coated 6-well plates seeded with iMEFs at 5 × 105/well right after the second viral transduction and medium was changed to MEF-CM supplemented with 16 ng/mL hbFGF, 0.5 μM PD0325901, 2 mM VPA, and 10 μM Rock inhibitor Y-27632 for 2 weeks. MnOFiPS cell colonies with typical ES cell morphology (well-defined edges and small cytoplasm/nuclei ratio) appeared on day 9 after viral transduction. Twenty-four randomly selected colonies were expanded by digestion with 0.5 mg/mL dispase or Collagenase IV and picked individually onto iHFF or iMEF feeders with coated Matrigel underneath on day 15. Cells were supplied with MEF-CM plus 16 ng/mL hbFGF. Reprogramming efficiency was calculated by dividing the number of alkaline phosphatase positive colonies by the number of seeded GFP+ fibroblasts. The use of mTeSR2 medium (STEMCELL Technologies) and XF/FF medium (Stemgent) without hbFGF was also tested on MnOFiPS cells without feeders with human ES (hES) cells serving as control. Expression of Oct4 and Tra-1-81 was quantified in both medium conditions without feeders.
Alkaline phosphatase and immunocytochemistry staining of ES cell markers
Approximately 3 × 104 human iPS cells and MnOFiPS cells per well were cultured for 4 days in 4-well chamber slides (Lab-Tek II) coated with Matrigel. Chamber slides were mounted and counterstained using Vectashield Mounting Medium with 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories). SSEA1, Oct4, SSEA3, and SSEA4 antibodies were obtained from Millipore. Tra-1-60 and Tra-1-81 antibodies were from Chemicon International. Oct3/4 and Nanog antibodies were obtained from Abcam. Alkaline phosphatase staining was done per the manufacturer's recommendations (Millipore).
Flow-cytometric analysis of Oct4, SSEA3, SSEA4, SSEA1, Tra-1-60, Tra-1-81, and CD34
Human iPS cells and MnOFiPS cells were treated in Collagenase IV for 7 to 10 min followed by cell dissociation buffer (Gibco) treatment for 10 min at 37°C. Cells were rinsed and repeatedly triturated to a single cell suspension in phosphate-buffered saline plus 3% fetal bovine serum (FBS); cell clumps were removed by 40 μm cell strainer filtration. Oct4-FITC, SSEA4-PE, and SSEA1 Cy5/PE staining was performed according to the manufacturer's protocol (Millipore). Rat anti-SEA3 antibody (mAb clone MC-631) was obtained from Develop Studies Hybridoma Bank, and rabbit anti-Nanog primary antibody (ab21603) was obtained from Abcam. Anti-human CD34-PE antibody was from BD Biosciences. Live cells were identified by 7-aminoactinomycin D exclusion and analyzed for surface-marker expression using flow-cytometric analysis (Calibur; BD Biosciences). The data were analyzed by FlowJo software (Tree Star).
Quantitative reverse transcription–polymerase chain reaction and genomic polymerase chain reaction analysis
Total RNA from MnOFiPS cells and MnOFs was extracted by RNeasy Mini Kit (Qiagen) according to the manufacturer's instructions. cDNA synthesis was performed with 5 μg total RNA by the first-strand cDNA synthesis kit (Amersham Biosciences) and 20 ng cDNA from each sample was used as template in triplicate reactions. Transgene-specific primers and endogenous gene-specific primers were designed to quantify relative expression levels between retrovirally expressed genes and endogenous genes, respectively. Genomic DNA (gDNA) from MnOFiPS cells and MnOFs was isolated by QIAamp DNA Blood Mini Kit (Qiagen), and 35 ng gDNA of each sample was amplified via transgene-specific primers in triplicate. Expression of OCT4, SOX2, c-MYC, and KLF4 was determined by quantitative polymerase chain reaction (PCR; Applied Biosystem) using SYBR green (Invitrogen) DNA-binding dye. Quantitative PCR reaction conditions were primary denaturation at 95°C for 10 min and 40 cycles of PCR consisting of 95°C for 15 s and 55°C for 1 min, followed by analysis of the amplified products using the dissociation curves. The signal intensities were normalized against GAPDH, and the 2-ΔΔCt equation was used to calculate the relative gene expression. The primers used are listed in Supplementary Table S1 (Supplementary Data are available online at
DNA microarray
Total RNA was isolated from MnOFiPS cells, MnOFs, MnES cells, and hES cells (H9) using RNeasy kit with DNase treatment (Qiagen). RNA probes for microarray hybridization were prepared and hybridized to Rhesus Macaque Gene expression kit (Agilent) according to the manufacturer's protocol. Microarrays were scanned and data were analyzed using Agilent Genomic Workbench software provided by our Shared Resources at FHCRC.
Short tandem repeat analysis
PCR was used to amplify across discrete genomic intervals containing highly variable numbers of tandem repeats (VNTR D4S2366) to verify the genetic relatedness of MnOFiPS cell lines relative to their parent fibroblasts. Primers were 6FAM-TCC TGA CAT TCC TAG GGT GA and AAA ACA AAT ATG GCT CTA TCT ATC G. gDNAs of MnOFiPS cells, hES cells (H9), MnES cells, and MnOF were isolated by QIAamp DNA Blood Mini Kit (Qiagen). A total of 50 ng of gDNA was used per reaction, preheated 94°C for 5 min, cycled 40 times through 94°C 10 s, 57°C 30 s, and 68°C 1 min, followed by 68°C 3 min, and analyzed by Applied Biosystems Genemapper 4.0 software. Qualitative determinations were made based on amplicon mobility for primer set: D4S2366, average heterozygosity 1.
Southern blotting
gDNA from MnOFiPS cells and MnOF was isolated by QIAamp DNA Blood Mini Kit (Qiagen), and was digested with the restriction enzymes BglI and EcoRI that cleave within the 389 probe region of the OCT4 gene and randomly within the gDNA, respectively, to produce detectable fragments. After digestion, ∼10 μg DNA was electrophoresed, transferred to a Hybond-N+ membrane (GE Healthcare), and hybridized using QuickHyb hybridization solution (Stratagene) following manufacturers' recommendations. The 389 bp OCT4-probe was generated via PCR amplification from 10 pg vector plasmid DNA and gel-purified. The probe was radioactively labeled using a Rediprime II DNA Labeling System (GE Healthcare) before hybridization.
Bisulphite genomic sequencing
gDNA from MnOFiPS cells and MnOF was isolated by QIAamp DNA Blood Mini Kit (Qiagen), and was treated by DNA methylation Bisulfite Assay Kit, CpGenome (Chemicon). PCR primers are listed in Supplementary Table S1. PCR was programmed as 95°C for 5 min, followed by 40 cycles of 95°C 1 min/58°C 1 min/72°C 1 min and 1 cycle of 72°C 10 min. PCR products were ligated into the pCR4.TOPO vector (Invitrogen) and sequenced with an M13F primer. Sequencing was analyzed using DNAdynamo software.
Karyotype analysis
Chromosomal studies were performed at the Cytogenetics Laboratory University of Washington Medical Center using standard protocols for high-resolution G-banding.
Proliferation assay, clonogenic assay, and telomerase activity
The total number of MnOFiPS cells was counted every 7 days and compared with the total number of hES cells (H9). MnOFiPS cells and hES cells were sorted for the Tra-1-81+ population, and 1 × 104 cells were seeded onto iHFF-coated plates. The number of typical ES cell-like colonies was counted 2 weeks later. Telomerase activity of both MnOFiPS cells and MnOFs was detected using a TRAPEZE telomerase detection kit (Millipore) according to the manufacturer's instructions. The samples were separated by TBE-based 10% acrylamide nondenaturing gel electrophoresis.
hbFGF dependence assay
MnOFiPS cells and hES cells H9 were maintained in MEF-CM with hbFGF (16 and 8 ng/mL, respectively) on Matrigel-coated plates. The presence of the stem cell marker Oct4 was quantified by flow cytometry every 7 days.
Formation of embryoid bodies
At 90% confluence, MnOFiPS cells maintained on Matrigel were harvested after collagenase IV treatment. The cell clusters were transferred at a ratio of 3:1 to low attachment 6-well plates in Dulbecco's modified Eagle's medium (DMEM)/F12 containing 20% knockout serum replacement (Invitrogen), 2 mM L-glutamine, 0.1 mM nonessential amino acids (NEAA), 0.1 mM 2-mercaptoethanol (Invitrogen), and 0.5% penicillin and streptomycin. The medium was changed daily. After 14 days as a floating culture, embryoid bodies (EBs) were paraffin-embedded and 4-μm sections were cut for immunohistochemistry staining using smooth muscle actin (SMA, #M0851; Dako), glial fibrillary acidic protein (GFAP, #Z0334; Dako), and α-fetoprotein (AFP, MAB1368; R&D Systems). Rhesus heart, brain, and human placenta were used as positive controls for SMA, GFAP, and AFP staining, respectively. Concentration matched isotype control slides were run to evaluate background staining.
Neural cell differentiation
PA6 feeder cells (Riken Cell Bank) were plated on gelatin-coated 6-well plates and incubated for 4 days. Small clumps of MnOFiPS cells were seeded on a PA6-feeder layer in Glasgow minimum essential medium (Invitrogen) containing 10% knockout serum replacement (Invitrogen), 0.1 mM NEAA, 0.1 mM 2-mercaptoethanol (Invitrogen), and 0.5% penicillin and streptomycin. Ten days after differentiation, A2B5 was used to detect neural precursors by immunohistochemistry with A2B5 antibodies (R&D Systems,) and Alexa fluor 488 goat anti-mouse IgM (Invitrogen). Chamber slides were mounted and conterstained using Vectashield Mounting Medium containing DAPI (Vector Laboratories).
Cardiomyocyte differentiation
MnOFiPS cells were maintained on a Matrigel-coated plate in MEF-CM supplemented with 16 ng/mL hbFGF and 30 ng/mL insulin-like growth factor (IGF) for 6 days. Cells were dissociated by Collagenase IV and cell clumps were resuspended in STEMPRO-34 SFM medium (Invitrogen) plus 200 mM L-glutamine supplemented with 5 μL/mL Transferrin (Roche), 0.039 μL/mL 1-thioglycerol (Sigma), 50 μg/mL ascorbic acid (Sigma), and 50 ng/mL bone morphogentic protein 4 (BMP4) (R&D Systems) to form EBs under 5% O2 hypoxic conditions. Growth factors were changed to 1 μg/mL BMP4, 6 ng/μL FGF, 5 ng/mL Activin A (R&D Systems), and 50 μg/mL ascorbic acid (Sigma) 24 h later and maintained for another 3 days. EBs were dissociated by 0.05% Trypsin via 20G syringe to single cells and plated at a density of 750,000 cells/6-well plate on growth factor reduced Matrigel (BD) in STEMPRO medium supplemented with 200 mM L-glutamine, 5 μL/mL Transferrin, 0.039 μL/mL 1-thioglycerol, and 50 μg/mL ascorbic acid for 4 days. Growth factors were removed after cells reached full confluence and the cell culture was switched to 20% O2 normoxia conditions. Cells were fed every 2 days for 9 days, and beating cells were video-recorded as well as stained by SMA (ab5694; Abcam) via immunohistochemistry.
Hepatocyte differentiation
MnOFiPS cells were maintained on 5% growth factor-reduced Matrigel (R&D Systems) to reach a confluence of 70%. The medium was replaced with RPMI1640 medium plus B27 supplement, 100 ng/mL activin A, and 50 ng/mL Wnt3a (R&D Systems) for 3 days (medium changed every day). Wnt3a was eliminated for the next 2 days. The medium was then changed to Knockout-DMEM containing 20% SR, 1 mM L-glutamine, 1% NEAA, 0.1 mM β-mercaptoethanol (all from Invitrogen), and 1% dimethyl sulfoxide (Sigma) for 7 days. The cells were finally cultured in L-25 medium supplemented with 8.3% FBS, 8.3% tryptose phosphate broth, 10 μM hydrocortisone 21-hemisuccinate, 1 μM insulin (all from Sigma), 2 mM glutamine, 10 ng/mL hepatocyte growth factor, and 20 ng/mL oncostatin M (both from R&D Systems). The medium was changed daily for 7 days during differentiation. Albumin was detected by immunohistochemistry (Abcam).
Hematopoietic differentiation
OP9 stromal cells were maintained in α-MEM supplemented with 20% FBS. OP9 cells were seeded on 0.1% gelatin-coated 10 cm plates until full confluence was reached. At this time, the medium was changed and cells were cultured for additional 4 days. The medium was then changed to α-MEM supplemented with 10% FBS just before MnOFiPS seeding. About 1.5–2 × 106 MnOFiPS cells were treated with 1 mg/mL collagenase IV and dispersed by scraping to maintain the cells in small clumps but not single cells and seeded per 10 cm dish. Unattached cells were aspirated the next day by adding 10 mL α-MEM supplemented with 10% FBS. Half of the culture medium was changed on the fourth, sixth, and eighth days.
Hematopoietic differentiation via EB differentiation was performed with MnOFiPS cells expressing GFP from a lentiviral vector. EBs of GFP+ MnOFiPS cells were supplemented with knockout-DMEM, 10% FBS, 10 ng/mL IL-3, 10 g/mL IL-6, 25 ng/mL BMP4, 50 ng/mL granulocyte colony-stimulating factor (G-CSF), 300 ng/mL SCF, 300 ng/mL FLt-3, and 1% pen-strep. The medium was changed every 4 days.
Cells co-cultured with OP9 were treated with 5 mL Collagenase IV for 20 min, followed by trypsin-EDTA for 15 min. EBs were dissociated with Collagenase B and cell dissociation buffer. Cells were analyzed by flow cytometry analysis (CD34 and CD45, both from BD).
Three thousand sorted CD34+ cells were plated into 35-mm plates MethoCult (Stemcell Technologies) for colony forming unit (CFU) assay. Twenty days after plating, colonies were picked, processed by cytospin, and Giemsa-stained for microscopy analysis.
Teratoma formation
MnOFiPS and human iPS cell clusters were prepared and injected into testis of male nonobese diabetic/severe combined immunodeficient (NOD/SCID/γc null) mice as previously described [42]. Briefly, 2 × 104 cells from various expanded iPS cell clones were injected into the testis capsules of mice aged weeks 5–6. Six weeks after the initial injection, formed teratomas were quantified by both size and weight. Teratomas were extracted and fixed with 10% buffered formalin and embedded in paraffin. Sample sections (5 μm thickness) were stained with hematoxalin and eosin for microscopic analysis. All experiments using mice have received approval from the Institutional Animal Care and Use Committee at FHCRC.
Statistical analysis
Results were presented as mean ± standard error of the mean. Statistical significance was determined using the unpaired Student's t-test and results were considered significant or highly significant when P was ≤ 0.05 or ≤0.01, respectively.
Results
Reprogramming of human and nonhuman primate cells with DNA plasmid and RNAs
We first attempted to use transient plasmid transfection by calcium phosphate, polyethylenimine, and other commercial reagents to generate iPS cells from human and nonhuman primate fibroblasts. We experienced unstable transfection and significant toxicity and, thus, were unable to establish iPS cells (Supplementary Fig. S1A). We also explored the use of Fugene6, with pHAGE-STEMCCA, a centivirus vector that expresses OCT4, SOX2, c-MYC, KLF4 which has been used to generate mouse iPS cells without viral integration [43]. Instead of generating iPS cells, use of pHAGE-STEMCCA/Fugene resulted in the reprogramming of fibroblasts into adipocytes as indicated by oil red O staining (Supplementary Fig. 1B). Similarly, transfection of M. nemestrina CD34+ cells with either synthesized RNAs (OSCK) or total mRNAs from MnES cells by Nucleofection yielded only multiple adipocytes and no iPS cell colonies (Supplementary Fig. 1C).
Establishing stable packaging cell lines for production of reprogramming factors
Gammaretroviral vectors have been widely used to generate iPS cells. In most cases, however, transient transfection was used to produce gammaretroviral vectors expressing reprogramming factors that lead to variable production and expression of the individual factors. To circumvent this problem we developed stable retrovirus-producing cell lines using Phoenix-RD114 and Phoenix-GALV cells [44] (Fig. 1A). Vectors produced by these 2 cell lines target different cell surface receptors, and thus, a wide range of different tissues can be infected. Vectors produced by the RD114 cell line have the additional advantage that they can be efficiently concentrated by high-speed centrifugation to improve titer and, thus, reprogramming efficiency. We first compared the transduction efficiency of Phoenix-RD114 and Phoenix-GALV-based viruses on IMR90 HFFs using a vector that expresses EGFP. At an MOI of 5, Phoenix-RD114 viruses yielded about 4 times higher transduction efficiency (∼89%) than Phoenix-GALV vectors (23%) (Fig. 1B). This transduction efficiency was also higher than using viruses produced by transient transfection (∼20%) [6]. Thus, gammaretroviral vectors produced from Phoenix-RD114 cell lines were used for subsequent experiments.
We generated 7 Phoenix-RD114 stable producer cell lines producing gammaretroviral vectors expressing OCT4, SOX2, KLF4, c-MYC, LIN28, hTERT, and SV40LT. These RD114-pseudotyped vectors could be efficiently concentrated to produce high-titer retroviral stocks (107 to 108/mL) (Fig. 1C). The generation of these retroviral stable producer cell lines eliminated the requirement for transfection reagents during vector production and allowed for a facile generation of concentrated virus-containing medium for reprogramming without contaminating residual transfection reagents. In addition, the availability of individual, defined producer cell lines for the different reprogramming factors allows for studies to replace individual reprogramming factors with small molecules.
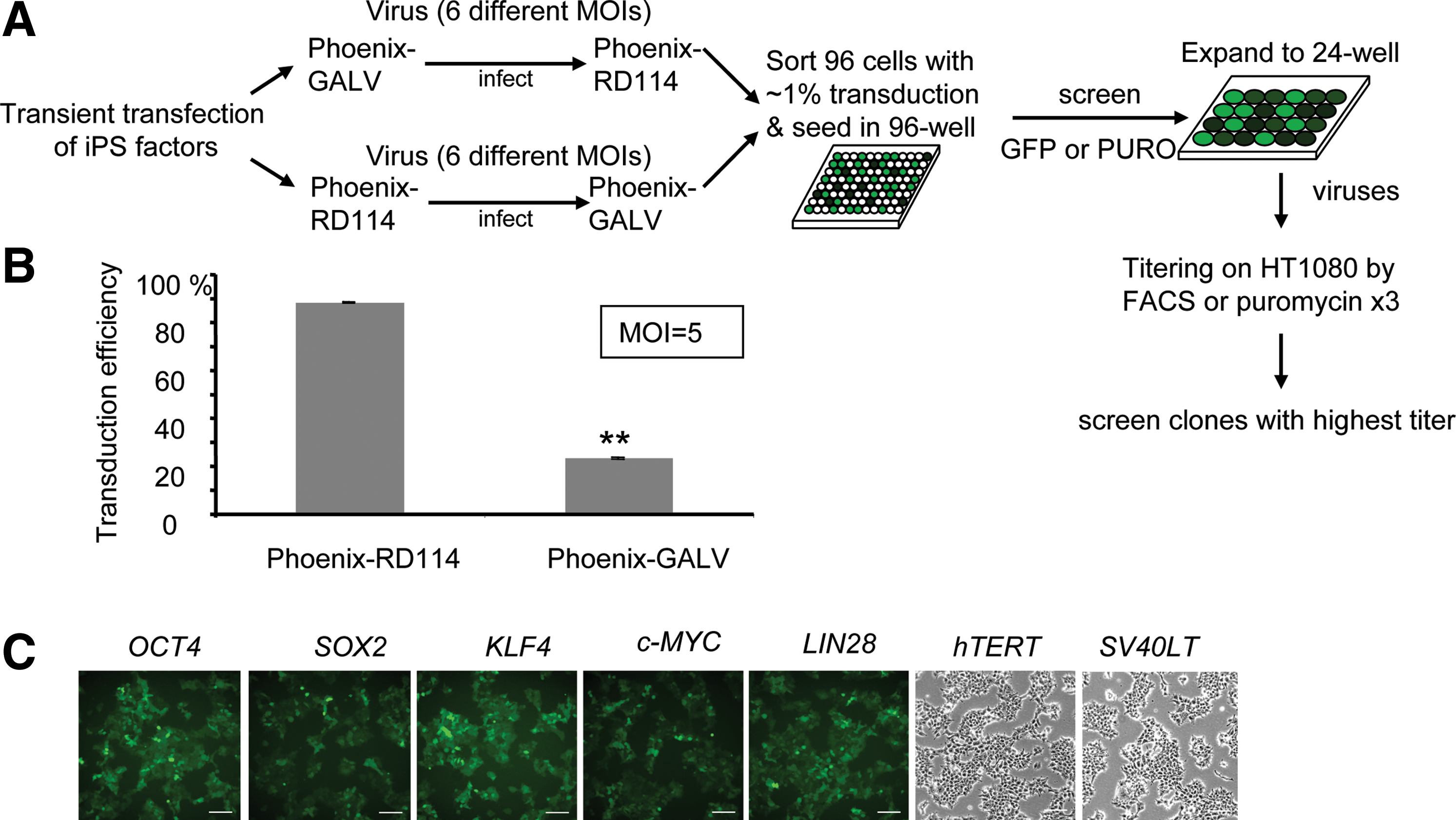
Generation of stable retrovirus producing packaging cell lines.
Generation of control human iPS cells
We first evaluated our reprogramming vectors and small molecules using human cells. Reprogramming vectors expressing OSCK produced by stable Phoenix-RD114 producer cell lines were able to infect 80% of human IMR90 fibroblasts when transduced at an MOI of 10 (Supplementary Fig. S2A, B). With the addition of VPA and Rock inhibitor Y-27632 for the first 7 days, human IMR90 cells yielded ES-cell like colonies by 32 days after transduction (Supplementary Fig. 2C, D). Addition of factors LIN28, hTERT, and SV40LT did not further increase the reprogramming efficiency. We also did not observe any improvement with the addition of BIX-01294, a selective inhibitor of G9a histone methyltransferase, which has been reported to enhance the reprogramming efficiency (data not shown) [28]. Three OSCK reprogrammed hES cell-like colonies were further expanded and analyzed for expression of stem cell markers and by teratoma assay to confirm their pluripotency (Supplementary Fig. 2E, G). Addition of several small molecules, which inhibit spontaneous differentiation in mouse ES cell culture, were evaluated for the generation of human iPS cells. Human iPS cells exhibited improved, well-defined, edge morphology when cultured without feeders with the addition of both 0.5 μM PD0325901 (a MEK inhibitor) and 3 μM CHIR99021 (a GSK3β inhibitor), suggesting their capacity to limit spontaneous differentiation (Supplementary Fig. 2F). ALK5, an inhibitor of the type 1 TGFβ receptor, had no noticeable effect with regard to preventing differentiation and was therefore not used in subsequent experiments. We noticed that using Phoenix-RD114 OSCK viruses in combination with PD0325901 and 3 μM CHIR99021 resulted in colonies without granules, which is indicative of true ES cell-like colonies [6]. This appeared to be an additional benefit of Phoenix-RD114 vectors for reprogramming.
Generation of nonhuman primate iPS cells
We next generated iPS cells from pigtailed macaques (M. nemestrina), a commonly used nonhuman primate model. Oral fibroblasts (Fig. 2A) transduced with the OSCK Phoenix-RD114 retroviral vectors had 40%–80% GFP+ transduction efficiency. In the presence of VPA and Rock inhibitor Y-27632 for the first 7 days plus inhibitor (0.5 μM PD0325901) and GSK3β inhibitor (3 μM CHIR99021), hundreds of ES cell-like colonies were first observed by day 9 after transduction and were picked (Fig. 2B) and passed to new iMEFS on day 15 (Fig. 2C). Compared with parallel reprogramming of M. nemestrina mesenchymal stem cells via pHAGE2-hSTEMCCA lentiviruses (gift from Dr. Gustavo Mostoslavsky) under the same conditions that yielded iPS cells with background granulated colonies within 9 days (unpublished data), every single colony generated by Phoenix-RD114 viruses showed a flat ES-cell like morphology without background granulated colonies. From the oral fibroblasts, we can get an average reprogramming efficiency of 0.2%–0.8% based on the ratio of the number of alkaline phosphatase positive ES cell-like colonies to the number of viral transduced fibroblasts (about 80 to 400 alkaline phosphatase+ colonies out of an average 5 × 104 GFP+ fibroblasts). Twenty-four MnOFiPS cell candidate colonies were randomly selected and screened for pluripotency. We also noticed that the addition of 0.5 μM MEK inhibitor (PD0325901) without GSK3β inhibitor (3 μM CHIR99021 was sufficient to prevent differentiation of MnOFiPS cell candidates in cultures with MEF-conditioned medium. This shortened reprogramming process was successfully repeated using mesenchymal stem cells isolated from 2 other pigtailed macaques (unpublished data), suggesting that this protocol is reliable and efficiently generates nonhuman primate iPS cells.
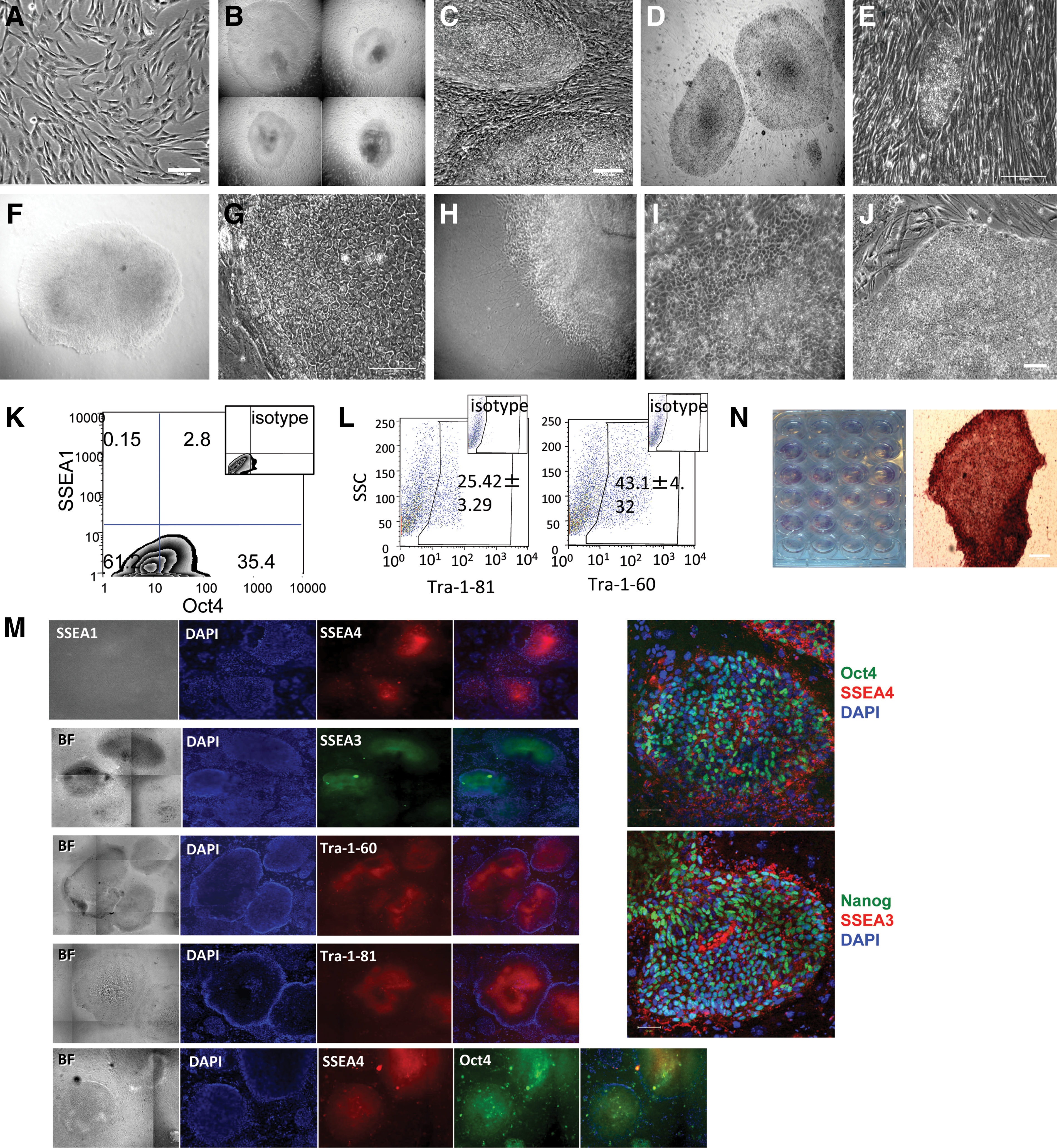
Generation of nonhuman primate (Macaca nemestrina) iPS cells.
Maintenance of nonhuman primate iPS cells
All nonhuman primate iPS cell candidate colonies maintained a well-defined edge when cultured on iMEF and iHFF through at least 20 passages (Fig. 2D, E). However, when transferred to feeder-free Matrigel culture conditions supplemented with either MEF-CM containing 0.5 μM MEK inhibitor PD0325901 and hbFGF (16 ng/mL) or mTeSR1, only a small proportion of nonhuman primate iPS cells maintained undifferentiated morphology (Fig. 2F, G), whereas a large proportion of cells changed morphology and spontaneously differentiated (Fig. 2H, I). Further, neither mTeSR2 (a newer defined medium for maintenance of hES cells) nor XF/FF medium (a defined xeno-free, low growth factor hES medium) supported the maintenance of an undifferentiated morphology of MnOFiPS cells and hES cells for >5 passages, when the cells were passed onto feeder-free Matrigel-coating plates (Supplementary Fig. S3A). The XF/FF medium, however, performed better than mTeSR2 medium with regard to preventing spontaneous differentiation of both MnOFiPS and hES cells as shown by the quantification of Oct4+ and Tra-1-81+ populations over time (Supplementary Fig. S3B). However, the addition of 30 ng/mL IGF or an increased concentration of hbFGF (200 ng/mL) in MEF-CM with the addition of 0.5 μM PD0325901 resulted in stable maintenance of an undifferentiated morphology of the MnOFiPS cells using feeder-free culture conditions (Fig. 2J).
Characterization of nonhuman primate iPS cells
Using a small tandem repeat assay, we confirmed that 3 randomly selected MnOFiPS cell clones shared the same DNA fingerprints with their parental oral fibroblasts, thereby eliminating the possibility of contamination with existing hES cells and monkey ES cells being cultured in the laboratory (Supplementary Fig. S4). Southern blot analysis revealed that each MnOFiPS cell clone had multiple transgenes with a unique integration pattern, demonstrating that these MnOFiPS cell clones were derived from individual fibroblasts (Supplementary Fig. S5).
MnOFiPS cell candidates were further analyzed using multiple pluripotent stem cell markers by flow cytometry and immunohistochemistry. We detected Oct4, Tra-1-81, Tra-1-60, SSEA3, and SSEA4, on every clone of candidate MnOFiPS cells, and all clones were negative for SSEA1 (Fig. 2K–M). Overlap staining confirmed Oct4, Nanog, SSEA3, and SSEA4 positivity (Fig. 2M). All candidates also exhibited active alkaline phosphatase activity (Fig. 2N). Reverse transcription–PCR analysis also confirmed the reactivation and expression of endogenous nonhuman primate OCT4, SOX2, and KLF4 (Fig. 3A–C) in clone 6 and 7. Transgene expression from the integrated gammaretroviral vectors were silenced in the established MnOFiPS cell candidates, as indicated by reverse transcription–PCR using transgene-specific PCR primers and diminished GFP signal (Fig. 3C). Expression of c-MYC did not vary markedly from the parental oral fibroblasts. PCR of gDNA from MnOFiPS using transgene-specific primers confirmed stable integration of retroviral OCT4, SOX2, KLF4, and c-MYC (Fig. 3D). Global messenger RNA expression analysis of MnOF, their reprogrammed derivative MnOFiPS cells, MnES cells, and hES cells (H9) revealed that MnOFiPS cells are transcriptionally highly close but not exactly identical to MnES cells, and both are dramatically different from their parental fibroblasts (Supplementary Fig. S6). MnOFiPS cells are also quite different from hES cells (H9), which may be due to the rhesus macaque-specific array we used. Specifically, the expression of OCT4, SOX2, and NANOG was substantially upregulated (16-fold, 1201-fold, and 176-fold increases, respectively) in the MnOFiPS cells in comparison with MnOF (Supplementary Fig. S6; Supplementary Tables S2 and S3).

Expression of transcription factors in MnOFiPS cells and MnOFs.
Reprogramming of somatic cells is accompanied by demethylation of promoters of pluripotency genes such as OCT4. Our bisulphate sequencing confirmed that multiple sites in the promoter regions of OCT4 are highly methylated in MnOFs, but little methylation was detected in the same regions of MnOFiPS cells and control ES cells, indicating that demethylation of promoters of pluripotency genes occurred in these iPS cells (Fig. 4).
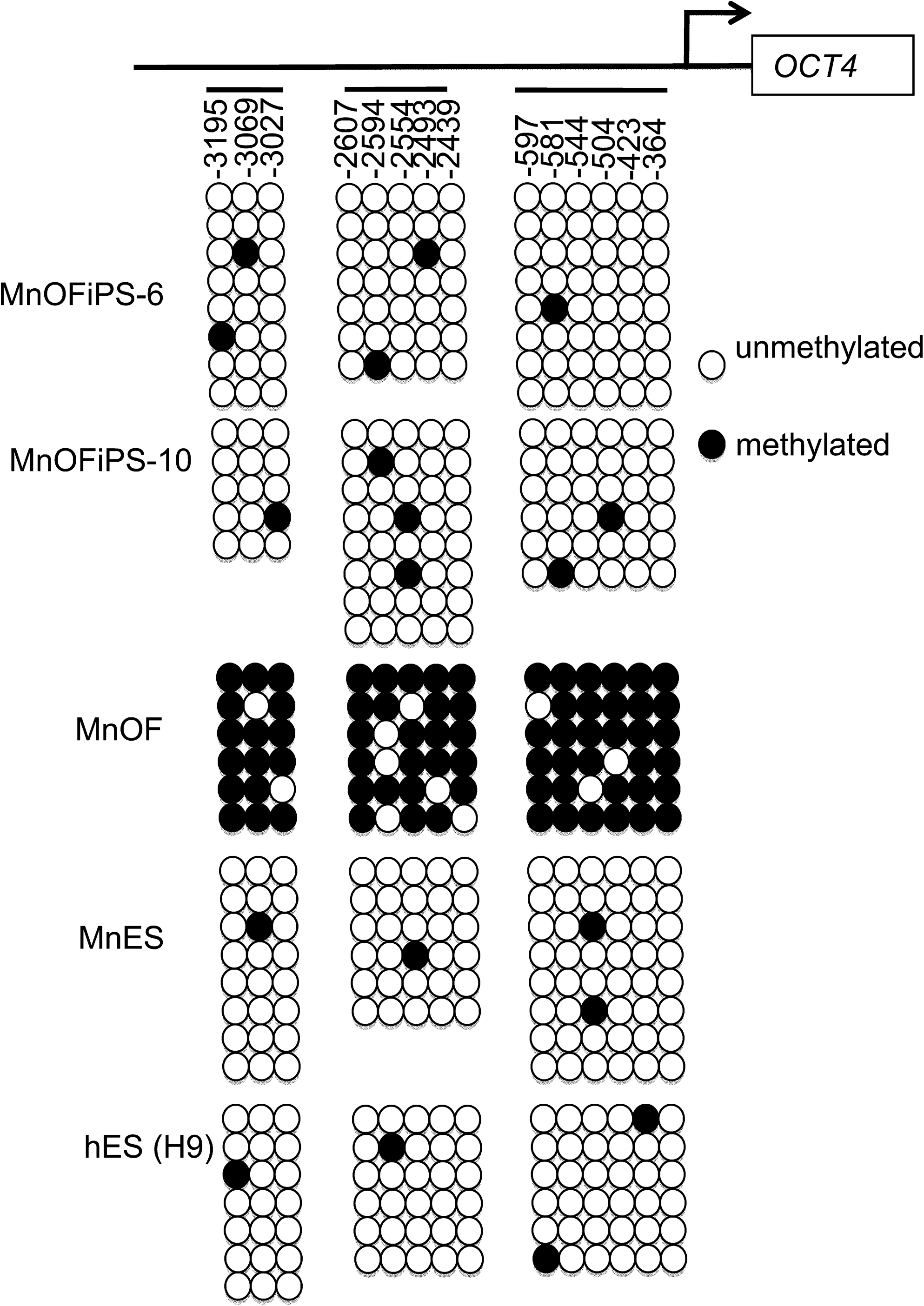
Bisulfite genomic sequencing of the promoter regions of OCT3/4 in MnOFiPS cells clone 6, clone 10, as well as in MnOF, MnES cells, and hES cells (H9). MnES, M. nemestrina embryonic stem.
Another feature of stem cells is the loss of telomerase activity as they differentiate toward somatic cells. As predicted, the reprogrammed MnOFiPS cells had restored high telomerase activity in comparison with their parental oral fibroblasts (Supplementary Fig. S7A). The self-renewal and cell cycle properties of these MnOFiPS cells were confirmed by cumulative growth curves and clonogenic assays. Both hES cells and MnOFiPS cells revealed a similar ∼4-fold increase in the number of cells accumulating over 21 days in culture (Supplementary Fig. S7B), and both cell lines have a similar 0.4% Tra-1-81+ clonogenic ability (Supplementary Fig. S7C). The MnOFiPS cells proliferated exponentially for at least 12 months and still exhibited normal karyotype (Supplementary Fig. S8A). In comparison, the parental fibroblasts lost the ability to efficiently proliferate after 4 passages. hES cells required 8 ng/mL hbFGF to maintain an undifferentiated state, whereas MnOFiPS cells required 16 ng/mL hbFGF to prevent spontaneous differentiation (Supplementary Fig. S8B).
Pluripotency of nonhuman primate iPS cells
To examine the developmental potential of nonhuman primate iPS cells, in vitro differentiation assays were performed. Differentiation of MnOFiPS cells via bulk EB formation yielded cells representing all 3 embryonic germ layers indicated by tissue-specific immunohistochemistry staining of SMA (cardiac muscle, mesoderm), GFAP (neurons, ectoderm), and AFP (liver, endoderm), which confirms the pluripotency of MnOFiPS cells in vitro (Fig. 5A–D).
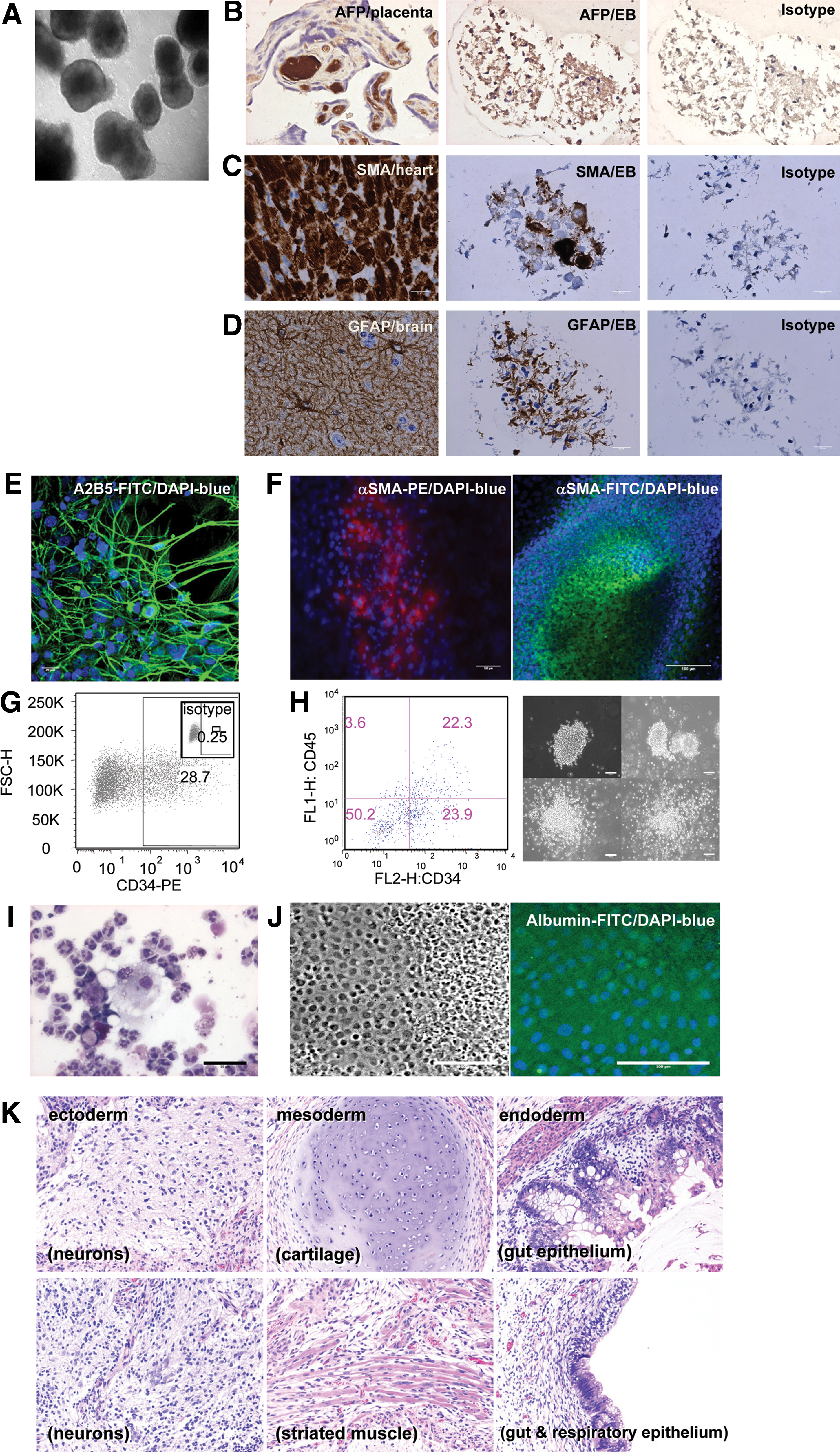
Pluripotency of MnOFiPS cells (clone 6).
Direct in vitro differentiation of MnOFiPS cells using culture conditions designed to generate neurons routinely generated A2B5-positive neural precursors (Fig. 5E). Coculture with OP9 stromal cells differentiated MnOFiPS cells toward a CD34+ cell type, and EB differentiation of MnOiPS cells with cytokines generated CD34+ CD45+ hematopoietic stem cells with CFU capacity (Fig. 5G, H), and the differentiated colonies contained white cells (Fig. 5I). MnOFiPS cells were also differentiated into SMA-positive cardiomyocytes (Fig. 5F, Supplementary Video S1) and albumin+ hepatocytes (Fig. 5J) in vitro. Further, after being transplanted into NOD/SCID/γc null mice, multiple MnOFiPS cell clones formed teratomas consisting of representative derivatives of all 3 germ layers, including neurons (ectoderm), cartilage, and muscle (mesoderm), and gut and respiratory epithelium (Fig. 5K), which confirmed their pluripotency in vivo. These in vitro and in vivo characterizations confirmed that our nonhuman primate iPS cells generated by viral vectors closely resemble ES cells in terms of pluripotency, marker expression, and differentiation potential.
Discussion
Here we demonstrate the efficient generation of nonhuman primate iPS cells from M. nemestrina using gammaretroviral vectors expressing OCT4, SOX2, c-MYC, and KLF4 (OSCK) produced by individual stable RD114-based vector producing cell lines. Nonintegrating methods such as transient transfection of plasmid DNA or RNA were inefficient in generating iPS cells in our study. The availability of defined, stable vector producing cell lines for the individual reprogramming factors will facilitate efforts to replace reprogramming factors with small molecules.
The inability to generate iPS cells using transient DNA or RNA transfection may be caused by inadequate/unstable transgene expression during the reprogramming process. We did repetitively observe adipocytes using this approach, suggesting that adipocytes can be a transient stage toward stemness during the reprogramming process, consistent with the recent success using adipocytes as a cell source to generate iPS cells [15]. Thus, we presume adipocytes could be a more appropriate target to investigate better nonintegrating methods of iPS cell reprogramming.
Our results show that nonhuman primate MnOFs can be efficiently reprogrammed into iPS cells using human stem cell transcription factors. We were able to show efficient reprogramming with 4 Phoenix-RD114 gammaretroviral vectors expressing OCT4, SOX2, KLF4, and c-MYC. The generation of these retroviral stable cell lines eliminates the requirement of transfection reagents and laborious DNA preparations, and more importantly, they reprogram both human and nonhuman primate iPS cells without generating granulated colonies. Thus, these stable gammaretroviral vector producing lines should be useful tools for generating iPS cells from many other species and tissue types.
We included a small molecule Rock inhibitor and histone deacetylase (VPA) based on reports from other publications [26,45]. We also found that the addition of both GSK3β inhibitor CHIR99021 and MEK inhibitor PD0325901 can reduce the spontaneous differentiation of human iPS cells. The GSK3β inhibitor can mimic the activation of Wnt signaling and maintain the pluripotent state of mouse ES cells. It has recently also been shown that the GSK3β inhibitor can replace SOX2 during the reprogramming of human iPS cells [30]. The MEK inhibitor has been shown to de-differentiate mouse pre-iPS cells into fully reprogrammed pluripotent cells [46]. In our study, we found that the MEK inhibitor PD0325901 can effectively reduce the spontaneous differentiation of nonhuman primate iPS cells when they are cultured on irradiated MEFs. In the presence of the PD0325901, the required concentration of hbFGF was 16 ng/mL, which is substantially less than the 200 ng/mL reported in the literature [47]. We also found that IGF can efficiently prevent the spontaneous differentiation of nonhuman primate iPS cells cultured without feeder cells, which will facilitate in vitro differentiation studies.
In summary, here we established stable, defined RD114-based retroviral vector producing cell lines for the generation of individual gammaretroviral vectors expressing the 4 main reprogramming factors, OCT4, SOX2, c-MYC, and KLF4. Transduction of human and nonhuman primate oral fibroblasts in the presence of small molecules resulted in the efficient and reproducible generation of iPS cells without any background granulated colonies, and the efficiency of reprogramming nonhuman primate cells (0.2%–0.8%) is particularly higher than other reports (0.03%) [7]. Our reprogramming protocol resulted in true nonhuman primate iPS cells within 9 days after viral transduction, which is 2 weeks shorter than reported by others (26–29 days [7] and 4–5 weeks [48]). Recently, with our viruses and protocol, fibroblasts from a p53-deficient patient were able to be reprogrammed into iPS cells within 6 days after viral transduction (unpublished data). Therefore, these shortened time frames suggest the possibility of developing nonintegrating methods with high reprogramming efficiency. In addition to the vector producing lines generated in this study, we also optimized the reprogramming conditions for nonhuman primate cells. The vector producing cell lines will also benefit studies attempting to find the optimal ratio of reprogramming factors by replacing individual factors with small molecules. The iPS cells generated in this study will also allow further transplantation of iPS cell-derived tissues in a clinically relevant large animal model.
Footnotes
Acknowledgments
We thank Anis Feki (Département de Gynécologie and Obstetrique, Hôpitaux Universitaires de Genève) for providing HFF feeders. We thank Carol Ware from the ISCRM Stem Cell Core at the University of Washington for providing hES cells and MnES cells. At FHCRC we thank Julie Randolph-Habecker in Experimental Pathology for assistance with the EB and teratoma immunohistology assays, Julio Vazquez Lopez at the Scientific Imaging Center, and the staff of the DNA array core facility. We also acknowledge the help from the Core Center for Excellence in Hematology (Michael Harkey and Cyd Nourigat).
Author Disclosure Statement
For all authors, no competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.