
Abstract
Stem cell-based spiral ganglion neuron (SGN) replacement therapy has been proposed to be a promising strategy to restore hearing either via replacing degenerated neurons or by improving the efficacy of cochlear implants which rely on functional neurons. However, lack of suitable donor cells and low survival rate of implanted cells are the major obstacles to successful implementation of therapeutic transplantation. The present study investigated the potential of mouse inner ear statoacoustic ganglion (SAG)-derived neural progenitors (NPs) to differentiate toward SGN-like glutamatergic cells and the influence to cell survival and differentiation when nerve growth factor (NGF) was supplied. We found that SAG-NPs could form neurospheres, proliferate, and differentiate into cells expressing neuronal protein neurofilament and β-III tubulin. NGF affected the cell fate of SAG-NP in a concentration-dependent manner in vitro. Low concentration of NGF (2–5 ng/mL) promoted cell proliferation. Medium concentration of NGF (20–40 ng/mL) stimulated cells to differentiate into bi-polar SGN-like cells expressing glutamatergic proteins. High concentration of NGF (100 ng/mL) could rescue cells from induced apoptosis. In the in vivo study, NGF (100 ng/mL) dramatically enhanced SAG-NP survival rate after implantation into adult mammalian inner ear. This finding raises the possibility to further induce these NPs to differentiate into SGN-like neurons in future in vivo study. In conclusion, given the capability of proliferation and differentiation into SGN-like cells with the supplement of NGF in vitro, SAG-NPs can serve as donor cells in stem cell-based SGN replacement therapy. NGF improved the survival of SAG-NPs not only in vitro but also in vivo.
Introduction
T
Despite the promising potential, many problems still need to be addressed in SGN replacement research. One of the dominant problems is to seek the appropriate donor cells. To functionally integrate into the auditory system, donor cells should have the capacity to differentiate into SGN phenotype. Embryonic stem cells (ESCs) [6 –10], ESC-derived neural stem cells (NSCs) [11,12], and brain-derived NSCs [13 –15] have been introduced into the adult mammalian inner ear. Currently, induction of these donor cells to differentiate into SGN-like cells usually requires gene transfection [9,13]. However, this is not recommended if clinical application is an eventual goal, as it could potentially cause genome mutation. Therefore, the donor cells that have intrinsic potential to become inner ear cells should be identified. This leads to intensive interest in inner ear-derived stem cells that are ready to adopt an inner ear cell fate without the need for gene transfection or viral involvement [16 –18]. Our current study explored the potential of statoacoustic ganglion (SAG)-derived neural progenitors (NPs), progenitors of SGNs, as donor cells, which has never been reported.
Another key problem in SGN replacement study is the low survival rate of implanted cells. The inner ear is a tiny fluid-filled compartment. Due to the anatomical size of the inner ear, only a few microliters of cell suspension could be delivered into the cochlea, which largely restricts the number of injected cells [13,19]. However, successful application of SGN replacement therapy requires a considerable number of implanted cells to survive for sufficient attempts to adopt SGN cell fate in the host microenvironment and subsequently replace the function of damaged or lost SGNs. Currently, the survival rate of implanted cells is very low. Less than 1% ESCs or NSCs were found to survive in the inner ear [7,20]. Thus, improving survival rate of implanted cells becomes a timely issue that needs work up. Lack of growth factors at the transplantation site may contribute to the low survival of implanted cells. Thus, it might be logical to speculate that providing exogenous growth factors to the inner ear may enhance the survival rate of implanted cells. In our previous study, we observed that exogenous nerve growth factor (NGF) could improve the survival of the embryonic neurons which have been implanted into the adult mammalian inner ear [21]. A recent study demonstrated that infusion of 2 growth factors into the inner ear led to robust survival of implanted ESCs [9]. However, it is uncertain whether a single neurotrophic factor could enhance stem cell survival after implantation into the inner ear.
In this study, we investigated (1) whether SAG-NPs could differentiate into SGN phenotype in vitro and in vivo and (2) whether NGF improved SAG-NP survival and differentiation in vitro and after implantation into the adult mammalian inner ear.
Materials and Methods
SAG-cell culture
All animal study procedures were approved by local Institutional Animal Care and Use Committee. SAGs were obtained from embryonic day 12.5 mice (E12.5, E0.5 was determined when vaginal plug was found in wild-type C57/BL6 or eGFP-C57/BL6 mice, Jackson Laboratory), as previously described [22]. The pregnant mice were euthanized by overdose of CO2. The SAGs were carefully dissected from embryos and transferred to a new culture dish containing Dulbecco's modified Eagle's medium (DMEM)/F12 (Invitrogen). To avoid potential contaminating cells, the surrounding tissue was carefully removed using a sapphire knife. The remaining central SAG tissue was subsequently rinsed twice in fresh DMEM/F12. The SAGs were cut into small pieces and dissociated in 1 mL DMEM (Invitrogen) containing papain (20 U), N-acetyl-
To obtain a cell clone, the SAG-cell number was evaluated using a hemocytometer. The cell density was adjusted to ∼100 cells/mL, from which a 10 μL cell suspension was added to each well of a 96-well plate containing 90 μL of serum-free medium (DMEM/F12, 1% B27, 2% N2, 20 ng/mL basic fibroblast growth factor, and 20 ng/mL epidermal growth factor, all from Invitrogen) [16]. The wells containing solitary cells were identified, and the cells in those wells were expanded to obtain a cell clone at 37°C in humidified 5% CO2 and 95% air. The cells were passaged using TrypLE (Invitrogen) when the neurospheres were over 50%–70% confluence in the suspension culture (usually weekly) for several months. The cell samples were taken at passage 4–6 and cultured on 0.1% gelatin (Millipore) coated on 96-well or 24-well plates to allow cells to attach for the following assays.
Proliferation assay
To explore whether SAG-cells could grow in serum-free medium, SAG-cells (passage 4) were cultured in the presence of 3 μg/mL 5-bromo-2-deoxyuridine (BrdU; Sigma) for 24 h during culture days 3–4 [16]. To study BrdU incorporation, the cells were fixed in 4% paraformaldehyde followed by DNA denaturization in 1 M HCl for 30 min at room temperature. The samples were blocked in 10% donkey serum and 0.2% triton X-100 for 30 min before mouse anti-BrdU antibodies (BD) were applied over night at 4°C. Cy3-conjugated secondary antibodies (Jackson ImmunoResearch) were added to the samples to visualize cells using a Leica epifluorescence microscope with appropriate filters.
Differentiation assay
To test the differentiation of cloned SAG-cells, the cells were cultured in serum-free medium containing DMEM/F12, B27, and N2 for 7 days. At culture day 7, the samples were fixed in 4% paraformaldehyde. Mouse anti-neurofilament-H (RNF402; Santa Cruz Biotechnology) and rabbit anti-β-III tubulin (Tuj1; Covance) antibodies were used to label the cells expressing β-III tubulin and neurofilament-H proteins, which are usually expressed in neurons [23,24]. Mouse anti-vesicle glutamatergic transporter 1 (VGluT-1; Millipore) antibodies were used to detect cells expressing glutamatergic protein VGluT-1 [9]. Mouse anti-nestin (Millipore) and rabbit anti-Sox2 (Abcam) were used to detect cells expressing multipotent NP protein nestin and Sox2 [25,26]. Mouse anti-glial fibrillary acidic protein (GFAP; Covance) antibodies were used to detect cells expressing glial cell protein GFAP. Cy3 or cy5 (Jackson ImmunoResearch)-conjugated secondary antibodies and appropriate filters were used to visualize cells.
Neurotrophic factor study
To test whether a single neurotrophic factor could affect the cell fate of SAG-cells, either NGF or brain-derived neurotrophic factor (BDNF) was added to the serum-free medium (DMEM/F12, B27, and N2) at concentrations of 0, 1, 2, 5, 10, 20, 30, 40, 50, and 100 ng/mL, or glial cell-derived neurotrophic factor (GDNF) at concentrations of 0, 5, 10, 20, 40, 50, 100, 150, and 200 ng/mL (all from Invitrogen). Each culture condition contains 6 replicates. The cells were dissociated, and the cell number was evaluated using a hemocytometer. The cell density was adjusted to ∼1,000 cells/mL. A 10 μL cell suspension was plated into each well of a 96-well plate containing 90 μL of serum-free medium (DMEM/F12, B27, and N2) with the above-mentioned neurotrophic factors. Half of the culture medium was replaced every 2–3 days. To study whether neurotrophic factors affected cell growth, the CyquantNF cell proliferation kit (Invitrogen) was used. The viable cell number was counted at the beginning of culture and 7 days after culture using either a microplate reader (Molecular Devices) or a Leica epifluorescence microscope. To explore whether neurotrophic factors affected cell differentiation, the above-mentioned differentiation assays were used to evaluate the differentiation of SAG-cells. One-way analysis of variance was used to analyze the statistical difference of cell growth or differentiation in various concentrations of each neurotrophic factor. P<0.05 was considered as statistically significant in this study.
Neurite outgrowth assay
To evaluate neurite projections, the newly generated neurite outgrowths of 3 randomly selected cells in the central area of each of the 6 samples were visualized using a digital camera under a 20×objective. The longest neurite outgrowth was measured if the cells had multiple processes. The neurites were determined using the following criteria: labeled with the antibodies against β-III tubulin or neurofilament-H; and being longer than 25 μm (usually>the cell body). SigmaScan Pro software was used to measure neurite outgrowths as was previously described [21]. The average length of neurite outgrowths was calculated in each neurotrophic factor group.
In vitro apoptosis and neuroprotective assay
To improve cell survival, it is crucial to rescue stem cells from apoptosis, because it is reported that ∼17% of implanted stem cells were apoptotic at 24 h after implantation into the nervous system [27]. In this study, we tested the ability of neurotrophic factor to protect SAG-cells from apoptosis in vitro, which may subsequently provide a cue to the in vivo study. Since the neurotoxin staurosporine (STS) has been reported to generate apoptotic condition in NSC culture [28], it was used in this study as an apoptosis inducer.
To evaluate whether STS-treatment time affected induced apoptosis, we incubated SAG-cells in serum-free medium (DMEM/F12, B27, and N2) containing 30 nM STS (Sigma) for 1 and 4 h, respectively. To test whether STS-treatment had a concentration-dependent effect on apoptosis, 50 nM STS was added to the culture medium for 4 h. At the end of STS treatment, the SAG-cells were rinsed with DMEM/F12, cultured for 4 h in fresh serum-free medium, followed by CyquantNF assay to evaluate the viable cell number using a Leica epifluorescence microscope with appropriate filters. APO-BrdU terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay kit (Invitrogen) was applied to determine the apoptotic cells according to the manufacturer's instructions.
To test whether NGF could rescue SAG-cells from induced apoptosis, 0, 5, 20, 50, 100, or 150 ng/mL NGF was added to the SAG-cell culture medium for 1 h before STS treatment. Then, the cells were cultured in the medium containing NGF and STS for another 1 h. At the end of STS treatment, the SAG-cells were rinsed with fresh DMEM/F12 and incubated in fresh serum-free medium for 4 h. The CyquantNF assay was applied to evaluate the viable cell number and to assess the neuroprotective effects exerted by NGF. Each culture condition contains 6 replicates. Two-way analysis of variance was used to analyze the variance of viable cells in different NGF concentration groups. P<0.05 was considered to be statistically significant.
Implantation surgery
Twelve 3-month-old female Sprague Dawley rats were used in the study, 6 rats in the NGF group and the other 6 animals in the control group. Under deep anesthesia, the left temporal bone was exposed and opened to visualize the basal cochlear turn. A small cochleostomy (diameter ∼200 μm) was made into the scala tympani at the basal cochlear turn [13,21]. Five microliters of cell suspension containing ∼50,000 eGFP-SAG-cells were transplanted into the scala tympani of each rat via a microsyringe. In NGF group, NGF (100 ng/mL, in artificial perilymph) was simultaneously infused into the inner ear through the round window via a catheter that was connected to a mini-osmotic pump subcutaneously secured to the back of rat [21,29]. The artificial perilymph that contained (in mM) NaCl, 137; KCl, 2.8; CaCl2, 1.5; NaH2PO4, 8.0; MgCl2, 1.0; KH2PO4, 4.7; glucose, 11.0; and pH 7.4 [21,29] was infused into the cochleae of animals in the control group.
Histology and immunohistochemistry
The animals were euthanized using an overdose of pentobarbital and were heart perfused at 2 weeks after surgery [7,13]. The cochleae were dissected out and cryosectioned at ∼12 μm thickness using established stereological methods [21,30]. The above-mentioned anti-neurofilament-H, anti-β-III tubulin, and anti-VGluT1 antibodies were used to detect the implanted cells that had differentiated into cells expressing neuronal or glutamatergic proteins.
Microscopy and data analysis
The eGFP-SAG-cells were detected by eGFP fluorescence or antibody specific for eGFP [7,13]. The cell survival rate was calculated as (number of surviving cells)/(number of implanted cells)×100% [7,13]. Student's t-test was used to determine whether NGF exerted neuroprotective effects on SAG-cells after implantation into the inner ear.
Results
Characterization of SAG-cells
Neurosphere formation
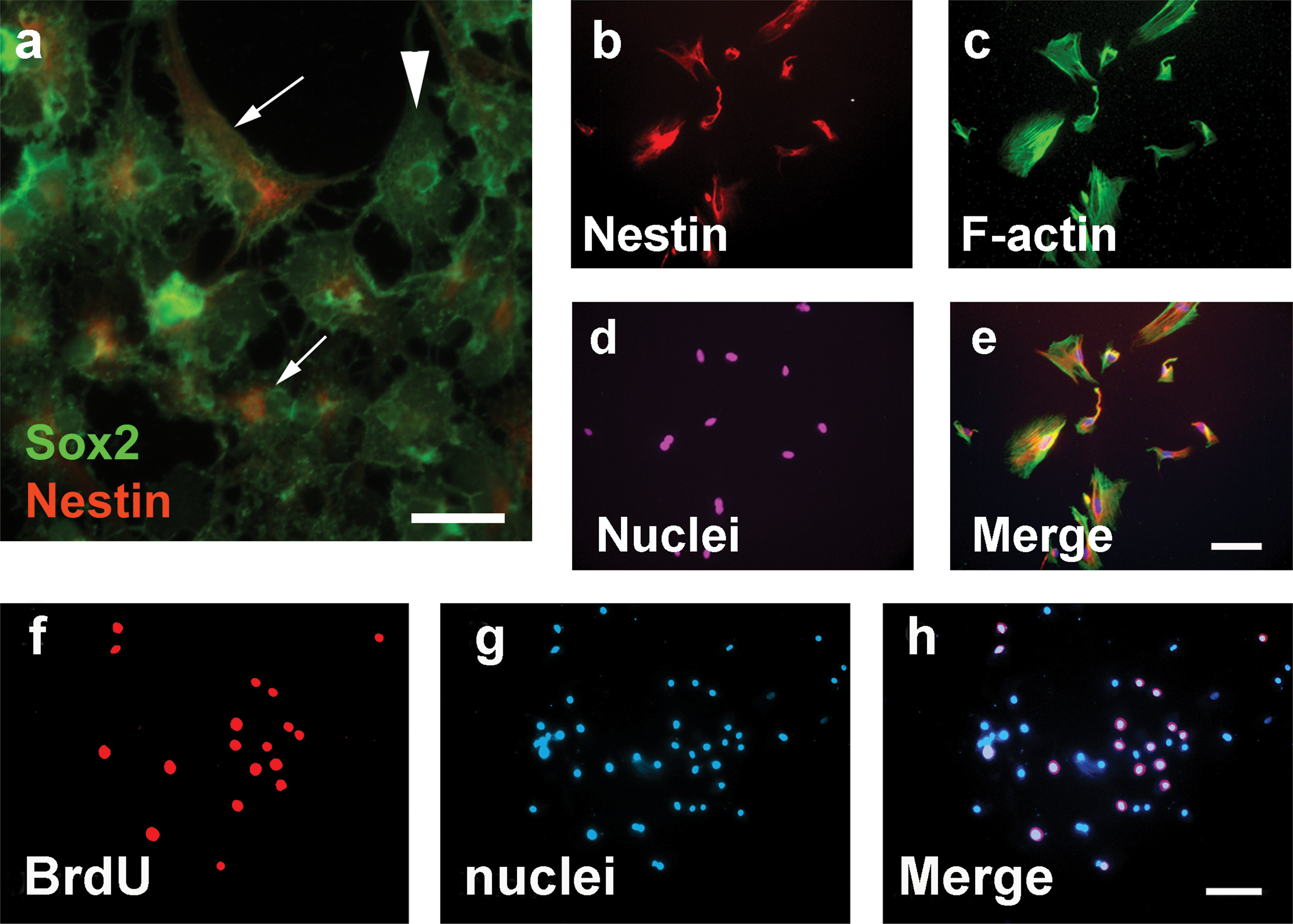
After being plated into a 96-well plate, solitary E12.5 SAG cells were identified in 8 wells, whereas multiple cells were found in 75 wells and zero cell in 13 wells. Among those 8 wells containing solitary cells, the solitary SAG cells in 4 wells were observed to grow and form spheres at days 6–8 in the suspension culture containing serum-free medium (Fig. 1). A subsequent study confirmed that dissociated, solitary cells from the late stage embryonic SAG or postnatal cochlear/vestibular ganglion could form spheres. When cultured in serum-free medium for 3 days, the cells from the sphere expressed nestin and Sox2 (Fig. 2a–e) proteins, which are usually expressed in multipotent neural progenitors [25,26].
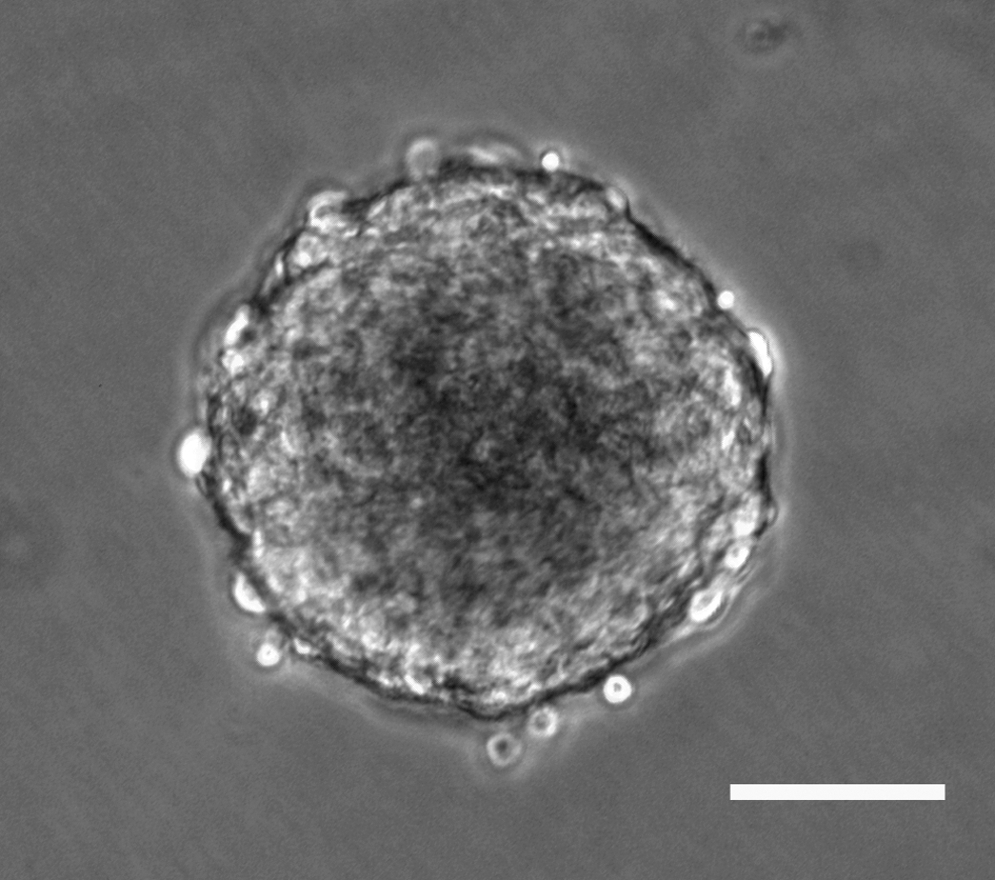
Solitary E12.5 statoacoustic ganglion (SAG) cells formed a sphere at days 6–8 in the suspension culture containing serum-free medium. Scale bar: 50 μm.
Proliferation
The SAG-derived cells could grow in the suspension culture for at least 9 passages (∼51–68 days). SAG-cells were found to incorporate BrdU (Fig. 2f–h), suggesting S-phase entry of these cells.
Differentiation
When SAG-derived spheres were cultured in serum-free medium containing DMEM/F12, B27, and N2 for 7 days, the majority of the cells were found to differentiate into cells expressing glial cell protein GFAP, whereas a small proportion of the cells (4%) differentiated into cells expressing β-III tubulin. We did not find any cells simultaneously expressing neuronal protein and glial cell protein. Since these SAG-cells could proliferate, form cloned spheres, express neural progenitor proteins (nestin and Sox2), and differentiate into cells expressing neuronal or glial cell proteins (refer to B2), they were termed as SAG-derived neural progenitors (SAG-NPs) in this study.
Neurotrophic factor affects SAG-NP cell fate in vitro
Low concentration of NGF enhanced SAG-NP cell growth in vitro
To determine whether a single neurotrophic factor could enhance stem cell growth, we cultured SAG-NPs in serum-free medium containing neurotrophic factors including NGF, BDNF, or GDNF at a series of concentrations. At the initiation of culture, ∼10 cells were found in each culture well. During culture days 3–5, no remarkable cell number difference was observed among the groups. On culture day 7, we found that low concentration of NGF (5 ng/mL) significantly enhanced the cell growth of SAG-NP in vitro, compared with BDNF or GDNF (P<0.05) (Table 1). After day 7, the cell confluence is usually over 70%–80%, which is too high to evaluate normal cell growth. Thus, all samples were fixed for histology study at culture day 7.
NFs, including NGF, BDNF, or GDNF, were added to SAG-NP culture medium (Dulbecco's modified Eagle's medium/F12, B27, and N2) for 7 days. The viable cell number shown in the table was evaluated by CyquantNF assay. It was found that 5 ng/mL NGF had a significant effect on cell growth (P<0.05).
BDNF, brain-derived neurotrophic factor; GDNF, glial cell-derived neurotrophic factor; NF, neurotrophic factor; NGF, nerve growth factor; SAG-NP, statoacoustic ganglion-neural progenitor; std, standard deviation; —, not applicable.
Medium concentration of NGF stimulated SAG-NPs to differentiate into cells expressing neuronal proteins
When cultured for 7 days in the presence of 30 ng/mL NGF, over 85% of SAG-NPs differentiated into cells expressing neurofilament-H and β-III tubulin proteins, which are usually expressed in neurons (Fig. 3a–c). Approximately ∼4%, 25%, 50%, 60%, and 25% of the cells differentiated into cells expressing β-III tubulin and neurofilament-H when the NGF concentration was 0, 10, 20, 40, and 50 ng/mL, respectively (Table 2 and Fig. 3g). This suggests that NGF may stimulate the neuronal differentiation of SAG-NPs in a concentration-dependent manner. BDNF or GDNF could stimulate neuronal differentiation as well but the effect was less notable than did NGF (Table 2).
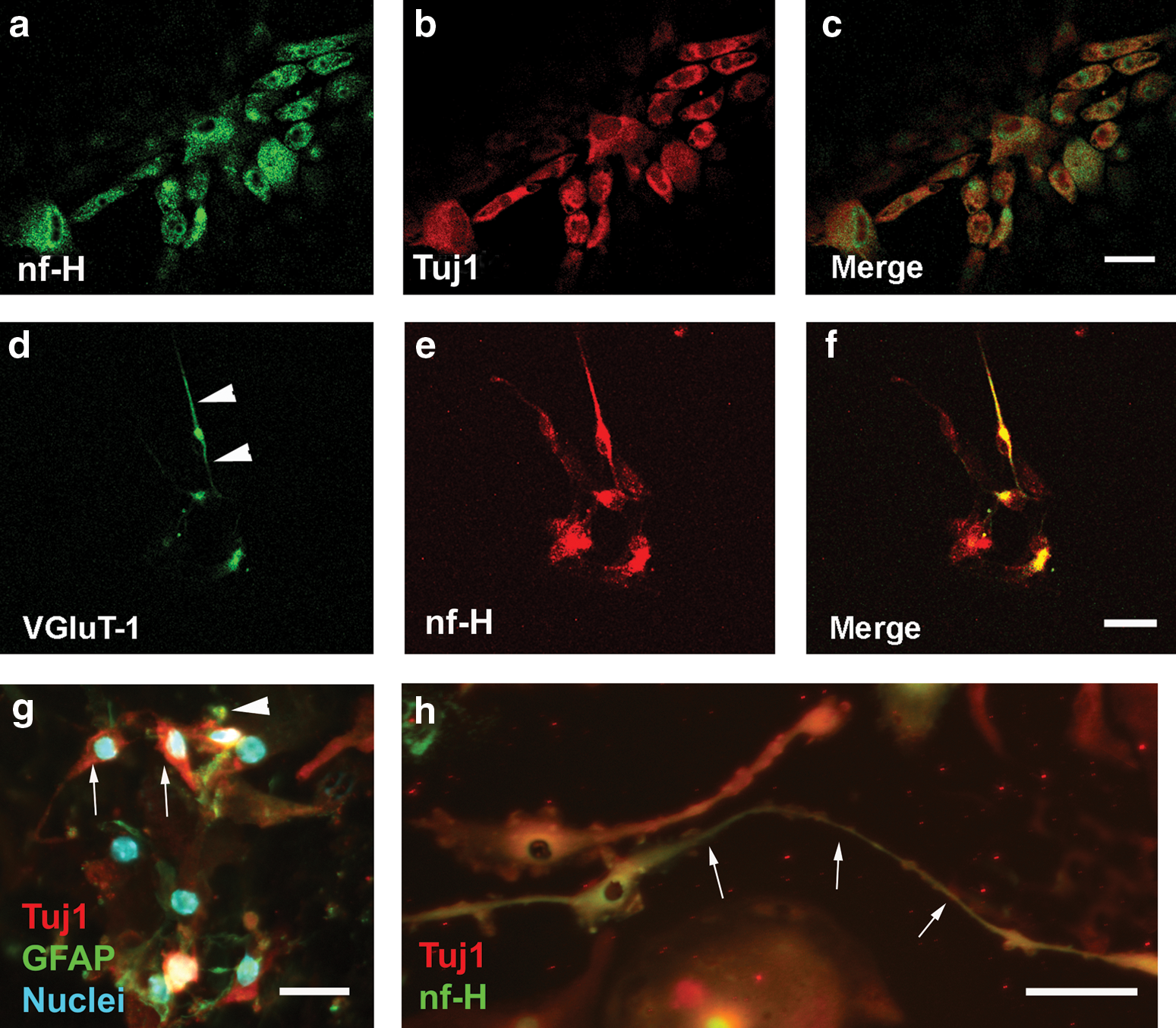
The neuronal differentiation of SAG-NP was determined by the expression of nf-H and Tuj1 proteins, which are usually detected in neurons. It was found that 30 ng/mL NGF induced a significant number of SAG-NPs to differentiate into cells expressing both nf-H and Tuj1 (P<0.05).
nf-H, neurofilament-H; Tuj1, β-III tubulin; Tuj1+, β-III tubulin positive; nf-H+, neurofilament-H positive; —, not applicable.
NGF stimulated SAG-NPs to differentiate into cells expressing glutamatergic neuronal proteins: To test whether NGF stimulated glutamatergic neuronal differentiation, we added 20 ng/mL NGF to SAG-NP culture medium for 7 days. We found that SAG-NPs differentiated into cells expressing not only neurofilament-H but also VGluT-1 (Fig. 3d–f), suggesting that NGF can either stimulate SAG-NPs to differentiate into SGN-like cells expressing glutamatergic proteins or maintain the survival of differentiated cells that express neurofilament-H and VGluT-1. BDNF or GDNF had less effect in terms of glutamatergic neuronal differentiation and/or survival.
NGF stimulated neurite outgrowth of SAG-NP-derived cells: To examine whether NGF stimulates neurite outgrowth from SAG-NP-derived cells, we cultured SAG-NPs in serum-free medium containing NGF, BDNF, or GDNF for 9 days. We found that significant outgrowths (labeled by neurofilament-H or Tuj1) were projected from the cells in NGF supplying group (20–40 ng/mL) (Fig. 3h), whereas no obvious neurite outgrowths were observed in the cultures supplied with BDNF or GDNF. The neurite outgrowth length (average±standard deviation) was 31±6, 45±14, or 34±7 μm when 20, 30, or 40 ng/mL NGF was supplied to the culture medium. This result suggests that NGF may stimulate neurite outgrowths from SAG-NP-derived cells expressing neuronal proteins.
High concentration of NGF rescued SAG-NPs from STS-induced apoptosis
To investigate whether NGF could rescue SAG-NPs from apoptosis and to optimize NGF concentration, we applied NGF to SAG-NP cultures that contained apoptosis inducer.
Generation of apoptosis using STS: STS was used to induce apoptosis, and the degree of apoptosis was assessed by TUNEL assay and cell counting. It was found that SAG-NP cell number decreased in STS-treated cultures in a time- and concentration-dependent manner. After the treatment of 30 nM STS for 1 h, ∼40% SAG-NPs were apoptotic based on APO-BrdU TUNEL assay (Fig. 4a–c), whereas the viable cell number was reduced by ∼60% when the cells were treated by 30 nM STS for 4 h. When SAG-NPs were cultured with 50 nM STS for 4 h, the viable cell number was decreased by ∼70%–80%. It seems that 30 nM STS treatment for 1 h would generate a mild apoptotic condition in SAG-NP culture, which would be applied to the following study.
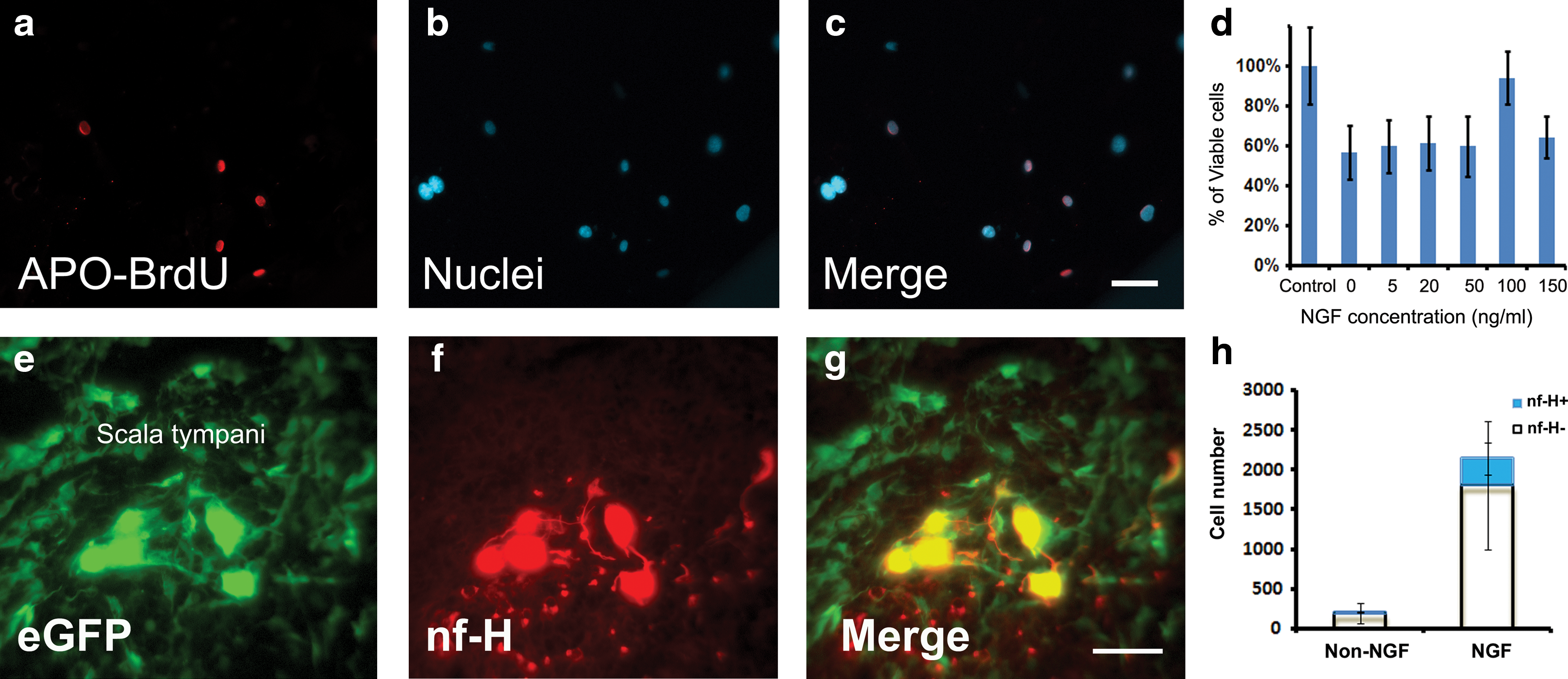
NGF rescued SAG-NPs from induced apoptosis: When 0, 5, 20, 50, and 150 ng/mL NGF was added to the culture medium for 1 h before 30 nM STS treatment, a significant SAG-NP cell number decrease was found compared with the control group without STS treatment (P<0.05), suggesting that these concentrations of NGF may not have remarkable neuroprotective effects. Compared with the control group without STS treatment, no significant cell number decrease was observed when SAG-NPs were pretreated with 100 ng/mL NGF for 1 h before 30 nM STS treatment (P>0.05), indicating that pretreating SAG-NPs with 100 ng/mL NGF might rescue SAG-NPs from STS-induced apoptosis (Fig. 4d).
NGF improves SAG-NP survival after implantation into the adult mammalian inner ear
Identification of implanted SAG-NPs
The implanted SAG-NPs were found in all 12 animals that had been implanted with SAG-NPs. These surviving cells were observed to emit strong green fluorescence using fluorescence microscopy with appropriate filters (Fig. 4e). The surviving SAG-NPs were located within the scala tympani at the implantation site. No obvious sign of infection or immune rejection was identified.
Survival
To investigate whether NGF enhanced implanted stem cell survival in vivo, we implanted SAG-NPs into the adult rat inner ears supplemented with 100 ng/mL NGF. We found that ∼10-fold more SAG-NPs (∼2,100 cells/ear) survived in the inner ears at 2 weeks after implantation (Fig. 4h) (P<0.05). The majority of surviving SAG-NPs were located within the scala tympani at the implantation site.
Differentiation
The immunohistochemistry study showed that ∼16% of the surviving cells differentiated into cells expressing neurofilament-H in the NGF group, whereas it was 7% in the non-NGF group (Fig. 4e–h) (P<0.05). However, no implanted SAG-NPs were observed to differentiate into cells expressing glutamatergic neuronal proteins.
Discussion
Our study demonstrated that SGN progenitors, SAG-NPs, could proliferate and differentiate into SGN-like cells expressing glutamatergic proteins in the presence of NGF in vitro. In the in vivo study, this is the first time that cochlea-derived SGN progenitor cells are introduced back into the cochlea. SAG-NPs have many features that make them highly promising as donor cells for a cell replacement therapy. First, SAG-NPs are biologically safe with low incidence of tumor formation or unlimited growth. The implantation of pluripotent ESCs has been reported to form teratomas in multiple organs, such as joint [31], heart [32], liver [33], pancreas [34], and brain [35], which significantly limited the application of this new therapy. The risk of tumor formation is closely related to the potency of implanted stem cells; thus, in vitro predifferentiation was reported to reduce the tumorigenecity of the implanted cells [36,37]. It is further supported by the observation that teratoma formation is often seen in pluripotent ESC implantation whereas rarely seen in multipotent tissue-derived stem cell implantation [38]. SAG-NPs are tissue-derived, multipotent neural progenitors that could grow for limited passages. Thus tumor formation or uncontrolled growth is not a concern. Second, SAG-NPs are inner ear-derived cells that are destined to differentiate into inner ear cell fate [16 –18]. Our current experiment confirmed that SAG-NPs could differentiate into cells expressing glutamatergic proteins in the presence of NGF in vitro without additional intervention. Third, inner ear tissue-derived stem cells are accessible. Inner ear tissues can be obtained from surgery by the translabyrinthine approach of acoustic Schwannoma. These tissues include utricle, ampulla, and vestibular ganglion. It is possible to identify stem cells from adult mammalian inner ear tissue [17], which could be utilized for in vitro research and implantation study.
The implanted stem cell survival remains a challenge in cell replacement therapy. We found that NGF affected SAG-NP cell fate in a concentration-dependent manner in vitro. Low concentration of NGF (2–5 ng/mL) promoted cell growth, medium concentration of NGF (20–40 ng/mL) stimulated cell differentiation, and high concentration of NGF (100 ng/mL) could prevent cells from STS-induced apoptosis. We are uncertain of the underlying mechanisms but speculate that low concentration of NGF may mainly work on high affinity NGF receptor TrkA, whereas medium or high concentration of NGF also reacts with low affinity receptors. Variable intracellular signaling pathways might also be involved in these events. In this study, STS, which has been used to induce apoptotic conditions in NSC culture [28], was applied to the NGF neuroprotective study. To confirm whether NGF-exerted neuroprotective effect is universal, other neurotoxins and apoptotic conditions should be tested in future investigations.
It is interesting that in contrast to BDNF or GDNF, NGF initiated better outcomes in SAG-NP cell growth and neuronal/glutamatergic differentiation in vitro, suggesting that NGF may be the lead neurotrophic factor in the induction of SAG-NPs to grow or to develop into SGN-like cells expressing glutamatergic proteins. However, this observation is not consistent with the previous studies showing that BDNF, NT-3, and their high affinity receptors TrkB and TrkC are critical to SGN development and/or survival [39 –41]. In these studies, the effect of BDNF and NT-3 was demonstrated in the mutant mice in which normal development of SGN might have been compromised. In addition, the relatively high concentration of NGF may interact with low affinity receptors to achieve cell growth and differentiation that is rarely seen in normal developmental studies.
In this study, we observed that NGF could stimulate SAG-NPs to become SGN-like bipolar cells expressing glutamatergic proteins in vitro, but no glutamatergic differentiation was detected after implantation into the adult mammalian inner ear. To induce SGN development in vivo, we will provide additional small molecules that will potentially guide implanted SAG-NPs to differentiate into cells expressing neuronal and glutamatergic proteins. A number of small molecules documented to be critical to SGN development can serve as good candidates including neurogenin and neuroD [42]. To confirm the neuronal differentiation, an electrophysiology study should be performed in the future study. Additionally, the surviving implanted cells were found mainly in the scala tympani at the implantation site; thus, guiding implanted cells to become integrated into functional spiral ganglion area will be one of the major challenges in the future study.
It is noted that infusion of NGF into the inner ear may also affect the other cell types, such as hair cells with TrkA receptors and Schwann cells with P75 receptors. In this study, no obvious hair cell or Schwann cell morphological change was observed on the histology sections between the NGF group and the control group, suggesting that NGF probably did not have adverse effect on these cell types. Further investigation is needed to characterize this.
In conclusion, our current investigation demonstrated that inner ear-derived SAG-NPs are suitable donor cells for stem cell-based SGN replacement therapy, which is supported by the observation that they could proliferate and differentiate into bipolar SGN-like cells expressing glutamatergic proteins in vitro. NGF could enhance the survival of SAG-NPs both in vitro and in vivo. The enhanced survival of implanted stem cells may provide a basis for future studies to demonstrate successful differentiation into functional SGN-like cells and the integration of these cells into spiral ganglion, which are the major challenges in our future study. Future studies will investigate the potential of multipotent stem cells cultured from human inner ear tissues as donor cells for cell replacement therapy and this part of work is ongoing.
Footnotes
Acknowledgments
This work is supported by American Academy of Audiology Research Foundation and Deafness Research Foundation. Dr. Hui Jiang is supported by Fundan University Jinshan Hospital, Shanghai, China.
Author Disclosure Statement
All the authors, including Drs. Lei Zhang, Hui Jiang, and Zhengqing Hu, agree that there are no commercial associations that might create a conflict of interest in connection with this manuscript.