
Abstract
We previously reported the differentiation of cells derived from porcine female fetal skin into cells resembling germ cells and oocytes. A subpopulation of these cells expressed germ cell markers and formed aggregates resembling cumulus–oocyte complexes. Some of these aggregates extruded large oocyte-like cells (OLCs) that expressed markers consistent with those of oocytes. The objective of the current study was to further characterize OLCs differentiated from porcine skin-derived stem cells. Reverse transcriptase (RT)-polymerase chain reaction and Western blot revealed the expression of connexin37 and connexin43, both of which are characteristic of ovarian follicles. The expression of meiosis markers DMC1 and synaptonemal complex protein, but not STRA8 and REC8, was detected in the OLC cultures. Immunofluorescence with an antibody against synaptonemal complex protein on chromosome spreads revealed a very small subpopulation of stained OLCs that had a similar pattern to leptotene, zytotene, or pachytene nuclei during prophase I of meiosis. Sodium bisulfite sequencing of the differentially methylated region of H19 indicated that this region is almost completely demethylated in OLCs, similar to in vivo-derived oocytes. We also investigated the differentiation potential of male skin-derived stem cells in the same differentiation medium. Large cells with oocyte morphology were generated in the male stem cell differentiation cultures. These OLCs expressed oocyte genes such as octamer-binding transcription factor 4 (OCT4), growth differentiation factor-9b (GDF9B), deleted in azoospermia-like (DAZL), VASA, zona pellucida B (ZPB), and zona pellucida C (ZPC). It was concluded that skin-derived stem cells from both male and female porcine fetuses are capable of entering an oocyte differentiation pathway, but the culture system currently in place is inadequate to support the complete development of competent oocytes.
Introduction
P
Several studies have shown the ability of embryonic stem (ES) cells to differentiate into primordial germ cells (PGCs) as well as oocytes [13 –15]. The ability of germ cell-like cells derived from ES cells to undergo meiosis was also investigated. The OLCs expressed the meiosis marker synaptonemal complex protein (SCP3), and a subpopulation of them showed first polar body formation, a feature of oocyte progression to metaphase of meiosis II [13]. A subsequent study by another group used fluorescence in situ hybridization to examine chromosome organization of the germ cell-like cells differentiated from ES cells, and reported that the SCP3-positive germ cell-like cells do not contain synapsed homologous chromosomes but instead display a chromosomal organization normally found in somatic cells [16]. However, via flow cytometry analysis, other groups have detected haploid gametes differentiated from ES cells [17,18]. The inconsistency of the results reported may reflect the different differentiation potency of different ES cell lines used, as well as variation in protocols for inducing differentiation. We have previously observed the morphology of first polar body formation in a small population of the OLCs generated from skin-derived stem cells. These OLCs, however, failed to become embryos after subsequent in vitro fertilization or electrical parthenogenetic activation. In the present study, we further characterized OLCs with regard to their meiosis status, their establishment of gap junctional communication with surrounding cumulus-like cells, and their epigenetic status on a differentially methylated region (DMR) of an imprint gene, H19. Our results confirmed the presence of these oocyte signatures and also identified some defects in the OLCs.
Materials and Methods
Isolation and culture of skin-derived stem cells
All animal material-related experiments in the study were conducted according to the Care and Use of Experimental Animals of the Canadian Council on Animal Care Guidelines, and have been approved by the University of Guelph Animal Care and Use Committee. Skin-derived stem cells were isolated and cultured as previously described [10] from the skin of porcine fetuses collected at days 40–45 of gestation (E40–E45). Cells were passaged twice before use. Stem cells from 4 to 6 fetuses were combined and used in each of the experiment. The results presented in this report were from 10 differentiations; therefore, 40–60 fetuses were used in the experiments.
Induced differentiation
Skin sphere cells were dissociated mechanically by pipetting and plated at 5–8 × 104 cells per 60-mm tissue culture dish (Fisher Scientific Corning) treated with 0.05 mg/μL of poly-
RNA isolation and reverse transcription
RNA was isolated using the Total RNA Kit (Norgen Biotek Corporation) according to the manufacture's protocol. RT-polymerase chain reaction (PCR) on differentiated cultures was performed as previously described [10]. Reverse transcriptase (RT)-PCR on groups of 15 large cells or oocytes was performed by freezing cells in 7 μL of lysis buffer containing 14 U of porcine RNase inhibitor (Amersham) and 5 mM dithiothreitol (DTT) (Invitrogen) at −80°C until use. Cells were then lysed by boiling for 1 min and vortexing for 1 min, repeated 3 times, and then stored on ice. Samples were DNase (Invitrogen) treated by adding 1 μL of 10 × DNase buffer and 1 U amplification-grade DNase (Invitrogen) and then incubated 15 min at room temperature. About 1 μL EDTA (25 mM; Invitrogen) was then added and the samples were incubated for 10 min at 65°C. RT was then performed by adding 0.5 μL H2O, 5 μL 5 × buffer, 1.25 μL of random hexamer primers (Applied Biosystems, Inc.), 6.25 μL 2 mM dNTPs (Invitrogen), and 1 μL MMLV reverse transcriptase (Invitrogen) to the sample. The samples were then incubated at 25°C for 10 min, 37°C for 50 min, and 70°C for 15 min.
Real-time PCR
Real-time PCR was carried out on a Smart Cycler (Cepheid) by using the Quantitect SYBR green PCR kit (Qiagen). DNase-treated cDNA (from a 25 μL RT reaction), 2.5 μL for cell populations, and 3.1 μL for groups of 15 cells were added to 12.5 μL of SYBR green mix and 0.3 μM each of forward and reverse primers (final volume 25 μL). Primers, expected product size, and accession numbers are presented in Table 1.
DAZL, deleted in azoospermia-like; GDF9B, growth differentiation factor-9b; SCP3, synaptonemal complex protein; ZPC, zona pellucida C.
Western blot
For immunoblotting, protein from differentiated cells and adult pig ovary samples were isolated using RIPA (radio-immunoprecipitation assay) lysis buffer with complete mini protease inhibitors (Roche) added fresh before use. About 30 μg of protein (as determined using a BSA protein assay kit; Pierce Chemical Co.) was mixed with 5 × reducing sample buffer, boiled for 5 min, and electrophoresed under reducing conditions on 12% polyacrylamide gels. Protein was transferred using an iBlot (Invitrogen) onto nitrocellulose membranes (Millipore). Membranes were incubated for 2 h in 5% nonfat dry milk blocking buffer at room temperature, followed by an overnight incubation at 4°C in anti-GAPDH antibody (1:5,000; Chemicon). After a 1 h incubation with anti-mouse IgG (alexa 680) (1:10,000) at room temperature, GAPDH protein was detected, after 3 washes, using the LI-COR Odyssey fluorescent scanner (LI-COR Bioscience). Blots were incubated in 1 × strip buffer (reblot plus mild; Millipore) at room temperature for 15 min and reblocked 1 h. Anti-connexin (CX)-43 antibody cross-reactivity and specificity to porcine cell types have been verified in [20] and antibody incubations were repeated for 1 h with 1:500 anti-CX37 antibody (Invitrogen) and 1: 2,000 anti-CX43 antibody (Sigma-Aldrich) by incubations with 1:10,000 anti-rabbit IgG (Alexa 680; Invitrogen). All blots were stripped, blocked, and reprobed between antibodies.
Immunofluorescence
Cells were washed twice with phosphate-buffered saline (PBS) and fixed in 4% paraformaldehyde (Fisher Scientific) in PBS for 20 min. Cells were then washed 3 times in PBS with 0.1% Tween 20 (Fisher Scientific) and incubated for 10 min, and then for 20 min in PBS with 1% Triton-X-100 (Fisher Scientific). Next, cells were blocked for 2 h in PBS with 5% BSA, and 0.05% Triton-X-100 (blocking solution), followed by an incubation with primary antibody: 1:400 anti-DAZL (Abcam), or 1:400 VASA (Abcam) for 2 h at 37°C, or overnight at 4°C. The cross-reactivity and specificity of deleted in azoospermia-like (DAZL) and VASA antibodies to porcine cell types have been verified previously in [21,22]. Cells were then washed in blocking solution and incubated with 1:250 phycoerythrin-conjugated goat anti-rabbit IgG (BD Bioscience) or 1:500 fluorescein isothiocyanate-conjugated rat anti-mouse IgG (BD Bioscience) for 1 h at room temperature. This was followed with a blocking solution wash (PBS-B) and incubation with 4′-6-diamidino-2-phenylindole (Invitrogen) for 1 min or Hoechst333258 (Sigma-Aldrich) for 5 min, followed by 3 washes with PBS-B. Cells were mounted using fluorescent mount medium (DakoCytomation) and viewed using an Olympus BX-UCB microscope and MetaMorph analysis software (Universal Imaging Corporation).
Chromosome spreading and SCP3 staining
Cells were trypsinized (Invitrogen) for 3 min at 37°C, washed once in Dulbecco's modified Eagle's medium H21 (Invitrogen) with 10% fetal calf serum (Gibco), and then resuspended in 0.5 mL PBS. Thirty microliters of the suspension was then placed on a slide and incubated for 5 min at room temperature to attach the cells to the slide surface. To spread the cells, 90 μL of 3% sucrose (Sigma-Aldrich) was gently added to the slide, which was then held for 20–30 min at room temperature. Fixative was prepared as 2% paraformaldehyde (Fisher Scientific) with 2 μL of 10% sodium dodecyl sulfate (Bio-Rad) per mL. The pH was adjusted to between 9 and 11 by adding 10 μL NaOH (1N) per 8 mL of fixative followed by 3 rinses in PBS-Tween20 (PBT), each for 10 min at room temperature. PBT was made by adding 0.15g BSA and 100 μL Tween-20 to 100 mL PBS. The primary antibody against SCP3 (1/100; Abcam) was then added and the slides were incubated in humidity chambers overnight at 4°C. The cross-reactivity and specificity of the antibody to porcine cell types has been verified previously in [23]. Before adding anti-rabbit fluorescein isothiocyanate (1/500; BD Bioscience), the slides were washed 3 times with PBT for 10 min in each wash. The slides were then incubated for 2 h at room temperature, and then washed twice with PBT for 10 min at room temperature. Slides were stained with Hoechst (Sigma-Aldrich) for 7 min, washed once in PBT, and then mounted using fluorescent mounting medium (Dako S3023; DakoCytomation). Slides were analyzed using a fluorescence microscope (Olympus).
Sodium bisulfite genomic DNA sequencing
DNA was isolated from 400 to 500 denuded porcine oocytes using DNeasy® Blood & Tissue Kit (Qiagen), a method based on lysis with proteinase K (Invitrogen). They were subsequently treated with sodium bisulfite from Methylamp™ DNA Modification Kit (Epigentek) according to the manufacturer's instructions. The bisulfite-treated DNA was amplified for the 260 bp sequence spanning the porcine H19 DMR1 with primary and nested PCR primers using PCR cycling parameters previously described [24]. Each 50 μL PCR included either 4 μL of bisulfite-treated genomic DNA (primary PCR) or 2 μL of primary product (nested PCR), 25 pmol of each primer, 1.25 units of Maxima HotStart Taq DNA polymerase (Fermentas, Burlington, ON, Canada), 2 μL of 2 mM dNTPs (Invitrogen), 10 × Hot Start PCR buffer (Fermentas), and MgCl2 (Fermentas) at a final concentration of 1 mM. The PCR products were purified with the WizardSV Gel and PCR Clean-Up System (Promega). The purified PCR products were cloned into a pGEM-T Easy Vector (Promega) according to the manufacturer's instructions. The positive clones were obtained by ampicillin and blue-white selection and each individual clone was DNA sequenced. The cytosine-to-uracil conversions were identified by sequence alignment.
Results
Upon being cultured in the differentiation medium, the skin sphere cells first grew and proliferated as a monolayer on the culture dish. At days 15–30 of differentiation, a population of shiny round cells formed (1%–5% of the differentiating cells), growing either as a single cell or in clusters (Fig. 1A). The clusters grew larger (Fig. 1B) and 5%–25% of them gradually detached from the rest as suspended aggregates. At approximately days 38–55, a rare subpopulation (0–5 per culture dish with the initial plating density of 5–8 × 104 cell per dish) of the suspended aggregates has large cells surrounded by smaller cells, similar to a COC of an ovarian follicle (Fig. 1C, D). OLCs were also found during differentiation (Fig. 1E, F), most not associated with cumulus-like cells. OLCs approaching 100 μm in diameter were regularly found at later stages of differentiation (Fig. 1F). These dynamic morphologies are consistent with what we have observed previously [11,22]. The frequency OLC generation varied from culture to culture, which also depended on the batches of follicular fluid and fetal calf serum stocks. It ranged from 0% to 100% (∼25% on average) of the dishes having OLCs. In the OLC-generating dishes, ∼3–8 OLCs can be harvested from each of the dish. One of the functional characteristics of COCs is the presence of gap junctions between the oocyte and surrounding cumulus cells and between cumulus cells. Gap junctions are intercellular channels formed by end-to-end docking of connexons between adjacent cells, each connexon being a cylindrical hexamer of CXs. Among the ∼20 members of the CX family in mammals [25], CX37 and CX43 are both essential for ovarian follicle development as revealed by analysis of the respective mouse gene knockouts [26,27]. To further characterize the COC-like structures generated from our differentiation protocol, real-time RT-PCR was performed to assess expression of these 2 CXs. As shown in Fig. 2A, transcripts of both GJA1 (encoding CX43) and GJA4 (encoding CX37) were detected. The levels of expression of these 2 genes were more than 4-fold higher than that of COCs isolated from porcine ovarian follicles. We next determined the presence of CX43 and CX37 proteins using Western blot. As shown in Fig. 2B, similar to the porcine ovarian control, both CXs were detected in our cells differentiated from the skin-derived stem cells. In the undifferentiated stem cell group, the expression was either undetectable (CX37) or at a much lower level (CX43). The data suggest the presence of gap junctions in the COC-like structures.

Dynamic morphology during cell differentiation from skin-derived stem cells.

CXs are expressed in structures resembling cumulus–oocyte complex.
Next, we sought to determine whether the germ cells generated from our differentiation can initiate and progress through meiosis, a function that is unique to oocytes and male germ cells at the later stages of their development. RT-PCR was performed to examine the expression of markers of meiosis: SCP3, REC8, DMC1, and STRA8. As shown in Fig. 3A, SCP3 was expressed at all time points tested, DMC1 were expressed in cells starting from 30 days of differentiation, REC8 was detected at day 35 of differentiation, whereas the transcripts of STRA8 were not detectable. Further, we analyzed the localization of SCP3 on chromosome spreads. The localization of SCP3 is characterized through different stages of prophase I. A very small subpopulation (3–5 cells/million) of our differentiated cells showed a chromosome staining pattern consistent with prophase in conjunction with positive SCP3 expression. Figures 3Ba and d show examples of staining resembling the leptotene stage, in which SCP3 is localized in punctate areas and chromosomes have yet not fully synapsed. At zygotene stage, the chromosomes progress synapsis and SCP3 is expected to be localized along the entire chromosome pairs. SCP3 staining that resembles the zygotene stage was detected in some cells from day 25 to 45 of differentiation (Fig. 3Bb, e). In addition, an SCP3 staining pattern similar to the pachytene stage was also detected in the cultures (Fig. 3Bc, f). However, the majority of the SCP3-stained cells (∼0.1% of the cells tested) appeared to show discontinuous staining patterns that do not resemble any typical prophase stage (Fig. 3C).

Detection of meiosis markers in germ cell-like cells.
During gametogenesis, it is well known that epigenetic imprints are established in a parent-specific manner [28 –30]. H19 is a maternally inherited imprinted gene where the DMR1 region is semimethylated (∼50%) in the somatic tissue and is unmethylated in the oocyte [24]. Sodium bisulfite sequencing was performed to analyze the methylation status within DMR1 of the H19 gene 5′ flanking region. As shown in Fig. 4A, 66.8% of the 29 CpG sites were methylated in skin-derived stem cells before induced differentiation. In contrast, over 90% of CpG sites in this region were unmethylated in the OLCs collected from our differentiation culture (Fig. 4). Consistent with what was reported previously [24], 98% of the CpG sites were found to be unmethylated in porcine oocytes (Fig. 4). These results indicate that OLCs have an imprint pattern similar to that of natural oocytes.

The DNA methylation profile in differentially methylated region 1 at the imprinted H19 locus (GenBank: AY044827) in OLCs, skin-derived stem cells, and oocytes from porcine ovaries. Open and black circles represent unmethylated and methylated cytosines, respectively.
PGCs enter to a default oocyte fate unless they are placed in the testis environment [31]. To determine if XY stem cells can also differentiate into OLCs in our differentiation medium, we investigated the oocyte differentiation potential of male skin-derived stem cells with our differentiation medium, using dissociated fetal pig ovary cells from day 45 to 55 of gestation as a positive control. Figure 5Ac illustrates PGC-like shiny cells at an early stage of differentiation, which is very similar to what was observed in the female stem cell differentiation culture (Fig. 1A) and the positive control, which were porcine ovarian germ cells (Fig. 5Aa). OLCs with zona pellucida-like membrane were also observed within the differentiation culture (Fig. 5Ad), again consistent with the oocyte morphology of the positive control. Similar to OLCs from the fetal ovary and those differentiated from female stem cells, RT-PCR revealed that these male OLCs express markers of oocytes: OCT4, DAZL, VASA, ZPA, zona pellucida C (ZPC), and GDF9B (Fig. 5B). Figure 6 illustrates the expression of DAZL and VASA at the protein level by immunofluorescence. These data suggest that our differentiation medium provided an ovarian-like environment to direct the male skin-derived stem cells into the female germ cell path.

Differentiation of OLCs from male skin-derived stem cells.
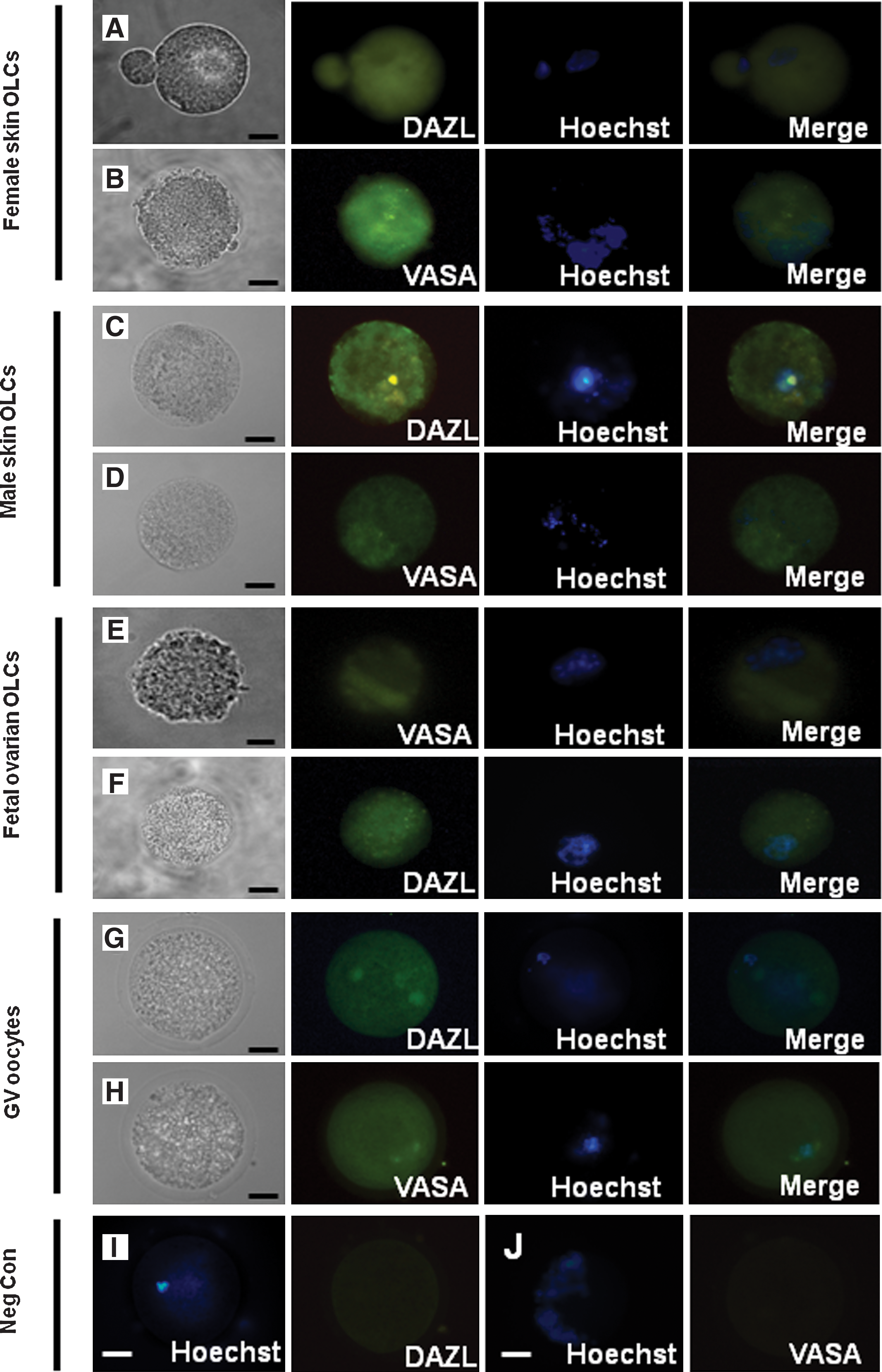
Analysis of germ cell protein marker expression in OLCs (DAZL and VASA) expression in OLCs from female
Discussion
One of the essential requirements for successful ovarian follicular development and oocyte growth is proper interaction between the oocyte and surrounding cumulus granulosa cells. Gap junctions allow for the bi-directional communication between closely associated cells by forming channels through which various bioactive molecules can be transported. Two adjacent cells form gap junctions by docking their connexons end to end, thereby forming an intercellular channel. In the mouse, CX43 forms gap junctions between cumulus and mural granulosa cells, whereas CX37 forms gap junctions coupling the oocyte with the adjacent cumulus cells. Loss of either CX disrupts folliculogenesis, preventing the development of competent oocytes [26,32,33]. In our findings, COC-like structures expressed both CXs, at both the mRNA and protein level, indicating that these 2 key gap junction components are physically established in the induced ovarian cell differentiation. The mRNA expression levels of both CXs 37 and 43 were significantly higher in the COC-like structures than in natural COCs. The expression levels of the 2 CXs in COC-like structures were quite variable across experiments as reflected by the large standard deviations (Fig. 2A). It is possible that, due to the suboptimal nature of our differentiation system, the expression levels of some of the genes in the COC-like structures are not as precisely regulated as those in true ovarian follicles. In addition, depending on the batches of FBS and follicular fluid used, the frequency of COC formation is highly variable, ranging from 0 to 30 COCs per culture dish. The majority of OLCs appeared as a single large cell lacking support cells. It is unknown if they have failed to physically associate with the cumulus-like cells, or they have formed COCs but dissociated spontaneously shortly after their formation. Regardless, this further indicates some development deficiencies in OLCs in our current in vitro system.
During meiosis, DNA is replicated once and then 2 successive cell divisions results in the formation of haploid gametes [34,35]. Prophase I is conventionally subdivided into leptotene, zygotene, pachytene, diplotene, and diakinesis stages. It is characterized by the pairing behavior of homologous chromosomes in a process called synapsis. During synapsis, the synaptonemal complex (SC), a meiosis-specific structure, is formed. The SC consists of several meiosis-specific proteins, including SCP3 [34]. At the leptotene stage, each pair of sister chromatids forms a meiosis-specific longitudinal axial core to which the chromatin loops are attached. SCP3 is one of the elements of these axial cores [36,37]. SCP3 is regarded as a marker for identifying meiotic transition as its expression is specific to meiosis and it is present from the initiation of meiosis [38]. Later at the zygotene stage, the axial cores of each pair of homologous chromosomes synapse in parallel, becoming the lateral elements of the synaptonemal complexes. During pachytene, the homologous chromosomes remain fully paired until completion of the crossing-over process. After this, the SC structure disassembles as the cell enters the diplotene/diakinesis stages. Our data on leptotene, zygotene, and pachytene SCP3 staining patterns in the germ cell-like cells indicated that at least a small subpopulation of these cells has initiated and reached certain stages of early meiosis. However, the majority of SCP3-stained cells appeared to show discontinuous staining patterns that do not resemble any typical prophase stage, suggesting that they failed to progress past the premeiotic stage. In addition to SCP3, several markers are known to be important in meiosis. The expression of STRA8 (stimulated by retinoic acid gene 8) is known to play an important role in the initiation of meiosis in germ cells. DMC1 is a meiosis-specific RECA/RAD51 homolog required for recombinational repair of meiotic DNA double-stranded breaks. It is thought to promote strand transfer between homologous DNA molecules in an ATP-dependent manner [39] and play a role during chromosome synapsis and homologous recombination [40 –42]. In addition, the meiosis-specific cohesin subunit REC8 is reported to be involved in homologous chromosome sister chromatid separation in mice [43]. REC8 is a meiosis-specific cohesin protein located in the centromeres and in adjacent chromosome arms. REC8 is suggested to play a role in maintaining sister-chromatid cohesion, where its presence at centromeres during meiosis I is important in the reductional pattern of chromosome segregation during meiosis [44,45]. In terms of SCP3 mRNA and protein level expression, our findings are consistent with previous reports on SCP3 expression in ES cell-derived germ cells in vitro [13,46]. The fact that STRA8 is not detectable in our system may explain why the majority of SCP3-stained cells failed to form typical meiotic patterns. Previous work has shown that SCP3 failed to be loaded onto chromosomes of STRA8-deficient mouse spermatogenic cells. In these STRA8 knockout mice male germ cells, SCP3 was localized in patterns reminiscent of those previously reported in premeiotic germ cells [47]. A recent study on the further characterization of in vitro-produced ES-derived germ cells showed that while SCP3 is expressed in these cells, it is not correctly localized; further, the chromosomes appeared more similar to somatic cell chromosomes than meiotic chromosomes [16]. A recent publication reported the deletion in azoospermia (DAZ) and related family members play import roles in germ cell meiosis. Whether the overexpression of the DAZ family in our differentiating cells will facilitate their meiosis progression should be investigated.
Igf2 and H19 are imprinted genes that have been widely studied in the mouse. Igf2 is paternally expressed, whereas H19 is transcribed from the maternal allele [48]. The reciprocal expression of both genes is regulated through Igf2/H19 DMRs situated 2–4 kilobases upstream from the H19 transcription start site. Previous work has revealed the methylation dynamics of the Igf2/H19 DMR in mouse gametes, suggesting that imprinting is established during gametogenesis [28,29]. In our finding, the stem cell DMR1of H19 is hemimethylated, which is consistent with a nongamete somatic methylation pattern. In addition, the CpG sites of DMR1 of our oocyte control were mostly unmethylated, which is also consistent with previous reports on pig and human oocytes [24,28]. The fact that DMR1 of H19 is highly hypomethylated in OLCs suggests that this imprint gene has undergone gamete methylation reprogramming. It is also noted that 1/10 of the DNA analyzed is partially methylated in OLCs. This again may reflect the suboptimal nature of the in vitro differentiation system, and thus the poor quality of the oocytes generated.
The finding that male skin-derived stem cells differentiated into OLCs is not surprising. In mammals, it appears that PGCs spontaneously enter the oocyte fate in the absence of a testis environment. XY PGCs in a fetal ovary will enter meiosis and develop as XY oocytes [31]. Retinoic acid (RA) is one of the meiotic initiators, which induces Stra8 expression and the subsequent meiotic entry of germ cells in the fetal mouse ovary [49]. However, somatic cells in the fetal testis express factors such as CYP26 that mediates the degradation of RA, thereby protecting male germ cells from RA exposure, and thus inhibiting entry to meiosis [50,51]. When placed in an appropriate female environment, PGCs were previously demonstrated to spontaneously proceed into the oocyte path in vivo [31,52]. In addition, male ES cells were shown to differentiate into oocytes in vitro [53]. In our experiment, the OLCs differentiated from male stem cells reached diameters and were morphologically very similar to OLCs derived from female stem cells. In addition, the cells expressed germ cell markers both at the mRNA and protein level. Our result is in line with these findings, further confirming that our differentiation system with FBS and ovarian follicular fluid can lead to oocyte development in a male cell model.
In summary, our study shows that a subpopulation of OLCs generated from differentiation of skin stem cells is able to associate with other cells within the culture to form COC-like structures. The generated COCs express CXs that are essential for follicular development. SCP3 and DMC1 were expressed, but STRA8 and REC8 were not detectable in the differentiated cells. A very small population of the cells showed an SCP3-stained chromosome pattern of meiosis at the early prophase, whereas the majority of the SCP3-positive cells failed to show a typical meiotic chromosome pattern, suggesting that they were not entering meiosis properly. Analysis of the methylation status of DMR1 of H19 revealed that the CpG methylation pattern of OLCs is largely consistent with those of natural oocytes. Our findings further confirm the development of cells with properties of oocytes from skin-derived stem cells. However, the incomplete meiosis progression indicates that the differentiation environment is suboptimal. This drawback of the system may provide opportunities for approaching certain types of infertility, as this in vitro model may mimic some cases of gonadal dysfunction. For example, deficiency in the expression of meiosis maker gene such as DMC1 caused male infertility and decreased female reproductive function [54]. In addition, prematurely condensed chromosomes and meiotic abnormalities are found after in vitro fertilization in unfertilized humans [55]. Future studies on manipulating the system by addition or removal of factors to and from the medium may help to identify factors that are critical for proper meiosis and successful oocyte development.
Footnotes
Acknowledgments
This research was supported by grants from the Canadian Institutes for Health Research (grant MOP-14150 to G.M.K.), National Science and Research Engineering Council, and the Ontario Ministry of Agriculture, Food and Rural Affairs, Premier's Research Excellence Award.
The authors wish to thank staff at the University of Guelph Arkell Swine Research Station for overseeing the sow-breeding program and staff at Conestoga Farm Fresh Pork for their assistance in ovary collection. We would also like to thank Ezra Hart for the help with optimizing chromosome spreading.
Author Disclosure Statement
No competing financial interests exist.