
Abstract
The domesticated pig has emerged as an important tool for development of surgical techniques, advancement of xenotransplantation, creation of important disease models, and preclinical testing of novel cell therapies. However, germ line-competent pluripotent porcine stem cells have not yet been derived. This has been a major obstacle to genetic modification of pigs. The transcription factor Oct4 is essential for the maintenance of pluripotency and for reprogramming somatic cells to a pluripotent state. Here, we report the production of transgenic pigs carrying an 18 kb genomic sequence of the murine Oct4 gene fused to the enhanced green fluorescent protein (EGFP) cDNA (OG2 construct) to allow identification of pluripotent cells by monitoring Oct4 expression by EGFP fluorescence. Eleven viable transgenic piglets were produced by somatic cell nuclear transfer. Expression of the EGFP reporter construct was confined to germ line cells, the inner cell mass and trophectoderm of blastocysts, and testicular germ cells. Reprogramming of fibroblasts from these animals by fusion with pluripotent murine embryonic stem cells or viral transduction with human OCT4, SOX2, KLF4, and c-MYC cDNAs resulted in Oct4-EGFP reactivation. The OG2 pigs have thus proved useful for monitoring reprogramming and the induction and maintenance of pluripotency in porcine cells. In conclusion, the OG2 transgenic pigs are a new large animal model for studying the derivation and maintenance of pluripotent cells, and will be valuable for the development of cell therapy.
Introduction
P
While porcine cells sharing many characteristics of true embryonic stem (ES) cells have been derived (ES-like cells) [7 –10], neither true (germ line competent) porcine ES cells nor porcine primordial germ cells have been produced. It is not known whether this is because existing in vitro culture conditions cannot support porcine ES cells or because the factors that regulate human and murine pluripotency are not functional in porcine stem cells [11]. The transcription factor Oct4 is germ line specific and has been widely used to identify pluripotent cells in the mouse and many other species [12 –15]. Oct4 protein is found in mouse, porcine, and bovine blastocysts [12], murine ES cells [16,17], murine embryonic carcinoma cells [18], murine germ line cells [19], and somatic stem cells from various species [20]. Lack of Oct4 expression is associated with failure to form the inner cell mass in murine blastocysts [21] and with apoptosis of murine primordial germ cells [22]. Oct4 is critically involved in the control of self-renewal and maintenance of pluripotency in murine ES cells [23,24]. Oct4 belongs to a group of transcription factors that are capable of inducing pluripotency in somatic cells of the mouse [3,25], rhesus monkey [26], rat [27], and humans [4,28]. Recently, putative porcine iPS cells were produced which displayed several characteristics of pluripotent cells [29 –32]. Production of these iPS cells from somatic cells was inefficient and required continued expression of exogenous transcription factors [29 –31]. However, germ line competence has not been reported, indicating that these cells do not possess the full arsenal of pluripotent properties [29 –31]. A recent study reported evidence of germ line chimerism, but solely based on polymerase chain reaction (PCR) results [32]. The Oct4-enhanced green fluorescent protein (EGFP) (OG2) mouse that indicates when and in which tissue Oct4 is expressed has proven to be extremely valuable for basic studies on pluripotency and differentiation [14,15] and for following experimental induction of pluripotency after cell fusion [33,34].
Here, we report the production of Oct4-EGFP transgenic pigs using the OG2 construct and show that expression is restricted to the inner cell mass and trophectoderm of blastocysts, porcine germ line cells, and testicular germ cells. It is re-expressed after reprogramming fibroblasts by somatic cell nuclear transfer (SCNT), by fusion of porcine somatic cells with murine ES cells, or by viral transduction with the classical 4 reprogramming factors [3]. We anticipate that the OG2 transgenic pigs and primary cells derived thereof will be a valuable tool in the fields of reprogramming, developmental biology, and cell therapy.
Materials and Methods
Generation and characterization of Oct4-EGFP (OG2) transgenic pigs
Isolation and primary culture of porcine fetal fibroblasts
Porcine fetal fibroblasts (PFFs) from a 25-day-old wild-type fetus, obtained by hysterectomy of a pregnant German landrace gilt, were established as primary culture [20] and maintained in Dulbecco's modified Eagle's medium (PAA Laboratories GmbH) supplemented with 30% fetal bovine serum (FCS batch no. 40F8240K; Invitrogen), 1 mM sodium pyruvate (Sigma-Aldrich), 2 mM glutamine (AppliChem), 0.1 mM mercaptoethanol (Sigma-Aldrich), 100 U/mL penicillin, 100 μg/mL streptomycin (PAA Laboratories GmbH), and 1% nonessential amino acids (PAA Laboratories GmbH). Subconfluent fibroblasts cultured in a T75 tissue culture flask were used for transfection.
Transfection of PFFs with the human phOCT4-EGFP construct
The ApalI linearized minigene construct phOCT4-EGFP was transfected into PFFs by electroporation with 250 V and 500 μF using a Gene Pulser II electroporator (Bio-Rad Laboratories). After 2 weeks of selection with G418 (800 μg/mL) 161 surviving cell clones were determined. The 17 expanded colonies were analyzed by PCR and Southern blot for transgene integration. Three cell clones were clearly positive, 5 questionable and 9 negative.
Transfection of PFFs with the murine Oct4-EGFP (OG2) construct
The Oct4-EGFP (OG2) genomic construct contains the Oct4 promoter (9 kb), EGFP cDNA, and ∼9 kb of the Oct4 exon/intron region. The plasmid was amplified in XL10 bacteria and purified with ion-exchange columns. The linearized construct was excised from its plasmid by digestion with NotI (New England Biolabs) and purified and used for cotransfection. Subconfluent PFFs were trypsinized and electroporated simultaneously with 21 μg of the OG2 construct and 2.25 μg of the neo construct with 250 V and 500 μF using a Gene Pulser II electroporator (Bio-Rad Laboratories). Selection with geneticin (800 μg/mL; G418; PAA Laboratories) was applied 48 h after cotransfection. Two weeks after selection, surviving clones were subcloned and expanded. Cell clones that had integrated the OG2 construct did not express EGFP and did not show green fluorescence. These were grown to confluence in 4-well dishes and then used in SCNT.
SCNT and production of cloned pigs
Oocytes were collected from abattoir ovaries, matured in vitro, and used in SCNT as described [35,36]. In vitro matured oocytes were enucleated by removing the first polar body along with the adjacent cytoplasm containing the metaphase plate. The donor cells were arrested at G0/G1 of the cell cycle by serum starvation (Dulbecco's modified Eagle's medium + 0.5% FCS) for 48 h [35]. A single fibroblast was placed in the perivitelline space in close contact with the oocyte membrane. Cell membrane fusion was induced in Ca2+-free medium (0.25 M sorbitol, 0.5 mM Mg-acetate, and 0.1% bovine serum albumin) by a single electrical pulse of 1.1 kV/cm for 100 μs (Eppendorf Multiporator). Reconstructed embryos were activated in an electrical field of 1.0 kV/cm for 45 μs in SOR2 activation medium (0.25 M sorbitol, 0.1 mM Ca-acetate, 0.5 mM Mg-acetate, and 0.1% bovine serum albumin) followed by incubation with 2 mM 6-dimethylaminopurine (Sigma-Aldrich) in NCSU23 medium for 3 h before embryo transfer to recipients. Reconstructed embryos were either transferred to recipients [37] or cultured in vitro to blastocysts. A total of 94–110 reconstructed embryos were surgically transferred into one oviduct of each recipient by midventral laparatomy under general anesthesia [37]. Pregnancies were confirmed by ultrasonography on days 25 and 35 and were allowed to go to term. Table 1 shows a summary of SCNT results. All animal experiments were carried out according to German Animal Welfare Laws and approved by an independent Animal Welfare Committee.
Two pregnancies were terminated for fetal cell culture and EGFP expression analysis.
EGFP, enhanced green fluorescent protein.
Integration of the OG2 transgene in pigs checked by PCR
Genomic DNA was purified from transfected cells and ear biopsies from transgenic piglets by treatment with 560 μL of lysis buffer (50 mM Tris pH 8.0, 100 mM NaCl, 100 mM ethylenediaminetetraacetic acid, and 1% sodium dodecyl sulfate) containing 40 μL from 20 mg/mL proteinase K (Roth). The DNA was then purified with phenol/chloroform (Roti-Phenol-Chloroform, Roth). Ethanol 100% was added for DNA precipitation and the pellet was washed twice with 70% ethanol before it was air-dried and dissolved in sterile water. PCR amplification was performed with 200 ng of genomic DNA template in final volumes of 50 μL using 1 U Platinum Taq Polymerase (Invitrogen), 1 × PCR buffer (50 mM KCl, 20 mM Tris-HCl pH 8.4; Invitrogen), 200 μM of each dNTP (BIOLINE GmbH), 1.5 mM MgCl2 (Invitrogen), and 1.0 μM of each primer. The PCR conditions and product lengths are presented in Table 2. DNA derived from nontransgenic (wild-type) pigs and water served as a negative control. PCR products were separated on 2% agarose gels. Amplified products were confirmed by sequencing (Agowa).
F, forward primer; R, reverse primer; RT-PCR, real-time reverse transcription–polymerase chain reaction.
Integration of the OG2 transgene checked by Southern blot
Genomic DNA (10 μg) from transgenic fetuses/piglets was digested using HindIII (New England Biolabs) and processed according to standard procedures using the DIG High Prime DNA Labeling and Detection Kit II (Roche Applied Science). DIG-labeled EGFP and an Oct4 promoter-specific probe were employed to confirm transgene integration in the porcine genome (Table 2). The DIG-labeled DNA probes used for Southern blotting were generated with a random primed labeling technique according to the Manual instruction. Both EGFP and Oct4 promoter-specific DNA probes were amplified from GOF18 plasmid DNA with the EGFP and murine Oct4 promoter-specific primers used also for genotyping of the Oct4-EGFP transgene (primer sequences and PCR conditions are listed in Table 2). The amplified DNA fragments (concentration 300 ng) were used for DIG-labeling. For the electrophoresis we used 1% agarose gel, which was loaded with genomic DNA samples and subjected to 80 V for 5 h. After denaturation and neutralization of the gel, DNA was transferred in 20 × SSC buffer into membrane (capillary transferring). After fixation with ultraviolet crosslinking, the nylon membranes were prehybridized for 4 h and then probes were added and left for hybridization. Twenty-four hours later, after washing and blocking, the hybridized probes were immunodetected with anti-digoxigenin-AP, Fab fragments. For observation, CDP-star (chemiluminescent substrate) was added and the membranes were incubated for 10 min at the room temperature. The membranes were exposed to an X-ray film for 1–4 h after removal of CDP solution.
RNA extraction and expression analysis of the OG2 transgene checked by northern blot
Total RNA was extracted from 200 mg of testis, heart, muscle, and kidney isolated from transgenic or wild-type piglets/pigs using TRIsure Reagent (TRIsure; BIOLINE GmbH) according to standard procedures [38]. Polyadenylated RNA (Poly[A]) was isolated from 70 to 250 μg total RNA using the NucleoTrap® mRNA kit according to the manufacturer's protocol (Macherey-Nagel). The quality of the RNA was evaluated with Agilent 2100 Bioanalyzer (Agilent Technologie). Poly(A)RNA (2.5 μg/lane) was loaded onto a denaturating agarose gel (1%) containing 2 mL of 37% formaldehyde/100 mL gel in 1 × MOPS buffer. After electrophoresis, RNA was transferred to a HybondN+-membrane (Roche) by a capillary transfer overnight in 20 × SSC and then fixed by cross-linking under ultraviolet light. Blots were prehybridized with DIG Easy Hybridization buffer (DIG Northern Starter Kit; Roche) and then hybridized overnight (68°C) with DIG-labeled RNA probes (concentration of the probes were 400 ng/μL). Hybridization was performed with 8 mL DIG Easy Hybridization buffer (DIG Northern Starter Kit; Roche) containing 2 μL DIG-labeled RNA probe. RNA probe specific for EGFP (900 bp) was obtained from pBKS vector (containing CMV promoter, EGFP gene followed by T7 promoter sequence on the 3′ end). The vector was linearized with AgeI enzyme before labeling. All RNA probes were labeled using T7 RNA polymerase with the DIG Northern Starter kit (Roche) as described in manufacturer's protocols. For the generation of RNA-specific probe for OCT4 (322 bp) and β-ACTIN (620 bp), we used the OCT4/β-ACTIN PCR products obtained by PCR using pig OCT4/β-ACTIN lower primers fused with T7 promoter (for primer sequences see Table 2). The hybridized probes were immunodetected with anti-digoxigenin-AP, Fab fragments and were observed with the chemiluminescent substrate disodium2-chloro-5-(4-methoxyspiro{1,2-dioxetane-3,2′-(5′-chloro) tricyclo [3.3.1.13,7] decan} 4yl) phenyl phosphate “CDP” according to the manufacturer's instructions (Roche). The CCD camera-based SL-7 chemiluminescence imaging system (Vilber Lourmat; Frankreich) was used for imaging.
EGFP detection by fluorescence microscopy
EGFP fluorescence was monitored in squash preparations of (1) cloned in vitro cultured Oct4-EGFP (OG2) porcine blastocysts from day 5/6, (2) in vivo derived F1-blastocysts from day 5, (3) parthenogenetic blastocysts (nontransgenic controls), (4) genital ridges and other tissues (intestine, brain, heart, lung, mesonephros, and skin) of day 25 fetuses (first trimester of pregnancy), (5) postnatal testis obtained from two 2–7-day-old transgenic piglets, and (6) a 7-month-old transgenic boar. EGFP fluorescence was detected with a fluorescent microscope (Olympus BX60F-3) equipped with a filter block containing an excitation filter of 470–490 nm, a dichroic mirror DM505, and a bandpass barrier filter BA515-550. UPlanFl 40 × air and UPlanFl 100 × oil objectives were used. Images were recorded with a DP71 digital camera (Olympus) and processed with CellP (Olympus) software.
Expression analysis of pig OCT4/mouse Oct4 genes in testis by real-time reverse transcription–-PCR analysis
Total RNA was isolated from wild-type and OG2 transgenic porcine testis using established protocols [for real-time reverse transcription (RT)–PCR details, see “Expression analysis of pluripotency related genes by real-time RT-PCR analysis”]. Primer sequences and PCR condition for pig OCT4/mouse Oct4 are summarized in Table 2. For normalization, the housekeeping genes GAPDH, EEF1A1, and pig β-ACTIN were amplified along with the porcine OCT4/mouse Oct4 genes. The expression ratio relative to the expression in piglet 1 (4 days old) or Oct4-EGFP (OG2) transgenic piglet (2 months old) was calculated by using the ΔΔCT method. The pig-specific OCT4 primer did not cross-react with the mouse Oct4 gene, and the mouse Oct4-specific primer did not cross-react with the pig OCT4 sequence.
Establishing OG2 transgenic pig lines, determination of germ line transmission of the Oct4-EGFP transgene, and isolation of F1 in vivo blastocysts
Three 7-month-old wild-type gilts were mated with 10-month-old Oct4-EGFP (OG2) transgenic boars. Blastocysts were recovered from 2 slaughtered animals on day 5 after mating by flushing the uterus with Dulbecco's phosphate-buffered saline containing 1% of newborn calf serum (PAA Laboratories GmbH) and were analyzed for EGFP expression. The remaining pregnant sow delivered 10 piglets that were analyzed by Southern blot for transgene integration.
Reprogramming of OG2 somatic porcine cells by fusion with murine ES cells
Cell fusion and hybrid culture
The murine RHN-ES cell line (Red fluorescent protein, Hygromycin resistance, and Nanog over-expression) and PFFs, isolated from day 25 transgenic fetuses carrying the Oct4-EGFP (OG2) and geneticin resistance constructs, were used to induce reprogramming of the porcine somatic cells by cell fusion. Mouse ES cells and fused mouse–pig hybrid cells were maintained on gelatin-coated dishes (0.1% gelatin; Sigma-Aldrich) without feeder cells in Glasgow MEM (G-MEM; Invitrogen) with L-glutamine (Invitrogen) supplemented with 10% FCS (batch no. 40F8240K, Invitrogen), 100 U/mL penicillin, 100 μg/mL streptomycin (PAA Laboratories GmbH), 1% nonessential amino acids (PAA Laboratories GmbH), 50 μM 2-mercaptoethanol (Sigma-Aldrich), and 1,000 U/mL Leukemia Inhibitory Factor (ESGRO; Millipore).
Inter-species cell fusion was performed as previously described [39] with minor modifications. Briefly, RHN-ES cells were dispersed by 0.025% ethylenediaminetetraacetic acid–trypsin treatment (PAA Laboratories GmbH) and ∼1.4–3.0 × 106 cells were plated on gelatin-coated 6 cm dishes in ES culture medium for 1–2 h. Subsequently, fetal porcine fibroblasts (2.5–5.0 × 106) were added to the attached RHN-ES cells and cultivated in ES medium for 10 min. Thereafter, the ES culture medium was replaced by serum-free G-MEM containing 100 μg/mL phytohaemagglutamine-P (Sigma-Aldrich). The cocultured cells were washed once with Dulbecco's phosphate-buffered saline (Sigma-Aldrich), and 1 mL of 50% polyethylene glycol (PEG) 1500 (Roche) was added to the attached cells for 2 min. After aspiration of PEG, fused cells were washed 4 times with serum-free G-MEM and then incubated in ES medium at 37°C. Hybrids were double-selected with geneticin (600 μg/mL; PAA Laboratories GmbH) and hygromycin (400 μg/mL; PAA Laboratories GmbH) 48 h after PEG treatment. Double-resistant clones were subcloned with cloning cylinders (Sigma-Aldrich) and expanded. Each coculture fusion experiment was performed with 9 independent replicates.
Expression analysis of pluripotency markers by endpoint RT-PCR
The mRNA expression of pluripotency markers in the reprogrammed cells was determined from total RNA using established protocols. Isolated total RNA from cell samples using TRIsure Reagent (TRIsure; BIOLINE GmbH) was treated with RNase-free DNase I (Epicentre Biotechnologies) and used immediately for RT. Poly(A)+ RNA was isolated from porcine matured oocytes by using the Dynabeads mRNA DIRECT Kit (Dynal) before reverse transcriptase treatment. Samples with sterile water instead of reverse transcriptase and RNAse inhibitor served as negative controls. The hot-start PCR was conducted with 120 ng of the cDNA template isolated from cells or 2 oocyte equivalents in a total volume of 50 μL using 1 U Platinum Taq DNA Polymerase (Invitrogen), 1 × PCR buffer (50 mM KCl and 20 mM Tris-HCl pH 8.4; Invitrogen), 200 μM of each dNTP (BIOLINE GmbH), 1.5 mM MgCl2 (Invitrogen), and 1.0 μM of each gene-specific primer pair. The PCR program consisted of an initial denaturation/activation step at 97°C for 2 min, followed by 28–36 cycles of 15 s at 95°C, 30 s at 57°C–63°C, 15 s at 72°C, and final elongation at 72°C for 5 min followed by cooling to 4°C. Primer sequences, annealing temperatures, and PCR conditions are summarized in Table 2. The pig-specific primers for pluripotency-related genes (OCT4, REX1, STAT3, SOX2, and NANOG) were designed and tested such that they did not cross-react with their mouse orthologs. For confirmation of the results, we used mouse ES cells (RHN-ES) as control, which showed no cross-reaction with all pig-specific primer used in this experiment. PCR products were separated on 2% agarose gels. As loading controls for endpoint RT-PCR primers specific for the hygromycin (contributed by the murine genome) were used.
Production of porcine iPS cells from OG2 transgenic fibroblasts
Lentivirus transduction of porcine Oct4-EGFP (OG2) fibroblasts and cell culture
Original lentivirus envelope protein (gp120) with a restricted host range was used as vectors. These were pseudotyped, and then encoated with heterogenous envelope protein from vesicular stomatitis virus glycoprotein. This envelope protein is highly stable and is reported to bind to cell surface phospholipids rather than a specific cellular protein receptor, thereby achieving a wide host range. The packaging vectors include Gag/Pol and RSV-REV.
Viral supernatants were collected every 24 and 48 h after transfection. Viral particles were concentrated by centrifugation at 14,000 rpm for 6–10 h at 4°C. Titration of the concentrated virus was done in SC1 cells (Mouse cell line). Eighty thousand SC1 cells were seeded and the individual virus was titrated using protamine sulfate. Day 4 after transduction the cells were fluorescent activated cell sorting (FACS) analyzed. The titer was calculated as virus per milliliters.
The cDNAs of human OCT4, SOX2, KLF4, and c-MYC, each cloned into a lentiviral vector, were used to produce pseudovirus in human 293T cells by transfection with each lentiviral vector along with the VSV-G envelope and packaging vector. Titrated viruses (multiplicity of infection-ratio of infectious virus particles to cells [MOI 5]) were used to infect primary porcine Oct4-EGFP (OG2) fibroblasts. On day 2 after infection, the cells were trypsinized and transferred to 10 cm plates seeded with mitomycin-inactivated mouse embryonic feeder layer (MEF). Subsequently, cells were maintained in human ES cell culture medium containing 4 ng/mL human FGF2. From days 10 to 14, colonies were monitored for the appearance of reprogrammed (iPS) cells showing EGFP fluorescence. Mechanical shearing was used to passage the cells.
Expression analysis of pluripotency-related genes by real-time RT-PCR analysis
Total RNA isolated from OG2 transgenic PFFs and 4 pig iPS cell clones using Trizol® (Invitrogen) was treated with RNase-free DNase (Epicentre Biotechnologies) and 0.5 μg RNA was used for cDNA synthesis using 20 U of MuLV Reverse Transcriptase (Applied Biosystems) in 20 μL volume according to manufacturer's instructions. The RNA quality was evaluated with Agilent 2100 Bioanalyzer (Agilent Technologies). Real-Time PCR was performed in an ABI 7500 Fast Real-Time System (Applied Biosystems) using SYBR Green–based 2 × master mix (Applied Biosystems). The following program was used: denaturation and activation of Taq Polymerase for 10 min at 95°C followed by 40 cycles of 95°C for 15 s and annealing, elongation, and data acquisition at 60°C/62°C/63°C for 1 min. Primer sequences, annealing temperatures, and PCR conditions are summarized in Table 2. The specificity of the PCR product was confirmed by dissociation curve analysis and size detection by agarose gel electrophoresis. Data generated by the Sequence Detection Software 1.3.1 were transferred to Microsoft Excel for analysis. For normalization, the housekeeping genes GAPDH and EEF1A1 were amplified along with the pluripotency-related genes. The expression ratio relative to the PFF cells was calculated by using the ΔΔCT method. The pig-specific primer did not cross-react with either human or mouse.
Results
Generation and characterization of OG2 transgenic pigs
Stable integration of an 18 kb murine OG2 construct in PFFs
Initial attempts to produce germ line reporter transgenic pigs using a minigene construct based on the 3.9 kb human OCT4 promoter (hOCT4) (Supplementary Fig. S1A; Supplementary Data are available online at
In the next round of experiments, female and male PFFs were cotransfected with the linearized 18 kb Oct4-EGFP and neomycin resistance constructs (Supplementary Fig. S1B). Integration of the OG2 construct was shown by PCR (Supplementary Fig. S1C) and positive cell clones were confirmed by Southern blotting with probes specific for the murine Oct4 promoter or for EGFP (Supplementary Fig. S1D). In total, 26 positive clones were identified.
Production of transgenic OG2 pigs via SCNT
Reconstructed embryos were either cultured in vitro up to the blastocyst stage or were immediately upon activation transferred (n = 811) to foster mothers. From 8 recipients 7 remained pregnant (Table 1). Two of the 7 pregnancies were terminated on day 25 of gestation to collect fetuses to establish fetal fibroblast cultures. The remaining 5 pregnant sows delivered 23 piglets at term. The OG2 transgene was detected in the 11 vital piglets by PCR and Southern blotting (Supplementary Fig. S1E) and in 11 of the 12 offspring lost perinatally (Supplementary Fig. S1F). The 11 other piglets developed normally (Supplementary Fig. S2A).
Germ line-specific expression of Oct4 in OG2 transgenic pigs
Germ cell-specific expression of the Oct4-EGFP marker was analyzed by RT-PCR, northern blotting, and fluorescence microscopy. EGFP fluorescence was found in embryos cloned from 3 different Oct4-EGFP (OG2) transgenic cell lines and cultured for 5–6 days to the expanded blastocyst stage (Fig. 1A). The intensity of fluorescence varied between blastocysts and EGFP fluorescence was detected in both the inner cell mass and the trophectoderm. From a total of 31 analyzed blastocysts, 27 (87.1%) displayed EGFP fluorescence. Parthenogenetic control blastocysts analyzed in parallel (Fig. 1B) did not exhibit EGFP fluorescence.

Reactivation of the Oct4-enhanced green fluorescent protein (EGFP) (OG2) transgene in pig.
EGFP fluorescent cells were detected in the genital ridges isolated from 12 of the transgenic fetuses on day 25 of development demonstrating germ line-specific expression of the transgene (Fig. 1C). Genital ridges isolated from wild-type fetuses were negative for EGFP fluorescence (Fig. 1D). In the pig, primordial germ cells settle in the genital ridge around day 24 of gestation [40]. EGFP-positive cells were not detected in other fetal tissues, including intestine (Fig. 1E), brain, heart, lung, mesonephros, and skin.
Porcine testis isolated from male transgenic piglets (two 7 days and one 2 months old) exhibited EGFP fluorescence in few isolated cells of the seminiferous tubules. Strong EGFP expression was detected in progenitor cells in adult testis from a 7-month-old adult transgenic boar (Fig. 2A). In contrast, FACS analysis and fluorescence microscopy of ejaculated spermatozoa collected from 3 mature OG2 boars revealed absence of EGFP fluorescence. Similar to OG2 mice, no EGFP expression was detected in other cell types or in testis isolated from wild-type piglets. The testis-specific expression of EGFP mRNA was confirmed by northern blotting, which revealed EGFP mRNA in transgenic testis isolated from young and adult pigs, but not in other organs, including heart, muscle, and kidney, isolated from these animals (Fig. 2B). Expression of EGFP mRNA was higher in testis isolated from adult transgenic animals than in testis isolated from neonates (Fig. 2B). Corresponding to the time course of EGFP expression in porcine testicular tissue, the porcine endogenous OCT4 transcript signal intensity was stronger in testis isolated from adult transgenic animals than in tissue from young transgenic pigs (Fig. 2C). This higher expression of the endogenous OCT4 in porcine testis was also found in testicular tissue isolated from adult wild-type animals (8 months, 2 years of age) when compared to young wild-type animals (4–7 days and 4 months of age) using real-time PCR analysis (Fig. 2D). Testis isolated from OG2 animals showed a stronger hybridization signal for the porcine OCT4 probe than testis isolated from wild-type animals (Fig. 2C). To verify whether or not or to what extent OCT4 expression in testis isolated from transgenic animals was due to the presence of the murine Oct4 gene, we carried out real-time PCR analysis with primers specific for the mouse Oct4 (Fig. 2E) and found also expression of murine Oct4 in testis from transgenic pigs.
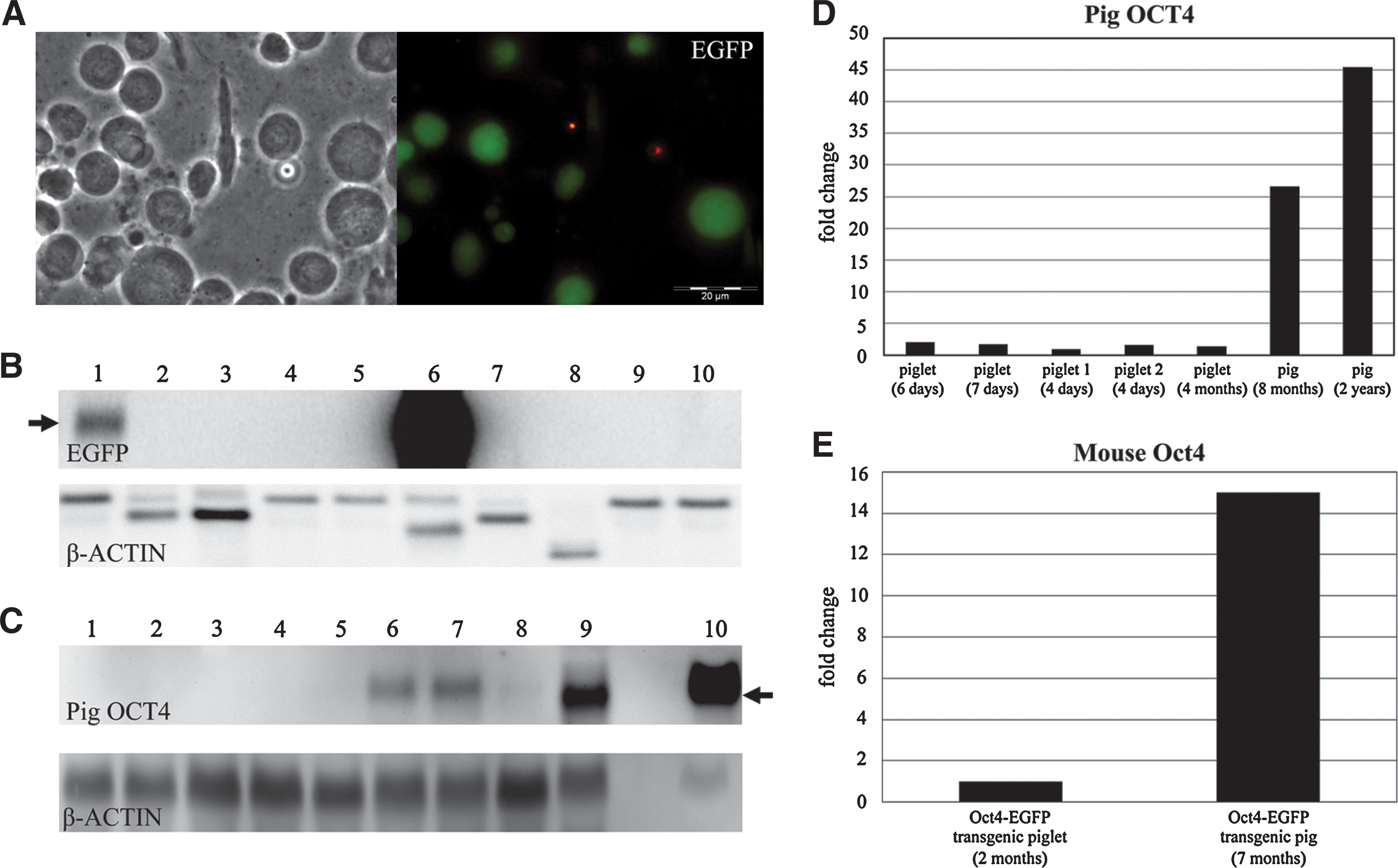
Reactivation of the Oct4-EGFP (OG2) transgene in the pig testis
Germ line transmission of OG2 transgene to F1-offspring
Germ line transmission of OG2 was tested by mating 2 transgenic boars with wild-type females. Approximately 60% (14 out of 23) of the blastocysts flushed on day 5 after fertilization expressed EGFP (Supplementary Fig. S2B, C).
One pregnant sow delivered 10 piglets at term with an average birth weight of 1.6 kg. Six of these piglets were viable and 4 piglets were either stillborn or too small to survive. An identical EGFP expression pattern as in the above cloned F0-males was observed in testis of one sacrificed neonatal F1-male. Stable transgene integration of OG2 was detected in 9 out of 10 piglets by Southern blot analysis (Supplementary Fig. S2D). The founder OG2 boar was estimated to have multiple integration sites of the OG2 construct, which apparently segregated in the offspring.
Transgene reactivation after reprogramming
Generation of inter-species mouse–pig hybrid cells by fusion
The usefulness of the transgene for monitoring reprogramming was demonstrated by fusion of porcine Oct4-EGFP (OG2) fibroblasts (Fig. 3A) with pluripotent murine ES cells (RHN-ES; Red fluorescent protein, Hygromycin resistance, and Nanog over-expression) (Fig. 3B). A total of 60 double-resistant hybrid colonies were subcloned, and 38 colonies showed EGFP fluorescence. The inter-species hybrids formed aggregated colonies typical for murine ES cells and showed a high proliferation rate (Fig. 3C).
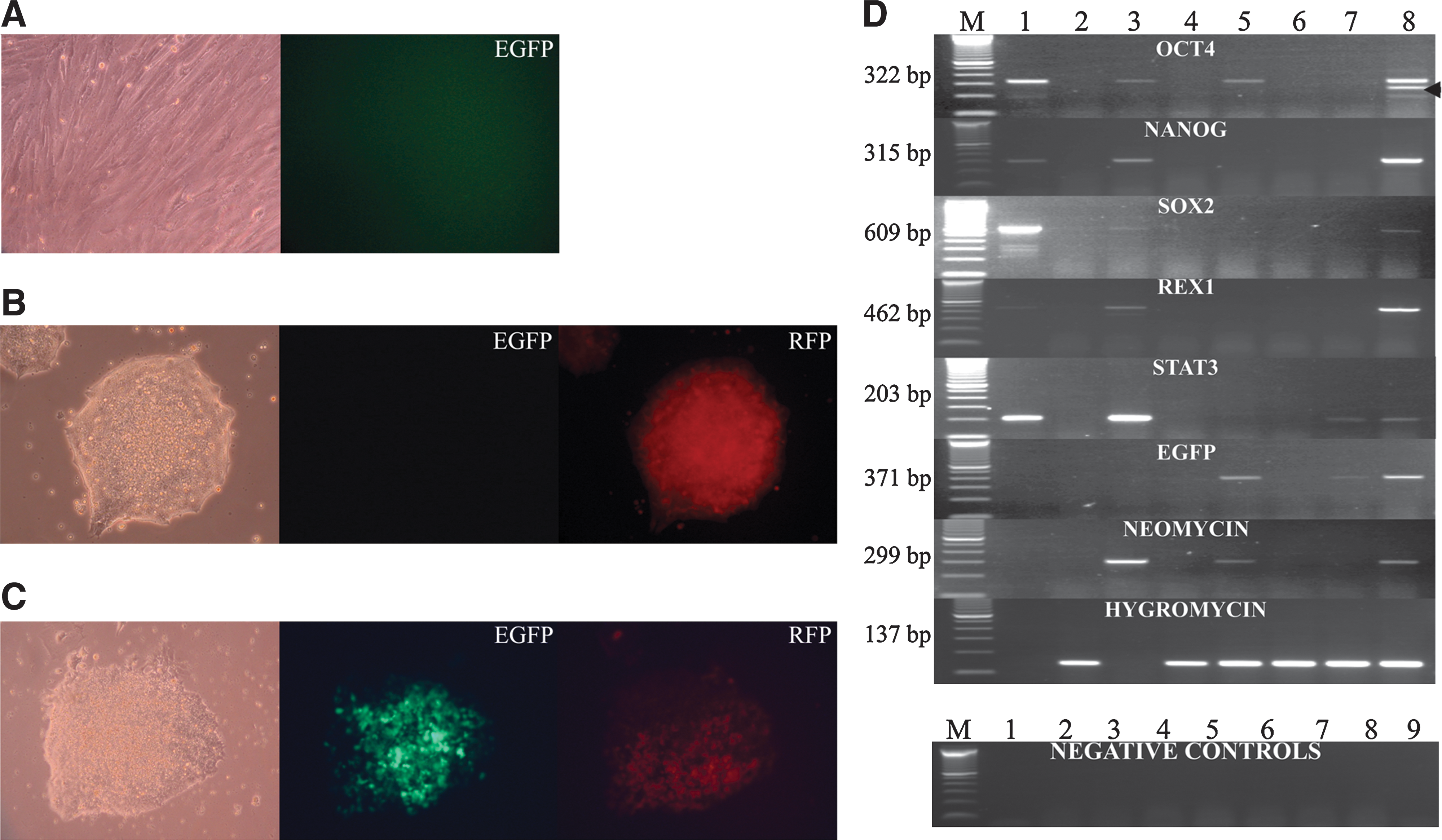
Reprogramming of porcine fibroblasts.
EGFP expression was first detected 3 days after fusion. Onset of EGFP expression in the fused mouse–pig cells (Fig. 3C) indicated that the OG2 transgene in the porcine genome was reactivated by factors from the murine ES cells. The mouse–pig hybrids rapidly lost the EGFP fluorescence during in vitro culture (Supplementary Fig. S2E). In a recent study from our laboratory using spectral karyotyping analysis, we had demonstrated that mouse–pig hybrids rapidly lose porcine chromosomes [41]. These incompatibilities between pig and mouse genome after inter-species cell fusion could cause splicing errors (Fig. 3D and Supplementary Fig. S3) and thus account for the loss of porcine chromosomes from hybrids.
Fluorescence intensity differed between hybrids and was asymmetrically distributed in various hybrid clones (Supplementary Fig. S2E). Porcine OCT4 sequences could not be amplified from hybrid clones 1, 3, and 4 (Fig. 3D). These clones did not express EGFP and the neomycin gene, likely due to the loss of porcine chromosomes carrying the transgenes from the hybrid cells. A similar rapid loss of Oct4 had been observed in primary outgrowths of mice and rat embryos before differentiation [42].
Gene expression analysis for markers of pluripotency was performed using pig-specific primers. Expression of porcine SOX2, STAT3, NANOG, and REX1 was detected in mouse–pig hybrid clone 5 (Fig. 3D). Hybrid clones 1–4 did not show expression of any of these genes, with the exception of hybrid 4, which expressed STAT3 and EGFP. Porcine OCT4 was expressed in hybrid clones 2 and 5; in addition to the expected 322 bp OCT4 amplicon, a smaller 257 bp product was observed in hybrid clone 5 (Fig. 3D). We hypothesize that the smaller product could be the result of a cryptic splice site in the porcine OCT4 transcript [41] (Supplementary Fig. S3). All hybrids expressed the hygromycin gene derived from the murine RHN-ES cells.
Generation of iPS cells from Oct4-EGFP (OG2) transgenic fibroblasts
Transduction of transgenic pig fibroblasts with 4 lentiviruses carrying human cDNAs for OCT4, SOX2, KLF4, and c-MYC resulted in EGFP-positive cells, which appeared as cell clusters 10–12 days after transduction (Fig. 4A). However, these cells could not be maintained over prolonged periods. Real-Time PCR of 4 other clones at passage number 6–8 revealed that the porcine OCT4, SOX2, and NANOG were upregulated (Fig. 4B) compared to nontransduced fibroblasts. Expression levels of pluripotency genes differed between individual pig iPS clones. The highest mRNA expression of SOX2 was detected in clones 2 and 4 (>80 and 100-fold higher than in PFF), the highest expression of OCT4 was found in clone 2 (>20-fold higher than in PFF), and the highest levels of NANOG were expressed at in clone 1 (5-fold higher than in PFF) (Fig. 4B). These differences between individual porcine iPS cell clones have also been found in human iPS cell lines and were mainly attributed to residual expression of the respective donor cells, thus reflecting different degrees of reprogramming [43].
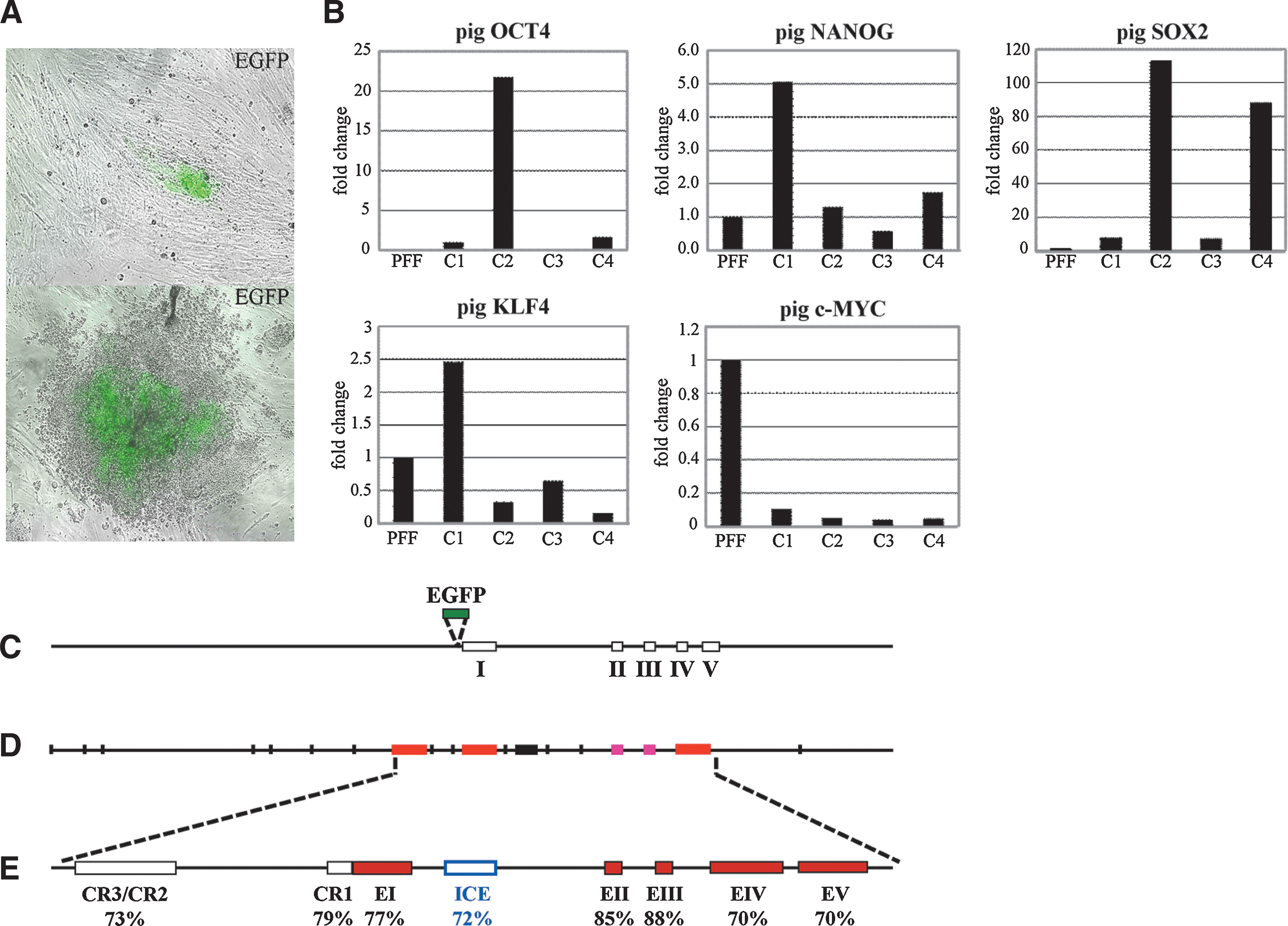
Generation of porcine-induced pluripotent stem (iPS) cells and upregulation of porcine pluripotent genes.
Discussion
The goal of this study was the production and characterization of transgenic pigs carrying the Oct4-EGFP transgene to allow successful monitoring of Oct4 expression by EGFP fluorescence after reprogramming experiments (SCNT, cell fusion, or generation of iPS approach). This article is the first report on germ line transgenic Oct4-EGFP pigs. We demonstrate that the fluorescent reporter is exclusively expressed in pluripotent cells during embryonic, fetal, and postnatal development. The successful reactivation of the Oct4 promoter after SCNT in reconstructed blastocysts, after fusion of Oct4-EGFP transgenic fibroblasts with pluripotent murine stem cells and after transduction with lentiviral vectors carrying the classical 4 reprogramming factors clearly demonstrates the usefulness and robustness of our approach. It has to be emphasized that until now, true germ line-competent, bona fide porcine ES cells have not yet been produced, and both morphology and culture conditions for true porcine stem cells are an enigma. The lack of optimized cell culture conditions for pluripotent cells in pigs impedes the proliferation and establishment of fully reprogrammed and stable pig cell lines after reprogramming experiments like cell fusion or transduction with lentiviral vectors and requires further study. The OG2 transgenic pigs provide a novel model for studying the fate of porcine germ cells during fetal development and will facilitate isolation and purification of porcine iPS and ES cells and development of reliable culture conditions for pluripotent porcine cells.
Previous attempts in our laboratory to produce germ line reporter transgenic pigs using a minigene construct based on the 3.9 kb human OCT4 promoter (hOCT4) were not successful. The Oct4-EGFP (OG2) construct is based on the 18 kb murine genomic Oct4 region harboring all regulatory elements of the Oct4 gene (Fig. 4C). Alignment of the murine, human, and bovine upstream promoter sequences of the Oct4 gene revealed a high level of sequence conservation [44]. In addition, a highly conserved intron region was identified in the Oct4 gene from mouse, pig, human, rat, and cattle (Fig. 4D, E). The OG2 transgene has been successfully used in previous studies on pluripotency in the mouse [14,19]. When we started this experiment and began to produce transgenic pigs, the porcine genome was not completely sequenced, which prompted us to use the mouse Oct4-EGFP construct in the pig. The pigs reported here are the first animals that are equivalent to the existing OG2 mouse model. The use of mouse Oct4-EGFP constructs in cattle and miniature pigs has not been successful with regard to germ line-specific expression and transmission [13,45]. The results presented here indicate that the genomic construct that harbors all regulatory elements of the Oct4 gene is required for successful production of germ line-specific Oct4-EGFP transgenic pigs.
F1-blastocysts collected on day 5 showed EGFP fluorescence in both compartments of the blastocysts, the inner cell mass and the trophectoderm (Supplementary Fig. S2C). In contrast to murine embryos, Oct4 protein persists in the trophectoderm of porcine [12,45] and bovine blastocysts [12,13,46], even though mRNA production ceases in trophectodermal cells [47]. This is attributed to prominent differences in preimplantation development between the laboratory mouse model and the domestic pig. Porcine blastocysts hatch from the zona pellucida around day 6/7 after fertilization and undergo rapid changes in morphology, finally resulting in a filamentous structure, primarily due to massive proliferation of trophectodermal cells. Porcine conceptuses do not attach to the endometrium before days 13–14 of gestation [40].
In the mouse, observation of EGFP fluorescence was used for identification of Oct4-positive migratory and postmigratory germ cells [14]. Primordial germ cells (PGCs) are the embryonic precursors of gametes originate from the pluripotent epiblast and migrate from the base of the allantois via the gut mesentery to the gonads where they differentiate [48 –50]. Here, we detected lower expression of the endogenous OCT4 gene in postnatal testis isolated from newborn transgenic or wild-type piglets than in testis isolated from adult animals. Similarly, EGFP expression was stronger in tissue from adult versus young testicular tissue isolated from transgenic animals. Previous studies had shown that Oct4 is expressed in mitotically arrested prespermatogonia of the mouse testis before birth and confined to type A spermatogonia in the testis after onset of spermatogenesis. It is downregulated in type B spermatogonia and spermatocytes in the adult testis. Oct4-EGFP fluorescent cells in the mouse testes were classified as primitive type A spermatogonia [15,19]. Results presented here demonstrate that the Oct4-EGFP (OG2) transgene is fully appropriate for observing pluripotent germ cells in the pig and that OCT4 is expressed in presumptive primordial germ cells of porcine fetuses and in progenitor cells in postnatal testes, but not in ejaculated spermatozoa.
A promising application of the Oct4-EGFP (OG2) transgenic pigs or cells thereof will be in reprogramming studies, where the EGFP expression can readily identify pluripotent cells. In reprogramming studies with murine cells, the Oct4-EGFP transgene has been used to determine de-differentiation of somatic cells to identify pluripotent cells by fluorescence microscopy, to purify pluripotent cells by FACS sorting, and to observe reprogramming of somatic cells by fusion with pluripotent cells [33,34,51 –54]. Here, we show the functionality of the Oct4-EGFP (OG2) transgene in the pig by demonstrating successful reprogramming of somatic cells by fusion with pluripotent mouse stem cells and by the production of iPS. Upregulation of endogenous genes associated with pluripotency, for example, OCT4, SOX2, and NANOG in most of iPS cells and fused hybrids (clone 5), is clear evidence for successful reprogramming. NANOG is critical for induction of pluripotency in fused hybrids and is required for complete reprogramming into a fully pluripotent state [55].
In conclusion, these data demonstrate the usefulness of Oct4-EGFP (OG2) transgenic pigs for reprogramming studies, rendering the pig an important model for regenerative medicine. The successful reactivation of the Oct4 promoter after SCNT in reconstructed blastocysts, after fusion of Oct4-EGFP transgenic fibroblasts with pluripotent murine stem cells or after transduction with lentiviral vectors carrying the classical 4 reprogramming factors, clearly demonstrates the usefulness and robustness of our approach. Somatic cells isolated from the Oct4-EGFP (OG2) transgenic pigs will facilitate detection of reprogrammed somatic cells after transfection with integrating and nonintegrating constructs expressing various combinations of transcription factors [29 –32] and for optimization the culture conditions for porcine iPS and ES cells. The OG2 cell lines provide a useful and reliable marker for positively identifying and isolating pluripotent cells, which show a reactivated OCT4 gene. These transgenic animals will be valuable for studying chimera formation and germ line contribution of porcine iPS/ES cells.
Footnotes
Acknowledgments
The authors thank Brigitte Barg-Kues, Petra Hassel, Anna-Lisa Queisser, and Stephanie Holler for excellent technical assistance and Dr. Nadine Hornen for work with the phOCT4-EGFP construct. We thank Dr. Tomo Saric (Institute of Neurophysiology, Medical Center, University of Cologne) for providing the RHN-ES cell line. Special thanks to the staff of the pig facility for preparing the recipient animals and care taking of cloned pigs. This study was supported by a grant from the Bundesministerium für Bildung und Forschung (BMBF, no. 01GN 0540).
Author Disclosure Statement
The authors declare no conflict of interest in connection with the submitted article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.