
Abstract
Hematopoietic stem cell (HSC) self-renewal is tightly regulated by a complex crosstalk between many cell-intrinsic regulators and a variety of extrinsic signals from the stem cell niche. In this study, we examined whether the p38 mitogen-activated protein kinase (p38) is one of the intrinsic regulators that can negatively regulate HSC self-renewal in vitro and whether inhibition of p38 activity with a small molecule inhibitor can promote HSC expansion ex vivo. The results from this study showed that sorted mouse bone marrow Lin−Sca1+c-kit+ cells (LSK+ cells) exhibited selective activation of p38 after culture in a serum-free medium supplemented with 100 ng/mL stem cell factor, thrombopoietin, and Flt3 ligand. The activation of p38 was associated with a significant reduction in HSCs and induction of apoptosis and cellular senescence in LSK+ cells and their progeny. Addition of the specific p38 inhibitor SB203580 (SB, 5 μM) to the culture inhibited the activation of p38 in LSK+ cells, which led to increase in HSC self-renewal and ex vivo expansion as shown by the cobblestone area forming cell assay, competitive repopulation, and serial transplantation. The increase in HSC expansion is likely attributable to SB-mediated inhibition of HSC apoptosis and senescence and upregulation of HoxB4 and CXCR4. These findings suggest that p38 plays an important role in the regulation of HSC self-renewal in vitro and inhibition of p38 activation with a small molecule inhibitor may represent a novel approach to promote ex vivo expansion of HSCs.
Introduction
T
Although p38 plays an important role in regulation of hematopoiesis during fetal development primarily via regulation of erythropoietin production [2,10], it is apparently dispensable for normal hematopoiesis in adult, as mice with conditioned knockout of the p38α (Mapk14) gene at age of 6–8 weeks were apparently normal until developing respiratory problems and dying at about 10 months of age [11,12]. In addition, HSCs from p38α null mice can reconstitute the hematopoietic system of a lethally irradiated recipient as efficiently as the cells from wild-type mice [10]. However, activation of p38 has been implicated in bone marrow (BM) suppression in various pathological conditions, including aplastic anemia and myelodysplastic syndromes [6,9,13]. Further, recently it was shown that mutation of the ATM gene and knockout of the FoxO3 gene induced premature exhaustion/senescence of HSCs [14,15]. The induction of HSC exhaustion/senescence was associated with an elevated production of reactive oxygen species, a selective activation of p38, and an upregulation of p16Ink4a (p16) in HSCs. Pharmacological inhibition of p38 activity rescured the defects of HSCs from ATM mutants and FoxO3 knockout mice. These findings indicate that p38 is dispensible for HSC self-renewal and normal hematopoiesis in a homeostatic condition, but it plays an important role in regulation of HSC self-renewal under stress conditions. Particularly, its activation by oxidative stress can mediate the induction of HSC senescence via up-regulation of p16 [14].
Currently, the use of HSC transplantation as an effective therapy for various hematological diseases is limited by the inability to obtain sufficient number of HSCs for transplantation [16,17]. Ex vivo expansion of HSCs could potentially generate ample HSCs to overcome the obstacle. Although considerable progress has been made in identifying positive regulators that can stimulate HSC self-renewal and ex vivo expansion [18], little is known about the inhibitory factors that limit HSC self-renewal. We suspect that p38 may be one of the potential inhibitory factors and its activation by oxidative stress resulting from cell culture in a normoxic condition (20% of O2) may inhibit ex vivo HSC expansion [14,15,19 –21]. To test this hypothesis, we cultured Lin−c-kit+Sca1+ (LKS+) cells isolated from mouse BM in serum-free medium supplemented with stem cell factor (SCF), TPO, and Flt3 ligand (STF) in the presence or absence of SB203580 (SB), a specific p38 inhibitor [22]. It was found that the LKS+ cells cultured without SB exhibited specific activation of p38, upregulation of p16, and significant reduction in HSCs. Addition of SB to the cell cultures abrogated p38 activation, inhibited the expression of p16, and increased the expansion of HSCs. These findings confirm that activation of p38 can inhibit HSC self-renewal in vitro and inhibition of p38 activity using a small molecule inhibitor can promote ex vivo expansion of HSCs.
Materials and Methods
Reagents
Phycoerythrin (PE)- or PE-Cy7-conjugated anti-Sca-1 (Clone E13-161.7, rat IgG2a); APC-conjugated anti-c-kit (Clone 2B8, rat IgG2b); biotin-conjugated anti-CD3ɛ (Clone 145-2C11, Hamster IgG1), anti-CD45R/B220 (Clone RA3-6B2, rat IgG2a), anti-Gr-1 (Clone RB6-8C5, rat IgG2b), anti-Mac-1 (Clone M1/70, rat IgG2b), and anti-Ter-119 (Clone Ter-119, rat IgG2b); purified rat anti-CD16/CD32 (Clone 2.4G2, Fcγ receptor blocker, rat IgG2b); and PE or fluorescent isothiocyanate (FITC) conjugated streptavidin were purchased from BD Pharmingen (San Diego, CA). Antibodies against phosphorylated-p38 (p-p38, Cat# 4631), phosphorylated-Erk (p-Erk; Cat# 4370) and phosphorylated-JNK (p-JNK, Cat# 9251) were purchased from Cell Signaling Technology (Beverly, MA). Alexa fluor-555–conjugated goat anti-rabbit IgG antibody (Cat# A21430) and Alexa fluor-488–conjugated rabbit anti-mouse Ig G antibody (Cat# A21204) were obtained from Invitrogen (Carlsbad, CA). Recombinant mouse SCF (Cat#: 455-MC), mouse TPO (Cat#: 488-Tpo) and mouse Flt-3 ligand (FL, Cat#: 427-FL) were purchased from R&D Systems (Minneapolis, MN). Stem Pro®-34 SFM (Cat# 10640) was obtained from Invitrogen. SB203580 (4-[4-fluorophenyl]-2-[4-methylsulfinylphenyl]-5-[4-pyridyl] 1H-imidazole or SB; Cat# 559389), a p38-specific inhibitor, was purchased from Calbiochem (San Diego, CA).
Mice
Male C57BL/6-Ly-5.2 (Ly5.2) and C57BL/6-Ly-5.1 (Ly5.1) mice were purchased from The Charles River Laboratories (Wilmington, MA) and The Jackson Laboratories (Bar Harbor, ME), respectively. Mice were housed 4 to a cage at the Medical University of South Carolina AAALAC-certified animal facility. They received food and water ad libitum. All mice were used at ∼8–12 weeks of age. The Institutional Animal Care and Use Committee of Medical University of South Carolina approved all experimental procedures used in this study.
HSC ex vivo expansion
HSC-enriched LSK+ cells were prepared and sorted as we previously reported [23]. Two hundred LSK cells were seeded in a well of 96-well plates and cultured in 100 μL of HSC expansion medium (Stem Pro-34 SFM supplemented with 100 ng/mL SCF, TPO, and FL) in the presence of vehicle [0.1% dimethyl sulfoxide (DMSO)] or SB (5 μM) at 37°C, 5% CO2, and 100% humidity. The cells were fed with 100 μL fresh HSC expansion medium along with the same concentrations of DMSO or SB after 3 days of culture. The cells were harvested after 7 days of incubation and pooled from a minimum of 5 wells for various assays.
Analysis of p38, Erk, and JNK activation by immunofluorescent microscopy
Approximately 5,000 fresh sorted LSK+ cells or the cells harvested from day-7 cultures of LSK+ cells were cytospun onto slides at 1,000 rpm×5 min. Cells were fixed with 4% paraformaldehyde and permeabilized with 0.2% Triton X-100. After block with 5% normal goat serum, cells were incubated with anti-CD-p-p38 (1:100), anti-p-Erk (1:100), or anti-p-JNK (1:100) overnight at 4°C. After extensive washing, the first antibodies were observed with Alexa fluor-555–conjugated goat anti-rabbit IgG antibody (1:200). Nuclei were counterstained with DAPI (1 μg/mL) for 2 min. Slides were mounted with Vecta shield; cells were viewed; images of the cells were acquired by a Zeiss Axio Observer (Carl Zeiss Microimaging Inc., GmbH, Jena, Germany). The images were captured using AxioVision (4.7.1.0) software (Carl Zeiss Imaging Solutions GmbH, Jena, Germany). The collected images were then displayed with the Adobe Photoshop V6.0.
Analysis of p38 activation in LSK+ cells by phospho-specific flow cytometry
Approximately 1×106 cells freshly isolated BM-mononuclear cells (MNCs) or equivalent number of cells harvested from day-7 cultures of LSK+ cells were first stained with PE-conjugated anti-lineage antibodies (B220, Thy 1, Ter-119, Gr-1, and CD11b) after blocking with anti-CD16/CD32 antibody. The cells were then fixed with CytoFix/Cytoperm (BD Pharmingen), permeablized with Cytoperm™ Plus (BD Pharmingen), and stained with APC-c-Kit, PE-Cy7-Sca-1 and mouse anti-p-p38 antibody (1:100) according to the manufacturers' instructions. p-p38 staining was detected by Alexa fluor-488–conjugated rabbit anti-mouse IgG (1:200). Cells were analyzed on a FACS Caliber (Becton Dickinson, San Jose, CA), and the data were analyzed using the CellQuest software (Becton Dickinson).
Cell phenotypic analysis by flow cytometry
Briefly, 1×105 cells harvested from day-7 cultures of LSK+ cells were incubated with biotin-conjugated antibodies against CD3ɛ, CD45R/B220, Gr-1, Mac-1, and Ter-119 and then with streptavidin-FITC. After incubation with anti-CD16/CD32 antibody, the cells were stained with anti-Sca-1-PE and anti-c-kit-APC antibodies. Cells were analyzed on a FACS Caliber (Becton Dickinson), and the data were analyzed using the CellQuest software (Becton Dickinson).
Colony-forming cell and cobblestone area forming cell assays
The colony-forming cell (CFC) assay was performed by culturing hematopoietic cells in the MethoCult GF M3434 methylcellulose medium (Stem Cell Technologies, Vancouver, BC) according to the manufacturer's instruction. Colonies of colony-forming unit (CFU)-granulocyte macrophage and burst-forming unit-erythroid were scored on day 7 and those of CFU-granulocyte, -erythrocyte, -monocyte, and -megakaryocyte on day 12 of the incubation. The cobblestone area forming cell (CAFC) assay was performed as described elsewhere [23]. The frequencies of CAFCs were determined at days 14 and 35 to represent the hematopoietic function of HPCs and HSCs, respectively.
Competitive repopulating assay
The cells harvested from a day-7 culture of 200 LSK+ cells (from Ly5.2 mice) were mixed with 2×105 competitive BM-MNCs (from Ly5.1 mice) and transplanted into lethally irradiated (9.5 Gy TBI) Ly5.1 mice by tail-vein injection. Peripheral-blood samples were obtained from the retro-orbital plexus using heparin-coated micropipets (Drummond Scientific, Broomall, PA) at 8 and 20 weeks after transplantation. After red blood cells had been lysed by 0.15 M NH4Cl, the samples were stained with FITC-conjugated anti-Ly5.2 and analyzed for donor-derived cells on a FACS Caliber (Becton Dickinson). Donor-derived cells (Ly5.2) in T-cell, B-cell, granulocyte, and monocyte/macrophage lineages were analyzed by staining the cells with PE-conjugated anti-Thy-1.2, PE-conjugated anti-B220, and PE-conjugated anti-Gr-1/Mac-1, respectively.
Secondary transplantation
BM cells were harvested from the primary recipient 20 weeks after transplantation as described above. They (5×105) were mixed with 2×105 competitor cells (from Ly5.1 mice) and transplanted into each lethally irradiated (9.5 Gy TBI) recipient (Ly5.1 mouse). Twelve weeks after the secondary transplantation, donor cell engraftment (Ly5.2) was determined by flow cytometric analysis as described above.
Apoptosis assay
The expanded cells were first stained with biotin-conjugated lineage antibodies and PE-conjugated streptavidin and PE-Cy7-conjugated anti-Sca1 and APC-conjugated anti-c-kit antibodies as previously described. The cells were then stained with FTIC-Annexin V from the apoptosis assay kit (BD Biosciences) according to the manufacturer's protocol. Apoptotic cells were determined by flow cytometric analysis on a FACS Caliber (Becton Dickinson) and the data were analyzed using the CellQuest software (Becton Dickinson).
Senescence-associated β galactosidase staining
Senescence-associated β galactosidase (SA-β-gal) activity in freshly sorted LKS+ cells and the cells harvested from a day-7 culture of LSK+ cells was determined using a SA-β-gal staining kit (Cat# 9860) from Cell Signaling Technology according to the manufacturer's instructions and our previously reported procedures [23].
Real-time reverse transcription-polymerase chain reaction
Total RNA was extracted using the RNeasy Mini Kit (Qiagen Sciences, Germantown, MD) according to the manufacturer's instructions. First-strand cDNA was synthesized from total RNA using Super Script II first-strand synthesis system (Invitrogen). Polymerase chain reaction (PCR) primers for p16, p19Arf, p21 and p53 were predesigned and commercially available from Applied Biosystems (Foster City, CA). All other primers were customer-designed and obtained from Integrated DNA Technologies (Coralville, IA) and the sequences of the primers were listed in Table 1. The threshold cycle (Ct) value for each gene was normalized to the Ct value of glyceraldehyde 3 phosphate dehydrogenase. The relative mRNA expression was calculated using the comparative CT (2−ΔΔCt) method as previously described [23].
Statistical analysis
The data were analyzed by analysis of variance. In the event that analysis of variance justified post hoc comparisons between group means, these were conducted using the Student-Newman-Keuls test for multiple comparisons. For experiments in which only single experimental and control groups were used, group differences were examined by unpaired Student's t-test. Differences were considered significant at P<0.05. All of these analyses were done using GraphPad Prism from GraphPad Software, Inc. (San Diego, CA).
Results
Culture of LKS+ cells in vitro selectively activates p38
Freshly isolated mouse BM LKS+ cells showed no detectable activation of p38, Erk, and JUN by immunofluorescent staining using antibodies specifically against p-p38, p-Erk, and p-JNK (Fig. 1A). After they were cultured in a serum-free medium supplemented with STF for 7 days, a significant increase in p38 phosphorylation was detected in the expanded cells (Fig. 1A, B). The activation of p38 could be inhibited by the addition of SB (Fig. 1B), which is a p38-specific inhibitor. However, the phosphorylation of other MAPKs such as Erk and JNK remained undetectable in the cells after in vitro culture (Fig. 1A). These findings suggest that culture of LKS+ cells in vitro can selectively activate p38. This suggestion is confirmed by p38 phospho-specific flow cytometric analysis that can measure p38 phosphorylation specifically in LKS+ population as shown in Fig. 1C.
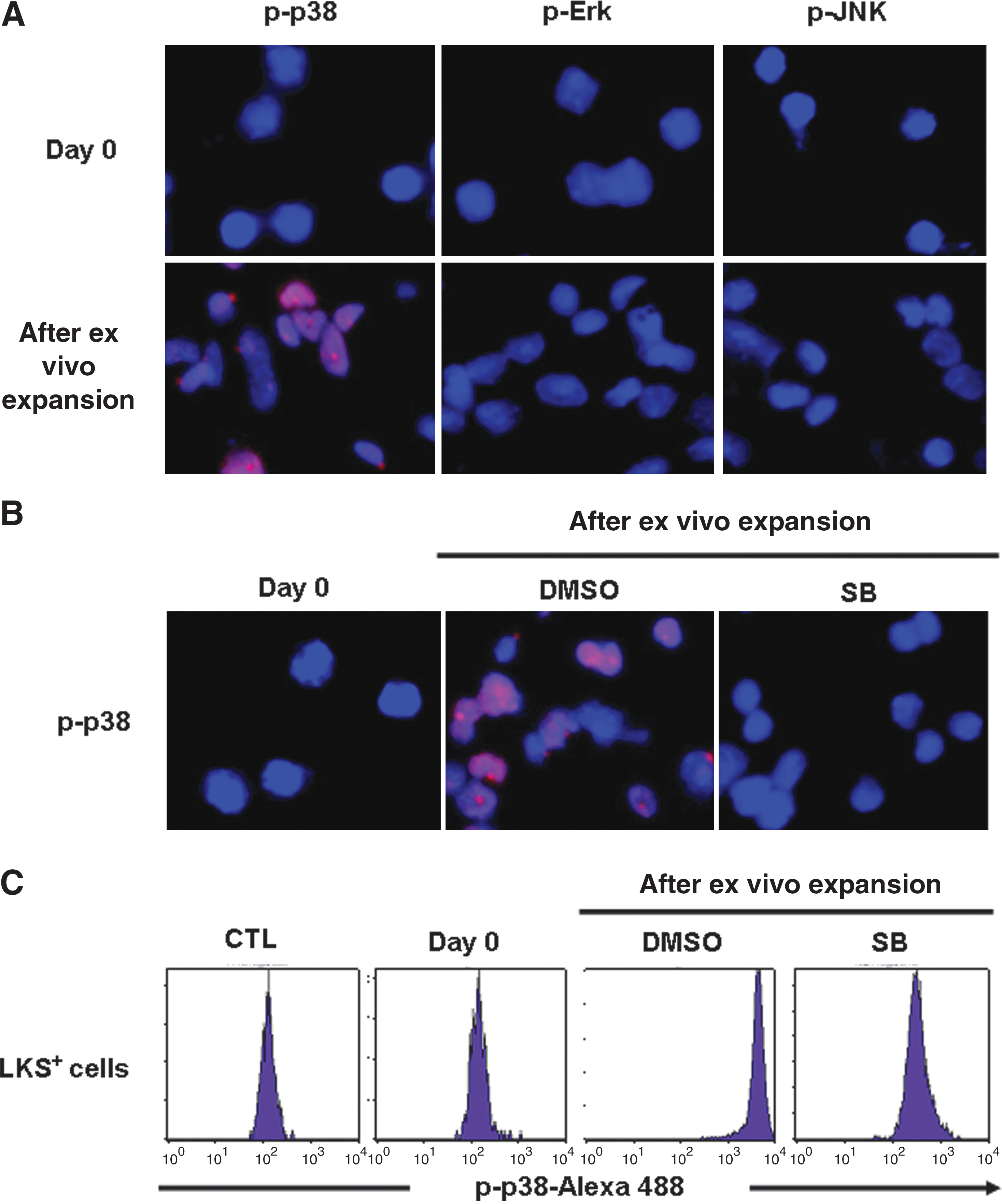
Culture of Lin−c-kit+Sca1+ (LKS+) cells in vitro selectively activates p38.
Inhibition of p38 significantly increases the production of LKS+ cells
To determine if activation of p38 inhibits HSC self-renewal and ex vivo expansion, we cultured LKS+ cells in the presence of vehicle (0.1% DMSO) or SB (5 μM) for 7 days. The cells harvested from the cultures were numerated to quantify total number of MNCs and phenotypically analyzed by flow cytometry to determine the frequencies of lineage negative cells (Lin− cells), Lin−c-kit+Sca1− (LKS−) cells and LKS+ cells. As shown in Fig. 2, LKS+ cells cultured with or without SB for 7 days produced a similar number of MNCs. However, cell cultures with SB contained significantly more Lin−, LKS−, and LKS+ cells compared to cultures without SB. Particularly, the number of LKS+ cells increased 8.0- and 4.9-fold after 7 days culture in the presence or absence of SB, respectively, compared to input (day 0), indicating that p38 inhibition increased the production of LKS+ cells.
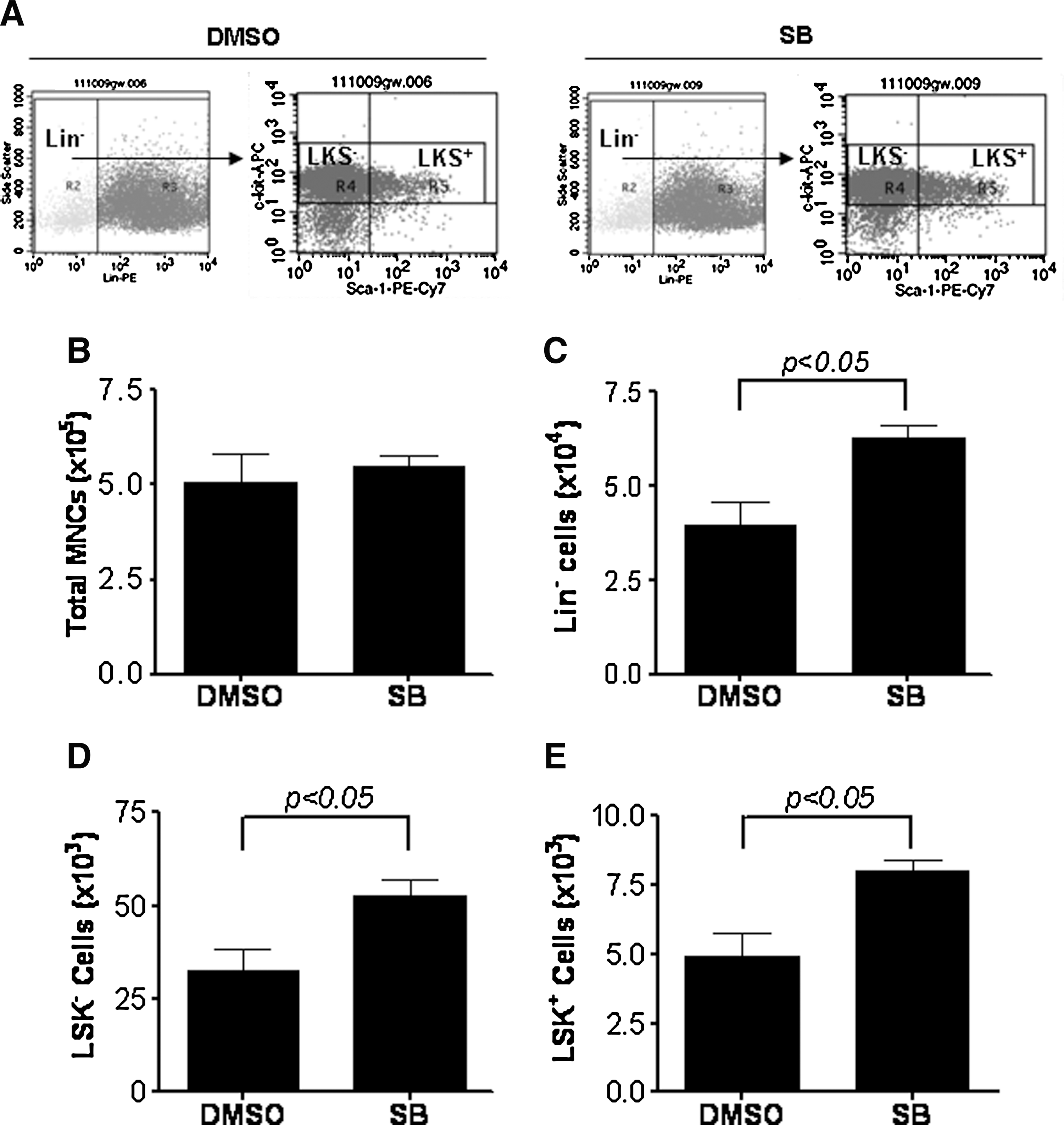
Inhibition of p38 significantly increases the production of LKS+ and Lin−c-kit+Sca1− cells (LKS− cells). LKS+ cells were cultured in vitro in the presence of vehicle (0.1% DMSO) or SB (5 μM) for 7 days. The cells harvested from the cultures were numerated and phenotypically analyzed by flow cytometry.
Inhibition of p38 increases the expansion of HSCs
To determine if the increase in LKS+ production induced by p38 inhibition leads to a greater expansion of HSCs, we analyzed day-35 CAFCs as an in vitro surrogate of HSCs [24]. In addition, day-14 CAFCs were analyzed to quantify HPCs [24]. As shown in Fig. 3, cell cultures with SB contained 7.6- and 2.1-fold more day-35 and day-14 CAFCs, respectively, than cultures without SB (Fig. 3). More importantly, when the numbers of day-35 and day-14 CAFCs in the cultured cells were expressed as a ratio to those in input, it was found that without p38 inhibition the majority of HSCs cultured with STF could not self-renewal during the culture, resulting in a net 74% reduction in day-35 CAFCs compared to input. This finding agrees with the previous observation showing that HSCs from mouse BM lose radioprotective and long-term engraftment potential after ex vivo expansion in the presence of multiple cytokine combinations [25]. In contrast, with p38 inhibition, HSCs underwent self-renewing proliferation, leading to an almost twofold increase in day-35 CAFCs compared to input (Fig. 3). Moreover, this also reflects a 7.6-fold increase in day-35 CAFCs compared to the cells cultured without SB, indicating that p38 inhibition promotes ex vivo HSC expansion.
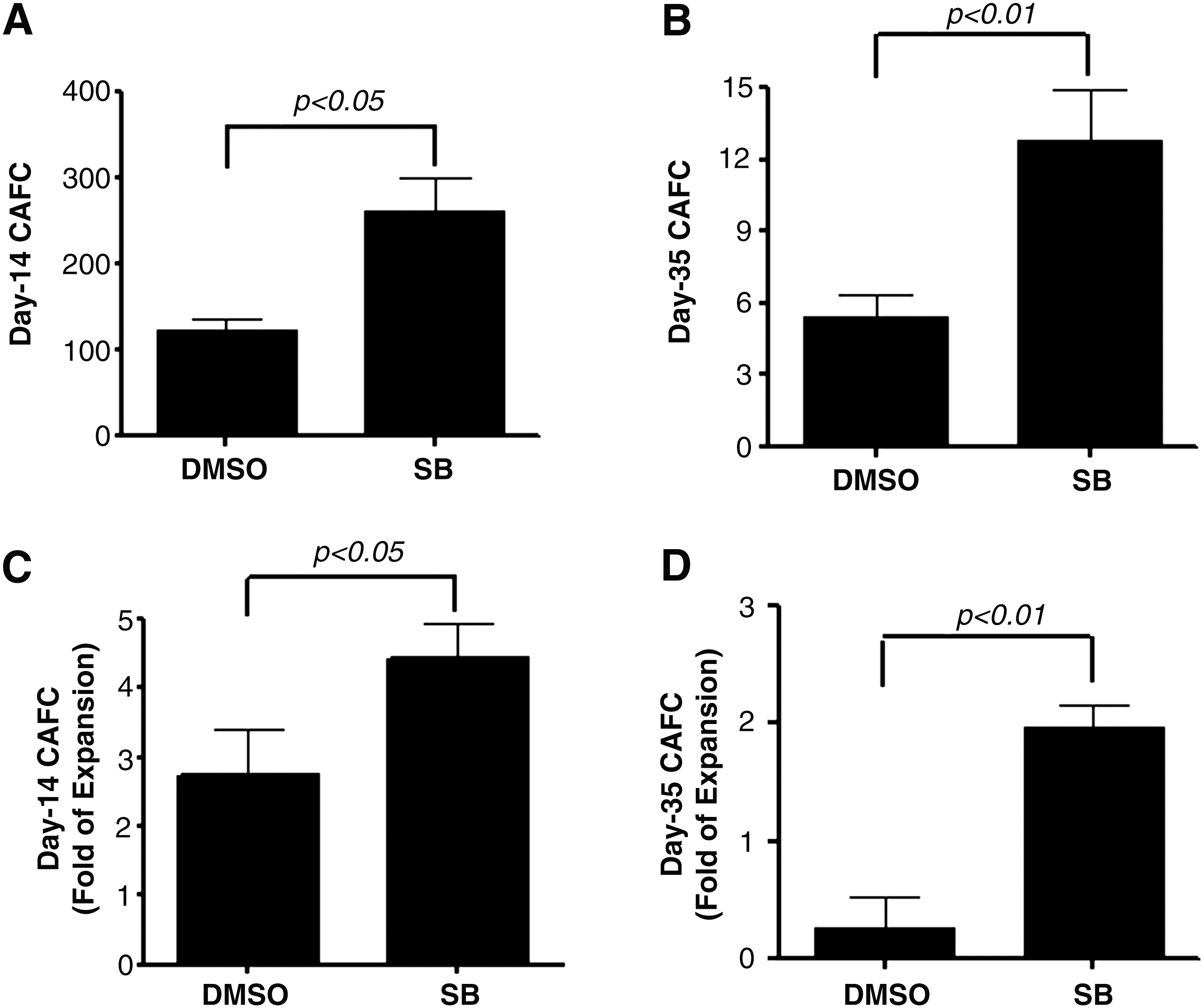
Inhibition of p38 increases the expansion of hematopoietic stem cells (HSCs). LKS+ cells were cultured in vitro in the presence of vehicle (0.1% DMSO) or SB (5 μM) for 7 days. The expanded cells harvested from the culture were numerated and analyzed by the cobblestone area forming cell (CAFC) assay to measure HSCs and hematopoietic progenitor cells by quantifying the frequencies of day-14 CAFCs and day-35 CAFCs, respectively.
Inhibition of p38 increases HSC long-term engraftment after transplantation
Because HSC transplantation is considered as the only golden-standard assay for long-term repopulating HSCs, we transplanted the cells harvested from LSK+ cell cultures with or without SB into lethally irradiated recipients to validate if inhibition of p38 promotes HSC self-renewal and ex vivo expansion. Peripheral blood from these recipients was analyzed for donor cell engraftment at 8 and 20 weeks after transplantation. As shown in Fig. 4 and Supplementary Figs. S1 and S2 (Supplementary Data are available online at
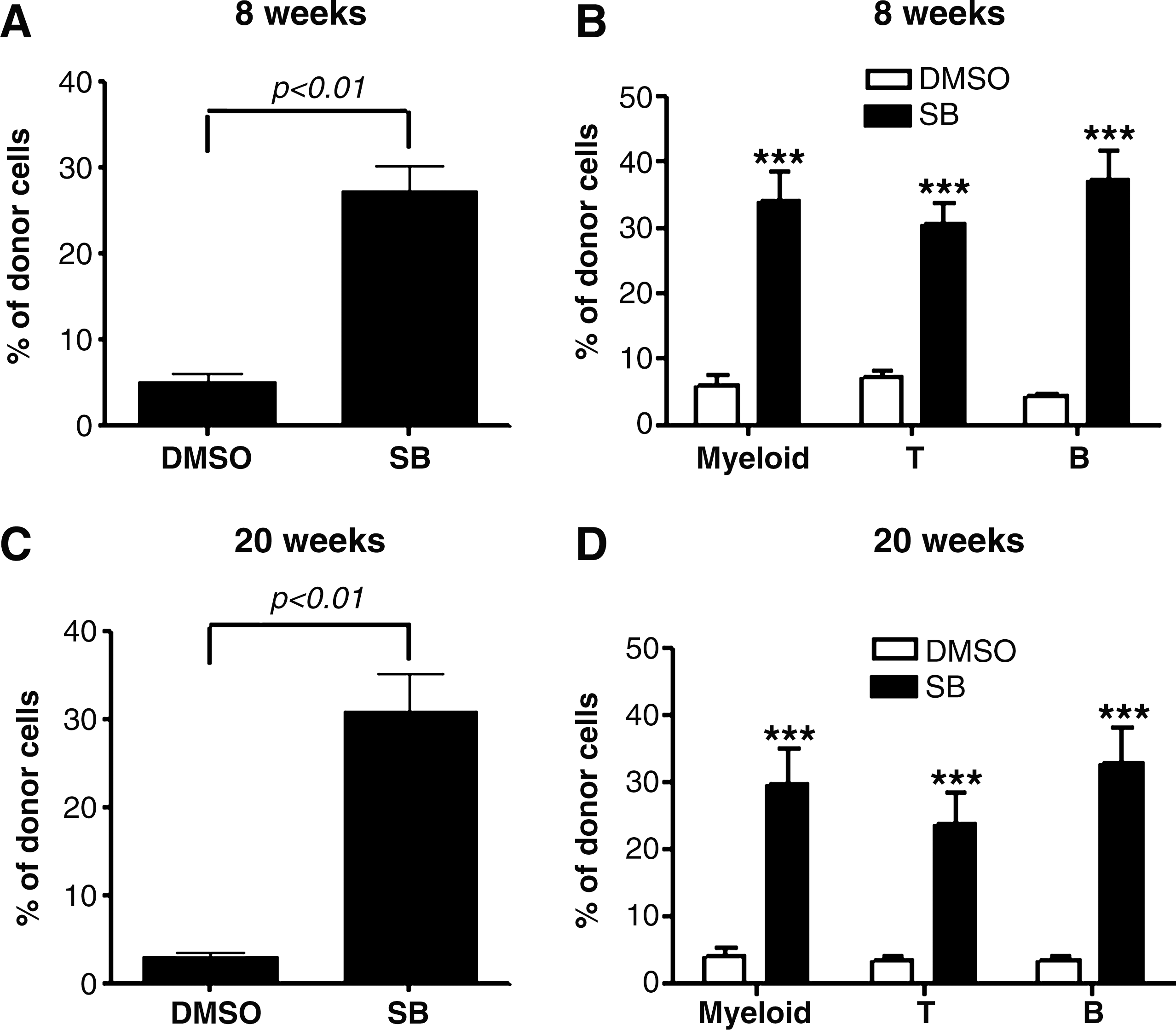
Inhibition of p38 increases HSC long-term engraftment after primary transplantation. LKS+ cells were cultured ex vivo in the presence of vehicle (0.1% DMSO) or SB (5 μM) for 7 days. The expanded cells harvested from the culture with 200 LSK+ cells were transplanted into a lethally irradiated mouse along with 2×105 competitors. Donor cell engraftment in peripheral blood was determined 8 and 20 weeks after transplantation.
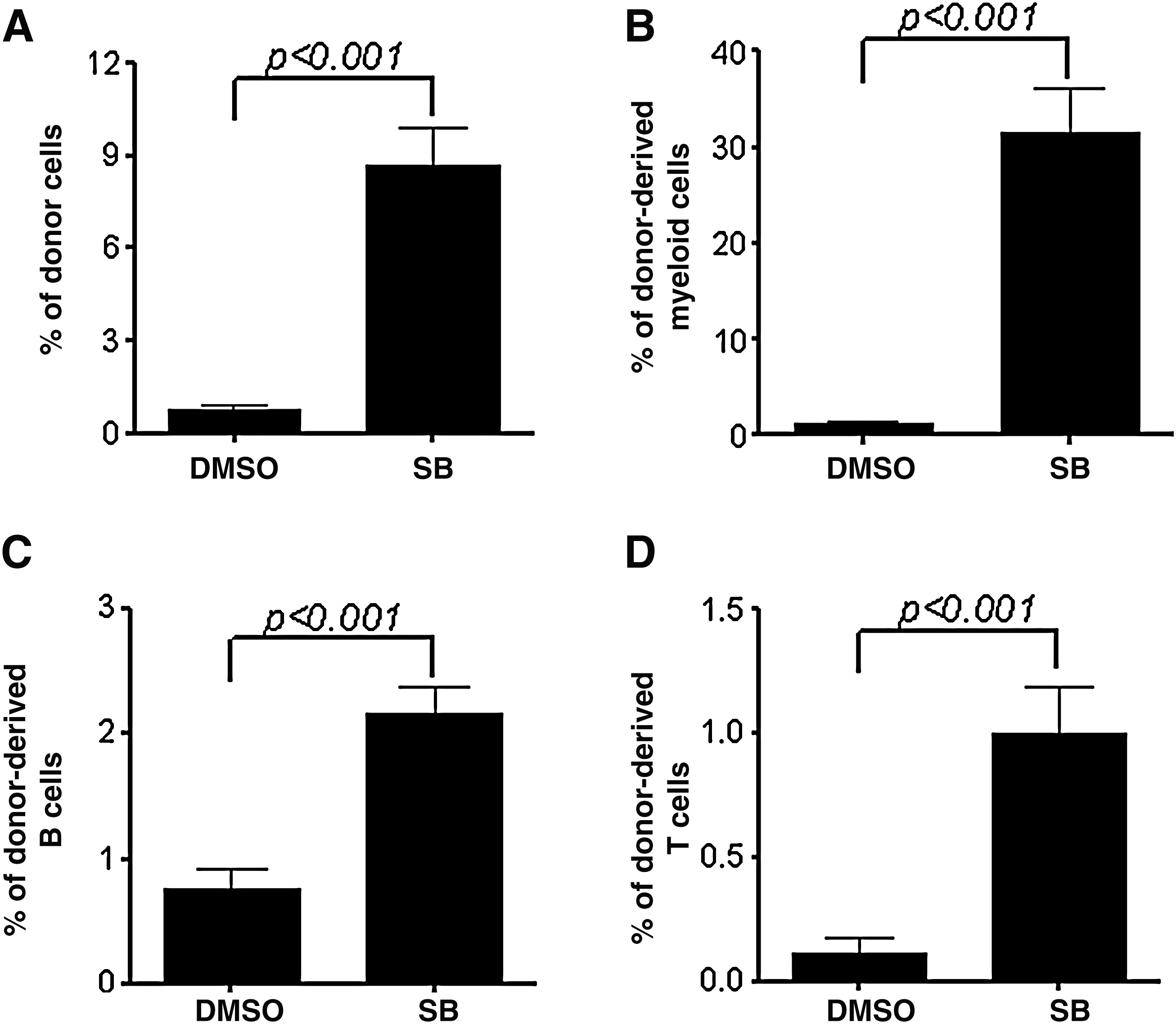
Inhibition of p38 increases HSC engraftment after secondary transplantation. Bone marrow MNCs were harvested from the primary recipients 20 weeks after transplantation. They (5×105) were transplanted into a lethally irradiated mouse along with 2×105 competitors. The donor cell engraftment in peripheral blood was determined 12 weeks after transplantation. The data are presented as mean±SE (n=7 mice/group) of percent of engraftment of total donor cells
Inhibition of p38 promotes HSC ex vivo expansion in part by inhibiting HSC senescence
To elucidate the mechanisms by which p38 inhibition promotes HSC expansion, we analyzed the effects of SB treatment on HSC differentiation, apoptosis, and senescence by CFU assay, Annexin V staining, and SA-β-gal staining, respectively. As shown in Fig. 6 and Supplementary Fig. S4, SB treatment had no effect on the formation of CFU-granulocyte macrophage, burst-forming unit-erythroid, and CFU-granulocyte, -erythrocyte, -monocyte, and -megakaryocyte from the progeny of HSCs, but significantly reduced the percentage of Annexin V positive cells in LSK+ cell population and SA β-gal staining in their progeny. These data suggest that inhibition of p38 promotes ex vivo HSC expansion not by inhibition of HSC differentiation but probably via suppression of HSC apoptosis and senescence. This suggestion is in agreement with the finding that incubation of LSK+ cells with SB reduced their expression of p53, p21, and p16 mRNA, since induction of p53, p21, and p16 in HSCs can mediate the induction of HSC apoptosis and senescence [26,27]. In addition, real-time RT-PCR analysis also revealed that inhibition of p38 promotes LSK+ cell expression of HoxB4 and CXCR4 mRNA. HoxB4 plays an important role in regulation of HSC self-renewal, whereas CXCR4 mediates HSC BM homing and engraftment [28,29]. Increases in their expression may also contribute to the increase in HSC ex vivo expansion after p38 inhibition and HSC engraftment after transplantation.
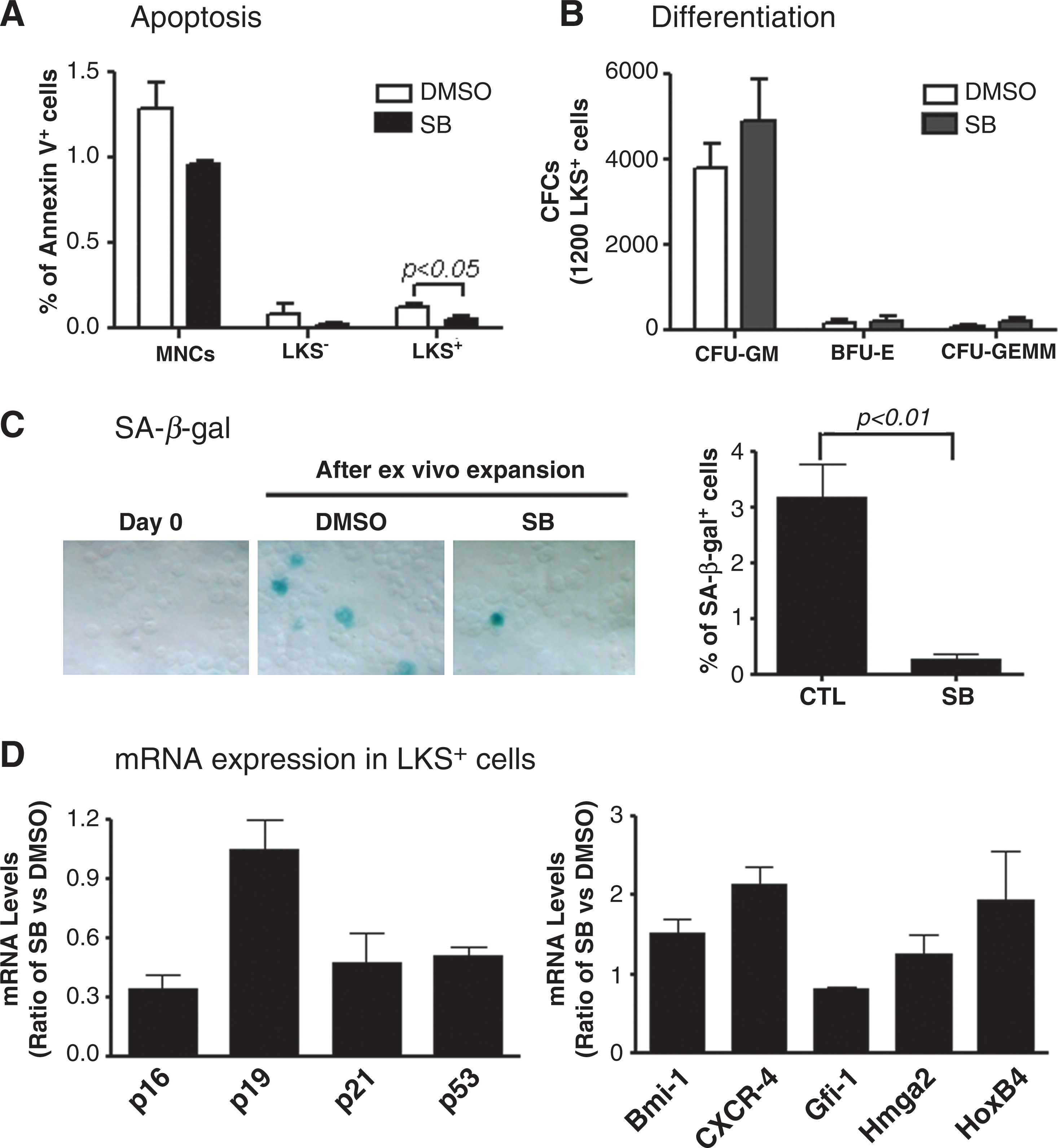
Effects of p38 inhibition on HSC apoptosis, differentiation, senescence, and gene expression. LKS+ cells were cultured ex vivo in the presence of vehicle (0.1% DMSO) or SB (5 μM) for 7 days. The expanded cells harvested from the culture were analyzed for apoptosis in MNCs, LKS−, and LKS+ cells by flow cytometry after staining with FITC-Annexin V
Discussion
Moderate ex vivo expansion of HSCs has been achieved by incubation of HSCs with various hematopoietic growth factors and cytokines, Notch ligands, Wnt3a, and angiopoietin-like proteins [16,17]. Coculture of HSCs with BM stromal cells and endothelial cells also increases the expansion of HSCs [16,17]. In addition, ectopical expression of the transcription factors HoxB4 can induce robust expansions of HSCs in vitro [28]. Recently, p38 has been identified as one of the intrinsic regulators that can negatively regulate HSC self-renewal [7,8,14,15]. Numerous small molecules of p38 inhibitors are widely available [22]. Some of them have passed Phase I and II clinical trial. Therefore, using p38 small molecule inhibitors to promote HSC self-renewal and ex vivo expansion has numerous advantages, because they are inexpensive and safe. The results from our present study provide the proof of concept that inhibition of p38 with a specific inhibitor can promote HSC ex vivo expansion in a mouse model system. First, we showed that culture of HSC-enriched mouse BM LKS+ cells in vitro selectively activated p38. This activation is likely attributable to oxidative stress resulting from cell culture in a normoxic condition (20% O2) [19 –21], because it has been shown that HSCs are sensitive to oxidative stress-induced p38 activation [14,15]. In addition, ex vivo expansion of HSCs inevitably generates numerous mature blood cells that are known to secrete hematopoietic negative regulators such as tumor growth factor-β which can activate p38 in HSCs [30,31]. Activation of p38 in HSCs has been shown to impair HSC self-renewal and cause HSC premature exhaustion in several human hematological diseases and in ATM mutant and FoxO3 gene(s) knockout mice [9,13 –15]. Similarly, we found that the activation of p38 in LSK+ cells and their progeny after they were cultured in vitro for 7 days with STF alone was associated with a 74% reduction in HSCs compared to freshly isolated LSK+ cells according to day-35 CAFC assays. Addition of the specific p38 inhibitor SB to the culture abrogated the activation of p38 and significantly expanded the number of HSCs, as the cells cultured with SB contained 2.0- and 7.6-fold more day-35 CAFCs compared to input (day 0) LKS+ cells and the cells cultured without SB, respectively. These findings suggest that inhibition of p38 with a small molecular inhibitor can promote HSC expansion ex vivo. This suggestion was confirmed by the observation that the progeny of LKS+ cells cultured with SB produced a significant higher long-term and multiple lineages donor cell engraftment after competitive repopulating assay and serial BM transplantation than those without SB.
The mechanisms by which p38 inhibition promotes HSC self-renewal and ex vivo expansion may be attributable primarily to the inhibition of cellular senescence, because it has been shown that p38 functions as a key molecule mediating diverse stimuli-induced cellular senescence via upregulation of p16 in a variety of cells, including HSCs [14,15,32,33]. This hypothesis is supported by the finding that the progeny of LKS+ cells cultured with SB exhibited significantly less SA-β-gal staining and expression of p16 mRNA than those without SB. SA-β-gal is a widely used biomarker of senescent cells [34] and increased expression of p16 has been implicated in the induction of cellular senescence down-stream of p38 activation [26,33]. In addition, a significant fewer apoptotic cells were detected in LKS+ cells cultured with SB than the cells without SB, indicating that p38 inhibition can also inhibit HSC apoptosis. Finally, real-time RT-PCR analysis revealed that the progeny of LKS+ cells cultured with SB expressed higher levels of HoxB4 and CXCR4 than those without SB. It has been well established that HoxB4 is a very important homeobox transcription factor that regulates HSC self-renewal [28]. Overexpression of HoxB4 or incubation of HSCs with HoxB4 promotes HSC expansion in vivo and ex vivo [28,35 –37]. Therefore, upregulation of HoxB4 expression by p38 inhibition may mediate SB-induced expansion of HSCs. In addition, CXCR4 plays an important role in HSC homing after transplantation via interaction with stromal cell-derived factor 1 [29]. Upregulation of CXCR4 expression by p38 inhibition may also contribute to the enhanced engraftment of the cells cultured with SB after transplantation. Collectively, these observations demonstrate that p38 inhibition has multiple effects on HSCs.
However, it has yet to determine if p38 inhibition can also lead to the expansion of human HSCs from BM, mobilized peripheral blood, and umbilical cord blood ex vivo. Since it has been shown that even human umbilical cord blood HSCs and adult mobilized peripheral blood HSCs prefer a hypoxic culture condition for ex vivo expansion [19 –21], this suggests that oxidative stress is likely a common denominator to limit human HSC self-renewal and expansion ex vivo, probably via activation of p38. Therefore, it is likely that p38 inhibition may also promote human HSC expansion ex vivo. This will be investigated in our future studies. In addition, the effect of p38 inhibition on HSC ex vivo expansion remains moderate. It will be interesting to determine if p38 inhibitors can be combined with other agents to more effectively promote HSC expansion ex vivo than individual agents by targeting different HSC self-renewal pathways [3,16]. Such a combined approach may lead to a greater expansion of HSCs than single agents, which could make HSC transplantation available to more patients and reduce the complications associated with HSC transplantation by decreasing engraftment times to allow more rapid immune reconstitution after transplantation.
Footnotes
Acknowledgments
The authors thank Mrs. Aimin Yang for her excellent technical assistance and Mr. Richard Peppler and Dr. Haiqun Zeng for the flow cytometric analysis and cell sorting. This study was supported in part by grants from the National Cancer Institute, National Heart, Lung, and Blood Institute, National Institute of Allergy and Infectious Diseases, National Center for Research Resources and the Winthrop W. Rockefeller Endowment for Leukemia Research.
Author Disclosure Statement
The authors declare no conflicts of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.