
Abstract
Thus far, autologous adult stem cells have attracted great attention for clinical purposes. In this study, we aimed at identifying and comprehensively characterizing a subpopulation of multipotent cells within human nasal septal cartilage. We also conducted a comparative investigation with other well-established stem cells such as bone marrow–mesenchymal stem cells, adipose tissue–mesenchymal stem cells, and unrestricted somatic stem cells. The isolated clonal population was characterized using immunofluorescence, flow cytometry, reverse transcriptase, and real-time polymerase chain reaction. Nasal septal progenitors (NSP) expressed critical pluripotency and mesoectodermal stem cell markers. They also shared many characteristics with MSC in expression of CD90, CD105, CD106, CD166, and HLA-ABC and lack of expression of CD34, CD45, and HLA-DR. NSP distinctly presented CD133 (Prominin-1). These cells could proliferate rapidly in vitro with a higher clonogenic potential and showed a longer lifespan than other studied cells. This population bears some other multipotent properties in showing a high capacity to be differentiated into other lineages including chondrocytes, osteocytes, and neural-like cell types. Another strong/positive feature of this population was their ability to be safely expanded ex vivo with no susceptibility to chromosomal abnormality or tumorigenicity both in vitro and in vivo. In conclusion, NSP could be considered as an alternative autologous cell source that can bring them to the top of therapeutic applications.
Introduction
F
Articular cartilage is a tissue with low self-renewal and poor regeneration potential. Current cell-based therapies for the treatment of cartilage defects usually involve autologous chondrocyte implantation. However, autologous chondrocyte implantation is not the best option because of chondrocyte dedifferentiation, limited lifespan, and also donor site morbidity [14 –16].
Although cartilage is a tissue with very low cell turnover, the occurrence of regeneration in healthy or diseased tissue raised the possibility that this tissue has a niche of stem-like or progenitor cells [17,18]. The presence of such mesenchymal progenitor cells was already addressed in articular cartilage with a role in its oppositional growth [8,19,20]. However, to the best of our knowledge, a progenitor-like population residing in nasal septal cartilage that requires a comprehensive study to investigate this population has not yet been fully characterized.
Unlike articular chondrocytes with a mesodermal origin, cells of nasal septum that are derivatives of neural crest have mesoectodermal embryonic origin [21]. The prominent characteristics of some cell types derived from neural crest tissue such as peripheral olfactory stem cells and olfactory ensheathing glia, similar to differentiation potential and high proliferation rate, prompted us to search for the presence of such valuable cells in nasal septal cartilage. Another advantage of septal cartilage as a potential autologous cell source relies on the ease of taking biopsy from septal cartilage in a routine septoplasty or rhinoseptoplasty that entails local anesthesia and less morbidity than harvesting biopsies from auricular, articular, or costal cartilages [22].
In this study, we showed that human nasal septum-derived cells not only posed progenitor-like properties (such as high self-renewal capacity, high colonogenecity, and multilineage differentiation potential with a capacity to osteogenic, chondrogenic, and neurogenic differentiation) but also had a stable chromosomal state with no sign of transformation over long-term in vitro culture. The aim of this study was to characterize this highly proliferative population that might be clinically valuable for future stem cell therapy studies; for this reason, cells harvested from 10 patients (in both sex) were evaluated for their expansion and differentiation potential, chromosomal stability, and also in vivo and in vitro tumorgenesis
Materials and Methods
Isolation and long-term culture of nasal septal progenitors
Human nasal septal cartilage samples were obtained from 10 patients aged between 18 and 34 years (mean=26.6±5.2) undergoing septoplasty or septorhinoplasty operations with their previous consent. All procedures were approved by the ethical committee of Stem Cell Technology Research Center (Tehran, Iran). The cartilage specimens were cut into small pieces, digested with 0.2% protease solution (Gibco) for 60 min, followed by incubation in 0.2% w/v collagenase type I and II (Coll-I and Coll-II; Gibco) and dispase (Sigma) for 16 h to separate the cells from their extracellular matrix. The digested cartilage samples were then passed through 70 μm filters (BD Biosciences). After rinsing and counting the cells of each individual, they were re-suspended in Dulbecco's Modified Eagle Medium (DMEM)-low glucose containing 15% fetal bovine serum (FBS; Gibco), 10 μg/mL ascorbic acid (Sigma), 1% penicillin/streptomycin (Sigma), and 1.25 μg/mL amphotericin-B (Sigma); plated in 75 cm2 polystyrene culture flasks at a density of 104 cell/cm2; and maintained in the standard conditions (37°C, 5% CO2). The resultant colonies were picked up and cultured in a separate flask until subconfluency; then, they were subcultured at a ratio of 1:3 using 0.05% trypsin/EDTA solution (Gibco). The cells were expanded in vitro until they reached their senescence. Some samples underwent freeze-thaw cycles before each passage. Cell morphology was continuously assessed throughout the in vitro expansion and differentiation. To compare the characteristics of nasal septal progenitors (NSP) with other previously studied cells, BM-MSC, adipose tissue-mesenchymal stem cells (AT-MSC), and unrestricted somatic stem cells (USSC), previously isolated and well characterized in this laboratory, were examined in parallel [23].
Clonogenic and proliferative potential
The routine colony-forming assay was done by seeding 100, 300, and 500 of single cells in 10 cm culture plates for assessing clonogenicity of NSP. After 14 days, colonies were stained with hematoxylin; and those with a larger diameter than 2 mm were scored as colonies. To assess the proliferation potential, the population doubling time (PDT) was calculated according to the following formula: DT=t(log2)/(logNt–logN0), in which t is the culture time and N0 and Nt represent the number of cells at time zero and t, respectively.
MTT assay
To optimize media composition for NSP culture, we assessed the viability of NSP under different culture conditions (DMEM/F12, high- and low-glucose DMEM; low-glucose DMEM supplemented with 2.5%, 5%, 10%, 15%, and 20% FBS) using methylthiazolyldiphenyltetrazolium bromide (MTT) reduction assay. The viability of NSP was also compared with BM-MSC, AT-MSC, and USSC.
Multilineage differentiation potential of NSP
Osteogenic and adipogenic differentiation
Osteogenic and adipogenic differentiation was performed according to the common protocols as previously described [23].
Chondrogenic differentiation
For chondrogenesis, 2×105 cells were pelleted by centrifugation for 4 min at 350 g. After 24 h, media was changed with 0.5 mL of chondrogenic induction medium containing DMEM-high glucose containing 1% ITS supplements (625 mg/mL insulin, 625 mg/mL transferrin, and 625 ng/mL selenious acid) (Gibco), 50 μg/mL ascorbate 2-phosphate (Sigma), 100 nM dexamethasone (Sigma), 10 ng/mL basic fibroblast growth factor-2 (bFGF-2) (Gibco), and 10 ng/mL transforming growth factor-β (TGF-β) (Sigma). The cell pellets were incubated/maintained in this medium for 3 weeks with medium exchange every 3 days; then, the aggregates were harvested and sectioned using standard histology protocols and stained with Alcian Blue (Sigma). To induce and maintain the chondrogenic features in the isolated primary population, some cultures were amended with 1 ng/mL TGF-β and 1 ng/mL bFGF-2 soon after isolation of the cells from the donor tissue. The resulting 2D cultured chondrocytes were then subjected to Alcian Blue staining and immunofluorescence for production of chondrocyte-specific Coll-II (see below).
Neurogenic differentiation
To induce neurogenesis, single NSPs were placed in nonadherent 6-well culture plates with a density of 100,000 cells per mL for 4 days in DMEM/F12 medium supplemented with 10% FBS, 1% non essential amino acids (Gibco), 2 mM L-glutamine (Gibco), and 500 nM retinoic acid (Sigma). The neurosphere-like colonies were then transferred to poly-l-lysine-coated flasks for another 4 days. Then, the medium was replaced with DMEM/F12 supplemented with 1% N2 (N2 is a serum-free chemically defined supplement to be used with neurobasal media for in vitro differentiation of stem cells) (Gibco) and 2% B27 (B-27 supplement is a serum substitute to support the cultures of neurons at low or high cell densities in both short and long-term cultures) (Gibco) for 12 days. The differentiated cells were used for RNA extraction and reverse transcriptase– polymerase chain reaction (RT-PCR) analysis of neural gene expression and immunocytochemisty of immature to mature neural markers such as β-tubulin III (β-TUB), microtubule-associated protein-2 (MAP-2), nestin, and neural-specific enolase (NSE).
Immunofluorescence
Cells were fixed in 4% paraformaldehdye and then processed for immunofluorescence staining. After blocking, cells were incubated at 4°C overnight with the following primary antibodies: anti-Oct4 (Abcam), anti-S100 (Chemicon), anti p75 (CD271) (Sigma), anti bone morphogenetic protein-2 (BMP-2) (Abcam), and anti GFAP (Chemicon) for non-treated NSP; anti-β-TUB (Sigma) and anti-MAP-2 for neurally induced NSP; and anti-Coll-II for immediate isolated population (P0) and chondrogenically induced cells. Cells were then incubated with appropriate FITC- or PE-conjugated secondary antibody (Abcam) for 3 h at room temperature. The cells nuclei were stained with 4′, 6-diamidino-2-phenylindole dihydrochloride (DAPI; 1:1,000) and visualized by a fluorescent microscope (TE2000-S; Nikon-Eclipse).
Flow cytometry analysis
Different cell types including NSP, AT-MSC, BM-MSC, and USSC were used to prepare a single cell suspension. To analyze intracellular markers, cells were permeabilized with 0.5% Triton X-100. To block nonspecific sites, a solution of 5% human serum in PBS was added to the cells for 30 min. Next, the cells were stained with the following mouse monoclonal antibodies against human CD105, CD106, CD90, CD166, CD45, HLA-ABC, HLA-DR (all from eBioscience), CD34, CD133 (Prominin-1) (Dako), and CD271, GFAP, OCT4 (all from Santa Cruz). In every experiment, the appropriate isotype matched antibody was used as control to detect nonspecific binding. Later, the cells were stained with relevant FITC- or PE- labeled secondary antibody, fixed with paraformaldehyde in PBS, and analyzed on an FACS Calibur cytometer (Becton Dickinson) with WinMDI 2.8 software. To determine the fraction of the cells in different cell division cycles, flow cytometric analysis of DNA content was employed for NSP and BM-MSC. Briefly, the trypsinized cells were fixed with ethanol for 2 h, permeablized with 0.1% Triton X-100, labeled with propidium iodide (PI), and assessed by flow cytometry as just described.
RNA extraction, RT, and real-time PCR analyses of gene expression
Total RNA was extracted using the Qiazol (Qiagen,), and cDNA synthesis was performed using Revert Aid kit (Fermentas). cDNA products were used for standard RT or real-time PCR. Real-time PCRs were performed using Maxima™ SYBR Green/ROX qPCR Master Mix (Fermentas) and monitored in Rotor-gene Q real-time analyzer (Corbett). The level of expression of BMP-2 in NSP and BM-MSC was evaluated with Real-Time PCR in triplicate, and then the average threshold cycle was estimated and normalized against HPRT. The fold change of each target gene was calculated with comparative ΔΔCt method. Primers used for the mentioned genes are listed in Supplementary Table S1 (Supplementary Data are available online at
Senescence assay
Senescence assay was performed in different passages using galactosidase staining kit (Cell Signaling Technology); after fixing the cells with 2% paraformaldehyde, the reaction solution composed of 1 mL of staining solution in citrate buffer at pH 6.0 was added to the culture and incubated overnight at 37°C. Cells with high galactosidase activity present characteristic blue products that can be examined by light microscopy.
Karyotype analyses
Metaphase cells at various passages were incubated for 2 h with 0.1 μg/mL colcemid (Gibco). The trypsinized mitotic cells were then lysed in a hypotonic solution of 75 mM KCl at 37°C for 15 min followed by fixation with 1:3 v/v acetone:methanol fixative. Seven day aged slides with fixed metaphase spreads were stained with trypsin-Giemsa and analyzed for karyotyping. For each time point, approximately more than 20 cells were examined.
Efficient transduction by enhanced green fluorescent protein-carrying lentiviral vector
Using classical lentiviral transduction procedure, NSP were genetically modified to express enhanced green fluorescent protein (eGFP). Cultures were maintained in the presence of eGFP harboring lentivirus at a multiplicity of infection of 10, and transductions were performed for 48 h. The efficiency of transduction was measured by flow cytometry as just mentioned. The stability of the transduction was assayed by examining the transduced cells after multiple passages.
Tumorigenic assay
A total of 6×106 eGFP expressing NSP at different passages were suspended in DMEM and then injected subcutaneously or intravenously into 2-month-old nude mice (Imam-Khomeini Hospital). They were under control for 4 months after the injection. MKN45 cell line was injected similarly as a positive control. During this period, animals were examined weekly for possible tumor incidence. After 4 months, mice were sacrificed, and their spleen, lung, BM, and liver were removed, fixed, sectioned, and stained with H&E. Sections were also evaluated by fluorescence microscopy for determination of any probable spread of engrafted cells. In vitro tumorgenecity and susceptibility to transformation were assessed using a well-established soft agar colony assay, which was followed up for 28 days.
Statistical analysis
All experiments were performed in triplicate. One-way analysis of variance was used to compare results. A P value of less than 0.05 was considered statistically significant. Data are reported as mean±SD.
Results
Isolation and characterization of human nasal septal-derived progenitors
In this study, human nasal septal-derived cells were isolated from 10 patients undergoing septoplasty or rhinoseptoplasty; and the expanded clonal population was designated NSP. We studied the phenotypic features of the isolated cells throughout the culture time. The morphology of the isolated cells at 3 and 7 days postisolation is shown in Fig. 1A and B, respectively. In monolayer culture, these cells are flat and have a spindle-like or polygonal shape. After reaching confluency (Fig. 1C), cells are in close contact but do not grow in more than 1 layer. At later time points, the cells tend to gain larger and more flattened fibroblast-like morphology (Fig. 1D–H).

In vitro morphology of human nasal septal-derived cells at different time points of culture. Representative photomicrographs of newly isolated cells at
To determine the immunophenotyping profile of NSP, we used flow cytometry to assess different cell surface antigens including MSC markers: CD105, CD106, CD90; cell adhesion marker CD166; hematopoietic and common leukocyte markers: CD34 and CD45; hematopoietic and neural stem/progenitor cells surface marker: CD133; low-affinity neurotrophine receptor: CD271; glial markers: GFAP; and stemness marker: OCT4; HLA-ABC, and HLA-DR. As expected, the isolated cells were negative for hematopoietic stem cell markers, that is, CD34 and CD45 (Fig. 2). We also monitored the expression profiles of 3 other available cell types, previously isolated and characterized in our laboratory (BM-MSC, AT-MSC, and USSC), to compare the expression profile of NSP with other well-known stem cells. As shown in Fig. 2, the expression profile of mesenchymal and cell adhesion markers follow the same trend in both NSP and BM-MSC. The immunophenotypic features of AT-MSC and USSC are available in Supplementary Fig. S1. The relatively high expression of CD133 in NSP is a distinct characteristic in surface marker profile between these cells and BM-MSC. The isolated population also showed some degree of expression of glial markers including GFAP and low-affinity nerve growth factor receptor (NGFR) (P75). The immunophenotypic features of NSP in long-term in vitro culture is illustrated in Fig. 3.The high expression of OCT4, as a stemness marker, was validated by both flow cytometry and immunostaining (Fig. 4A).
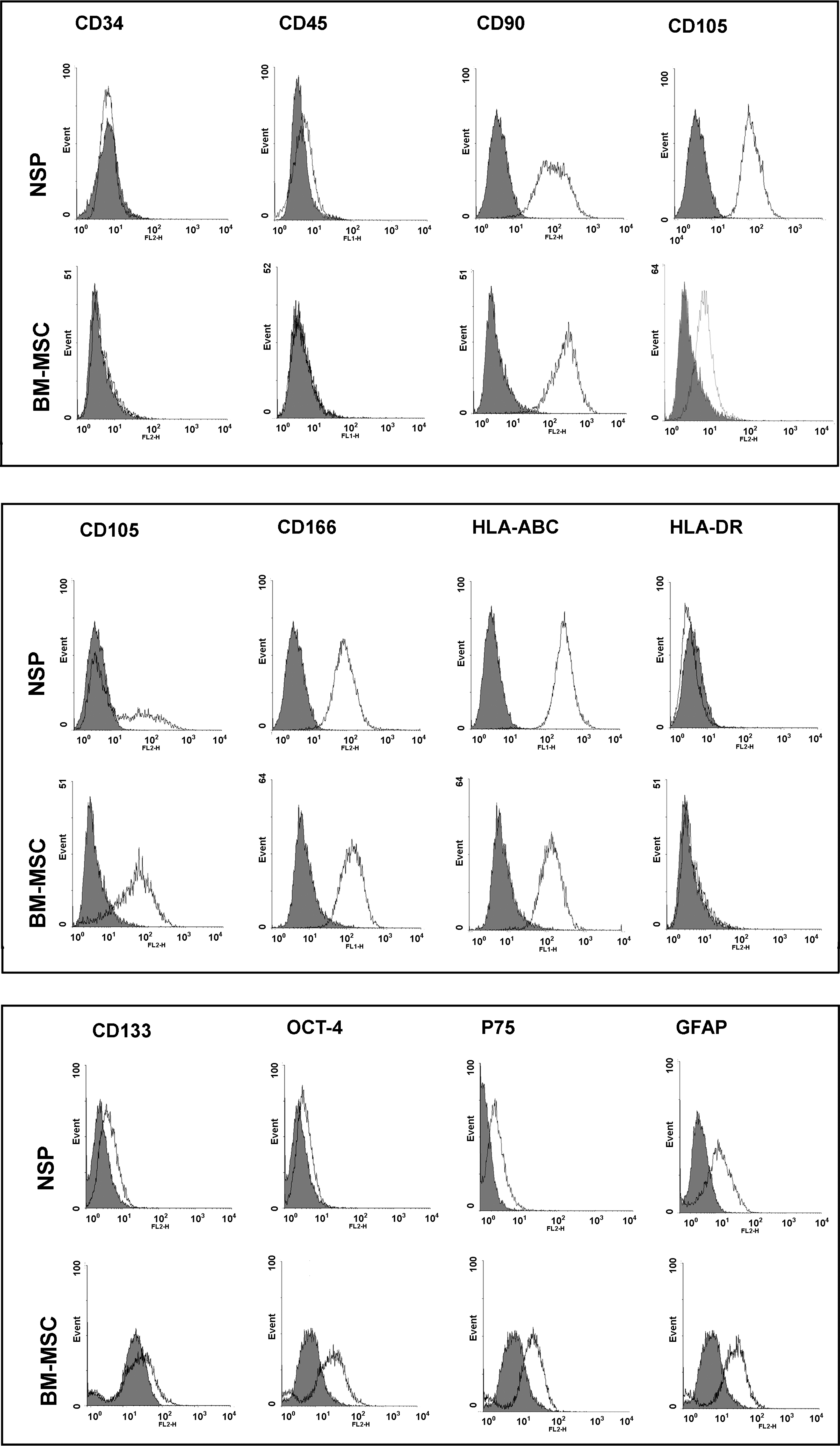
In vitro assessment of cell surface, stemness, and ectomesenchymal markers as well as HLA-ABC and DR in clone-derived passage 3 nasal septal progenitors (NSP) in comparison with bone-marrow-derived mesenchymal stem cells (BM-MSC) using flow cytometry. The histograms confirm negative expression of hematopoietic and common leukocyte markers: CD34 and CD45 and expression of MSC markers including CD90, CD105, CD106, and CD166 in both cell types. Expression profile of HLA-ABC and HLA-DR looks similar in NSP and BM-MSC. Expression of CD133 as a hematopoietic and neural stem/progenitor cells surface marker, OCT4 (stemness marker), CD271 (P75), and GFAP (ectomesenchymal markers) is depicted in the lower panel.
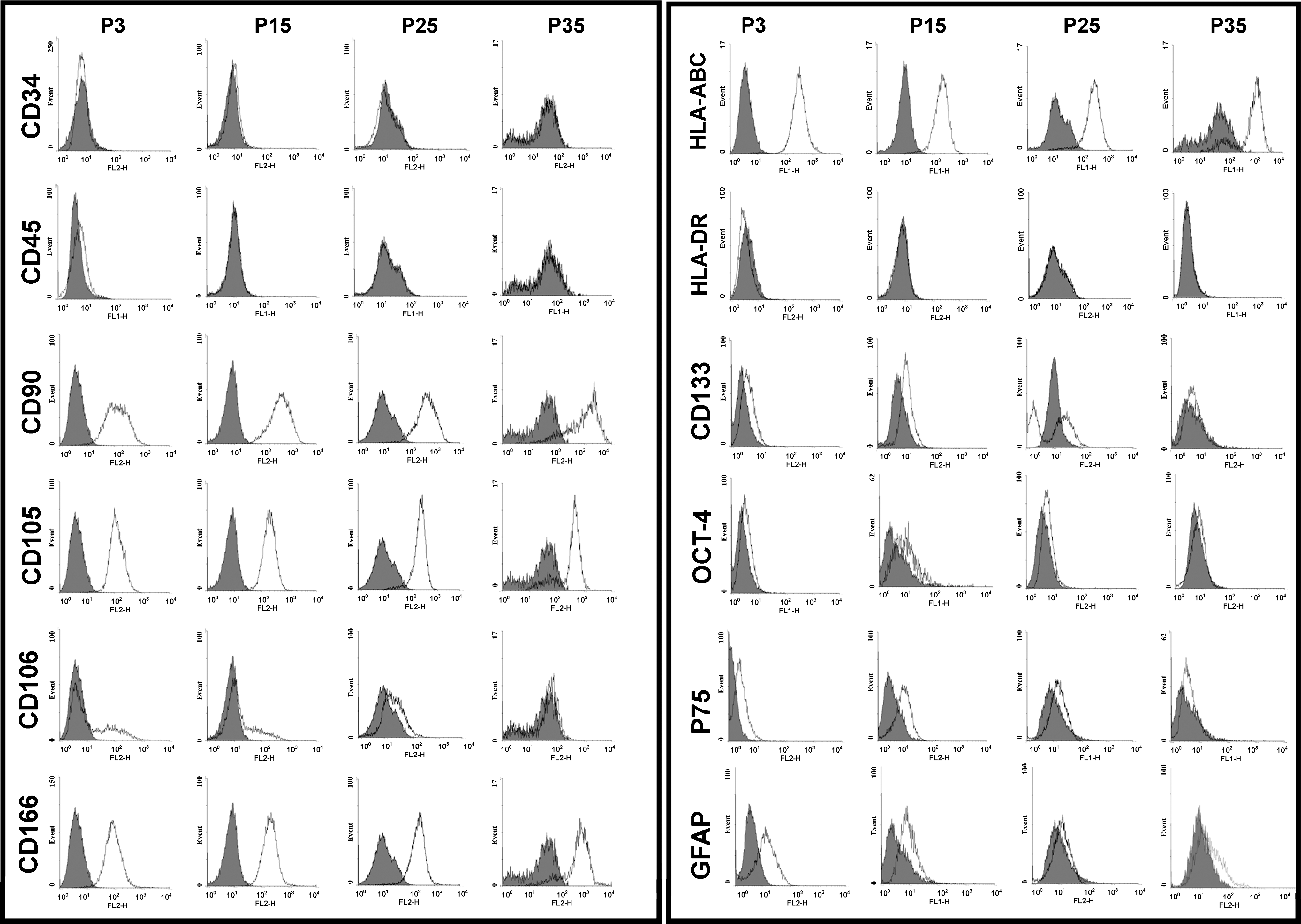
Comparison of different cell surface, stemness, and ectomesenchymal markers among different passages of NSP. Although some markers show a consistent pattern as cells age, ectomesenchymal (GFAP and P75), stemness (OCT4), and CD106 and CD133 markers show a decreasing trend with higher passages.
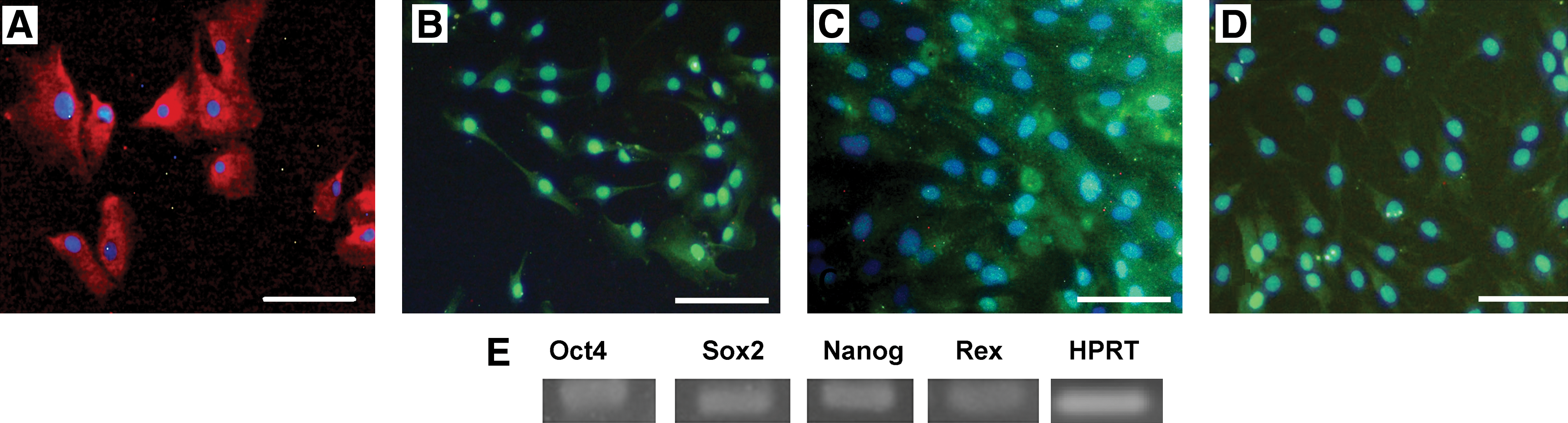
Representative immunofluorescence analysis of monolayer cultures of NSP at passage 3. Cells were fixed and stained with antibodies against OCT4
To further extend this study, we investigated the expression of ectomesenchymal stem cell markers (S100, P75, and GFAP) in NSP using immunostaining. As illustrated in Fig. 4B–D, all the mentioned markers showed a strong expression in the isolated cells. Using gene-specific primers and RT-PCR, the presence of 4 embryonic stem cell markers, that is, OCT4, Nanog, Sox-2, and Rex, was detected in this population (Fig. 4E).
NSP have a high self-renewal and clonogenic potential
After the surgery, a very small portion of the septal cartilage (≈5×5×1.5 mm) was used for cell isolation. Depending on the initial sample size, 300,000 to 600,000 cells were recovered, interestingly with a viability of 92% (±4.3%) as estimated by trypan blue staining. No significant correlation was observed between the donor age and the number of recovered cells. First colonies appeared after an initial lag phase of 3 to 5 days, whereas in BM-MSC it takes ∼2 weeks for the first colonies to appear. Even for highly proliferative cells such as USSC, it takes between 6 and 20 days for the first colonies to appear, with 4 colonies per cord blood sample in average. A representative colony of NSP stained with hematoxylin is shown in Fig. 5A; the colonies of NSP are morphologically similar to those of BM-MSC. Colony-forming unit (CFU) assay was used to assess the proliferative potential of isolated cells at different passages. The number of adherent colonies (diameter ≥2 mm) was 6±1.9, 73±3.4, 73±5.6, 25±6.2, and 18±4.7 per 100 cells seeded in 10 cm plates for freshly isolated cells (P0), P2, P3, P15, and P35, respectively. The colony assay was also repeated with 300 and 500 initial seeding cells, confirming the same results. Unlike MSC, we found a direct proportion between the number of seeded cells and CFU-F as already stated for 100 cell seeding density [24].The size of colonies diminished as a function of continued passaging, which was correlated with decreasing cell number in the corresponding colony (data not shown).

Cell seeding was carried out in both high and low confluency cultures, as previously described. Proliferation assay showed faster DT for low confluency cultures. However, there was no significant difference in their growth rate for the 2 sought strategies (data not shown). As illustrated in Fig. 5B, the NSP have high proliferative capacity, especially in the low passages. The mean PDT of NSP shows an increasing trend from 30±3.2 h in starting cultures to 380±14 h in late passages (P43). The presented results in Fig. 5B reflect experiments performed on NSP derived from donor 2; however, the PDT estimated for other donors followed the same range and trend. Typically, NSP recovered from a 2×2×2 sample can be expanded to more than 35 million cells over about 2 weeks of the isolation time.
In vitro long-term culture
As the cultures age, the cells enter a stationary phase with a morphology transformation from small, polygonal to larger and flatter fibroblast-like ones, which was concurrent with formation of granule-like bodies in their cytoplasm occurring nearly after passage 13. No growth arrest was observed in low passages in any of the donors; instead, NSP showed a progressive decrease in growth rate until reaching their senescence phase (Fig. 5B). Unlike BM-MSC, which are so sensitive to high density cultures [25], NSP are more resistant to high confluent cultures. There was no significant variation between cells of different donors in terms of their proliferation capacity in low passages. However, regarding their in vitro lifespan, some variations were observed among different samples. The estimated lifespan of different donors is summarized in Table 1. In general, regardless of the age and gender of the donors, all the cultures reached 30 DT in about 2 months. We noted that even after reaching high density, NSP still retain their proliferative potential, which was confirmed by keeping some P3 and P5 cultures for 2 months without any subculturing but with refreshing growth medium every other time. Under such conditions, the cells entered the stationary phase with some granulation and morphological alterations from bipolar, spindle like to a more broadened, flat appearance. In the mean time, no detachment from culture plates and also no sign of spontaneous differentiation were noticed. After this period, the dormant culture proceeded with usual 1:3 subcultivation. However, the progeny cells of this culture did not acquire their rapid growth rate, as their DT increased to nearly 2.5-fold more than that of the equivalent cells that had undergone routine passaging procedure.
The specimens were taken from different donors aged 18 to 34, both males and females. Cells obtained from each individual were evaluated for fibroblast colony-forming capability (at P0) and their in vitro lifespan.
CFU, colony-forming unit.
Cell cycle analysis
To estimate cell cycle phase distribution of different cell types, cells were fluorescently labeled with DNA-incorporating propidium iodide. As shown in Fig. 5C and D, it can be deduced from direct DNA measurements that although for both cell types the majority of the cells are in G0/G1 phase, the percent of the cells in G2/M phase in NSP is nearly twice that in BM-MSC.
Cryopreservation capacity
Another promising feature of NSP was their ability to keep their propagation potential after frequent cryopreservation and thawing cycles, which was investigated by comparing the duplication time of cells undergoing a period of cryopreservation before each passage with those routinely subcultured. The self-renewal capacity of cryopreserved NSP cultures was not significantly different from the parallel unfrozen cultures (P= 0.05) (data not shown), with a viability greater than 88% after each thawing step.
MTT assay
We compared the proliferation of NSP with other available well-characterized cell lines in our laboratory, using MTT assay. As outlined in Fig. 6A, there is an overall increasing trend in the proliferation and viability of 4 examined cell types, that is, BM-MSC, AT-MSC, USSC, and NSP. After 1 week, all the examined cells showed a decline in OD540, with NSP being the only exception that continued their upward increase throughout the experiment. Additionally, we determined the effect of growth media (DMEM-high glucose, DMEM-low glucose, and DMEM/F12) and serum supplementation on the proliferation of NSP (P3). As depicted in Fig. 6B, media richer in serum led to a better proliferation rate at all time points. DMEM-low glucose showed the best effects on culturing NSP enriched population (Fig. 6C).
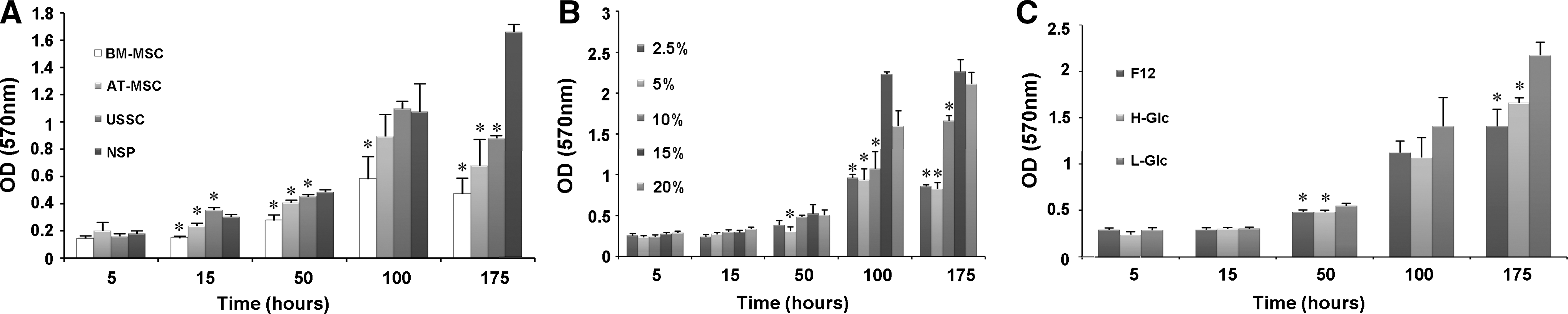
Methylthiazolyldiphenyltetrazolium bromide (MTT) assays were performed at indicated time points after cell seeding to measure the viability and proliferation rate of the cells.
Differentiation potential
Differentiation capacity of NSP populations was assessed by in vitro ability to produce osteocytes, adipocytes, chondrocytes, and neurons.
In vitro osteogenic assay
Osteogenic differentiation was assessed at different time points (P5, P15, P25, and P35). Osteogenesis was accompanied with creation of mineral aggregates as nodule-like structures that were stained with Alizarin Red S (Fig. 7). No mineralization of calcium phosphate was observed for control cells that were not induced by osteogenic medium (data not shown). To visualize the osteogenic potential of NSP with other stem cells, we repeated the same test with other cell types at the same passage. The results are presented in Fig. 7.
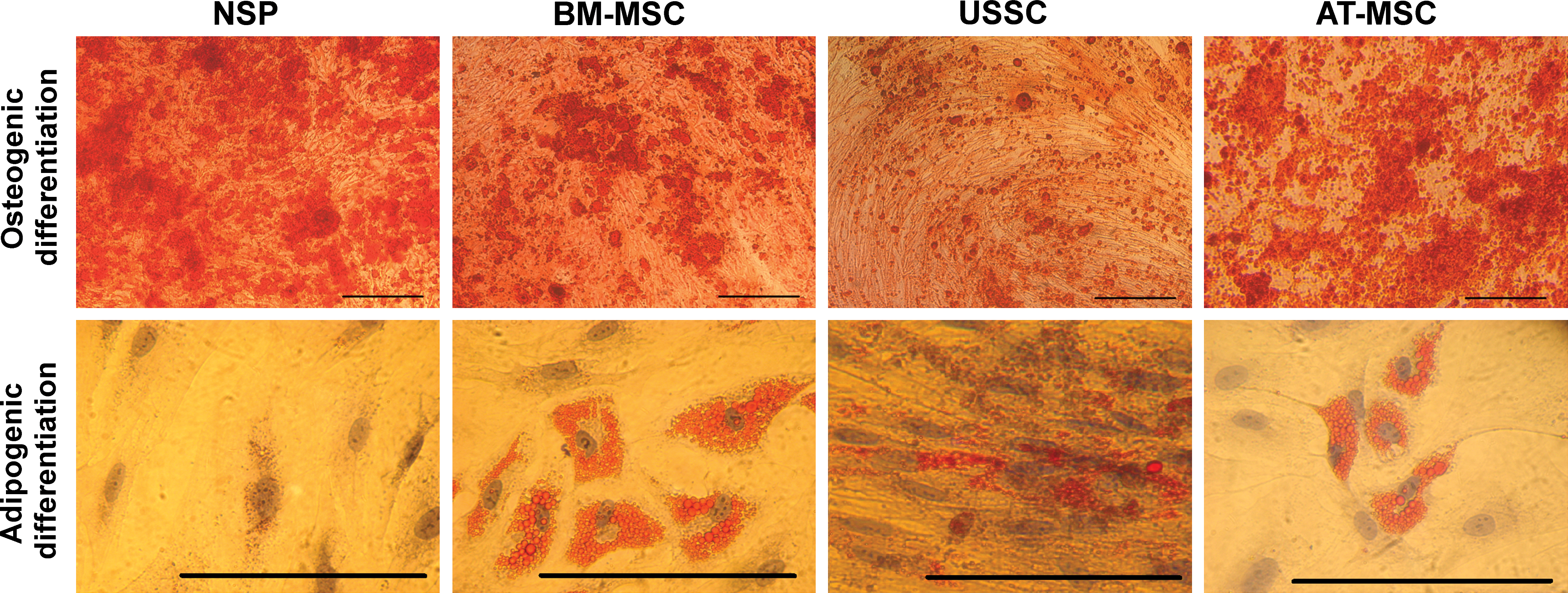
Osteogenic and adipogenic differentiation capacity of NSP in comparison with BM-MSC, AT-MSC, and USSC. All cultures were maintained under induction medium for 3 weeks and stained with Alizarin Red S (upper panel) or Oil Red O (lower panel). The expanded clonal population of nasal cartilage cells did not differentiate to adipocytes after 3 weeks in adipogenesis medium. Scale bars: 200 μm. Color images available online at
In vitro adipogenic assay
After 21 days of adipogenic induction, only few and small oily vesicles were observed in the induced cells (Fig. 7). Such lipid-containing cells were observed only in 1 sample (sample 6). The lack of adipogenic potential was persistent throughout the whole culture period till P35. The Oil Red stained cultures of differentiated NSP, BM-MSC, USSC, and AT-MSC are shown in Fig. 7. To figure out the lack of adipogenic potential of NSP despite their multipotent differentiation capacity, we evaluated the level of BMP-2 expression in NSP in comparison with BM-MSC, given that BMP-2 has a role in inhibition of adipogenesis [26]. Real-Time PCR analysis revealed a 30±1.2-fold increase in NSP relative to BM-MSC. We also assessed the differently expressed BMP-2 in NSP and BM-MSC in protein level using immunoassays (Fig. 8).

In vitro expression analysis of bone morphogenetic protein-2 (BMP-2) in NSP contrasted to BM-MSC.
In vitro chondrogenic assay
We examined the expression of cartilage-specific Coll-II in untreated primary cells isolated from septal cartilage. However, using immunofluorescence we could not detect any expression of Coll-II in 7 and 14 day old cultures of isolated cells in media devoid of any external stimuli (data now shown).
To analyze whether the isolated clonal population form septal cartilage is able to differentiate into chondrocytes and has the ability to form cartilage, we examined their chondrogenic potential by adding low concentrations of TGF-β and bFGF-2 (1 ng/mL each) to culture medium. These cells showed their ability to secrete Coll-II after 2 weeks of isolation in monolayer culture under low concentrations of chondrogenic induction factors (Fig. 9A, B). Alcian Blue staining proved the ability of these cells to secrete proteoglycans (Fig. 9C). The expression of collagen-specific markers was also confirmed by RT-PCR for constitutive 2D monolayer induction (Fig. 9D) and micromass pellet cultures (Fig. 9E). We also used the micromass pellet culture technique for chondrogenic induction of all the cell types. After 21 days of induction in chondrogenic medium, the pellet sections were assessed for the formation of chondrocyte phenotype. The presence of rounded and enlarged cells was obvious in H&E staining of BM-MSC and NSP, whereas USSC and AT-MSC turned to form narrow and closely packed morphology (Fig. 10, low and high magnification). The strong expression and accumulation of acidic proteoglycans was monitored in chondrogenic committed NSP by Alcian Blue staining. The 3 other cell types stained variably positive for Alcian Blue staining (Fig. 10). There was a poor and heterogeneous staining for AT-MSC. In contrast, NSP, BM-MSC, and USSC showed a uniform staining pattern. In the late phase of cell culture, these cells still maintain their ability to differentiate into chondrocytes (P5, P15, P25, and P35) (data not shown).

Chondrogenic potential of NSP.
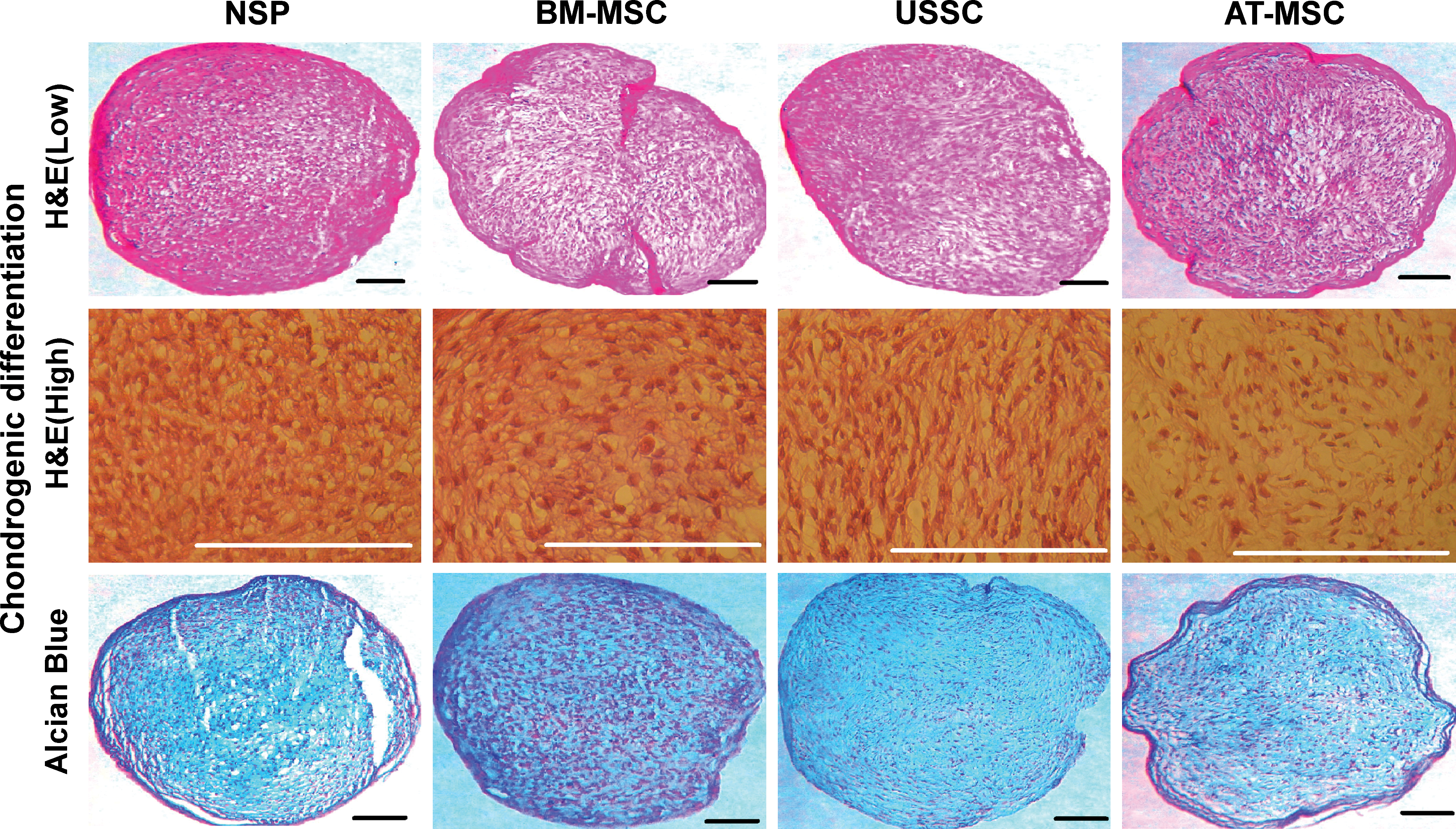
All cell types were cultured in micromass pellet for 3 weeks, fixed and stained with H&E and Alcian Blue (low, low magnification; high, high magnification). Scale bars: 100 μm. Color images available online at
In vitro neurogenic assay
To explore the differentiation potential of NSP to other lineages rather than classical mesenchymal cell types, neurosphere-like floating colonies were induced to differentiate to neural lineages. The normal morphology of neurosphere-like floating colonies and the neurites extended from cells are depicted in Fig. 11A and B. The expression of β-TUB and MAP-2 was assessed in protein level by immunofluorescence (Fig. 11C, D). It seems that the employed protocol has triggered the cells toward neural fates, as deduced from positive expression of mature neural markers in differentiated cells. Expression of Nestin, β-TUB, MAP-2, and NSE was further confirmed in RNA level. RT-PCR analysis showed expression of the 4 neural markers in induced cells contrasted to the untreated starting population (Fig. 11E, F). The upregulation of neural-specific markers, as deduced from RT-PCR analysis in neurally induced BM-MSC, AT-MSC, and USSC, is presented in Supplementary Fig. S2. The expression pattern of β-TUB, MAP-2, and NSE showed upregulation in all studied cell types.

Neurogenic potential of NSP
Senescence and transformation assay in long-term culture
A senescent cell specifically expresses galactosidase at pH 6. NSP cultures from all donors entered senescence as recognized by presence of blue staining in the cytoplasm. The proportion of aged cells in passage 1 of NSP and BM-MSC was about 2% (Fig. 12A, D, E, H), which is consonant with previous studies on MSC [27]. BM-MSC reached senescence phase nearly after 10 in vitro passages (Fig. 12G). In contrast, in NSP at the passage 15, nearly 10% of the cells expressed galactosidase (Fig. 12B). The senescence level showed an increasing trend as cultures aged (Fig. 12A–C and E–G for NSP and BM-MSC, respectively).
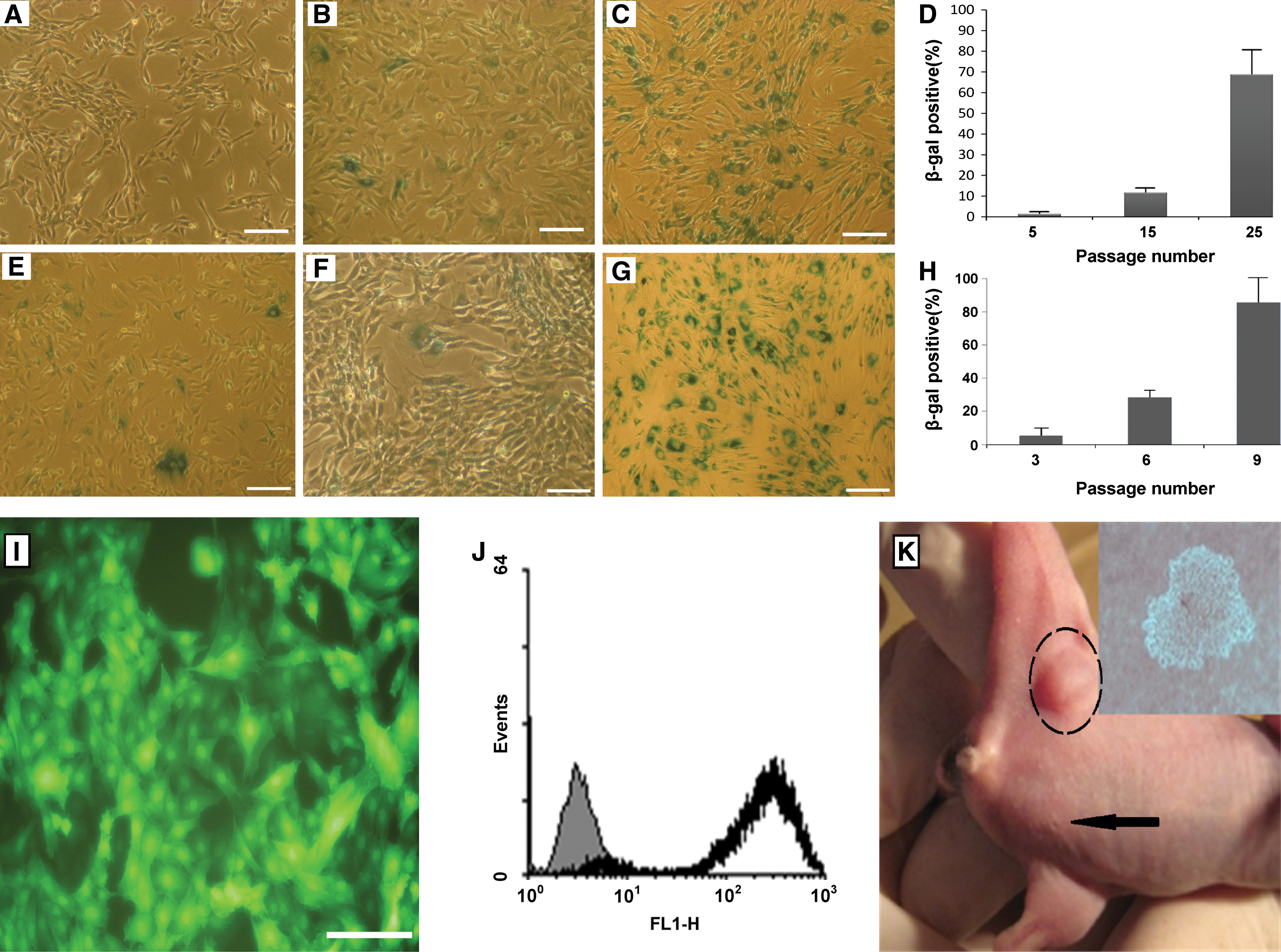
Karyotyping was also assayed to evaluate the chromosomal stability and unwanted transformation of NSP at different time points of in vitro culture, that is, at P5, P15, P25, and P35 postisolation. In all cases, NSP retained a normal karyotype (2n=46) during long-term culture (data not shown), and no chromosomal abnormalities were envisaged. None of the samples showed choromosomal abnormality.
After lentiviral transduction of eGFP into subconfluent cultures of NSP, 85%–95% of the cells expressed GFP (Fig. 12I, J). The proliferation rate of the transduced cells were not affected, except for the first days after transduction. In in vivo tumorigenesis assay, NSP and MKN45 were injected into the left and right legs of NOD-SCID mice, respectively (Fig. 12K). As expected, no sign of tumor formation or illness or cell spread was detected in the inoculated mice, which further verifies the lack of transformation of NSP during ex vivo expansion. In addition, the in vitro expanded NSP could not proliferate in soft agar, which means that the expanded population kept its anchorage dependant growth without any sign of transformation (Fig. 12K). These results confirm the chromosomal stability of the cells along with the safety of transduction procedure, which opens new frontiers to safely employ this invaluable autologous cell source for in vivo implications incase cell tracking or gene therapy is needed.
Discussion
To date, the presence and characteristics of stem and/or progenitor populations within various adult tissues has been widely studied [28,29]. However, for the translation of any potent cell source to clinical reality, the desired cell number should be obtained within the least time; a shortcoming associated with BM-MSC [1]. According to the literature and our own experiences, reaching a population size of up to 1 million of BM-MSC would need at least 10 to 24 days. In this regard, the worldwide scientific and medical community has been trying to find an autologous highly proliferative, genetically stable safe cell source, with nasal septal cartilage being underappreciated. The frequency of progenitors in the pooled population from septal cartilage was 6±1.9 in 100 plated cells, which is surprisingly far more than stem cells present in aspirated BM (1 in 25,000 to 1 in 100,000) or stem cells derived from adipose tissue (∼2%) [1,30]. Since relatively few cells can be obtained from aspirated BM or cord blood or even adipose tissue, these cells should be vastly expanded in vitro to give rise to sufficient cell numbers before differentiation [1]. It has been previously shown that primary MSC were about 60% clonogenic [24,27,31,32]. However, the expanded clonal population from septal cartilage showed a clonogenicity of 73±3.4. The ability to form high number of colonies from a few seeding cells is indicative of the prevalence of progenitor-like cells in the expanded population, as is the case for other tissues that contain a stem cell population [33]. Although CFU values decline at later time points, NSP are still highly clonogenic in comparison with other MSC at the equivalent passages. Human BM-MSC lose their self-renewal potential after about 40–50 population doublings, this deficiency is accompanied with gradual loss of their differentiation capacity [34].
When considering propagation potential and PDT, a comparison with other cells sharing the same embryonic origin as NSP, that is, neural crest-derived stem cells such as dental pulp stem cells and olfactory ensheathing cells, again confirms the striking self-renewal ability of this population [35 –37]. Besides, cell cycle analysis using flow cytometry revealed that the percent of NSP in G2/M phase was twice than that of MSC, which signifies the higher proliferation rate of these cells relative to MSC. Resistance of NSP to frequent freeze-thaw cycles adds to the value of these cells for prolonged cryopreservation, a crucial criterion for any potent clinical cell source. The apparent high transduction efficiency of these cells, as deduced from flow cytometry results, makes these cells formidable candidates when in vivo cell tracking or gene therapy is sought.
Rotter et al. demonstrated that although the biochemical and biomechanical properties of nasal cartilage are age dependant, the isolated cells presented no significance dependence on the age of the donor [38], signifying the fact that the in vivo microenvironment of the cells before harvest (which is vastly affected by the age) might not have effects on subsequent cell properties ex vivo. In our study also, though in a narrower range, we observed the same independency between donor age and in vitro cellular properties. The isolated progenitors showed the capability to form small floating clusters similar to neurospheres formed from neural stem cells [39]. We successfully exploited this ability to imitate the neurogenic pathway of embryonic and neural stem cells. In fact, the ability to form free floating aggregates is an attribute of undifferentiated stem and cancer cells [40,41], which further validates the stemness properties of the expanded population.
Although CD105 (Endoglin) has been known as a specific marker for MSC, recently the higher chondrogenic potential of CD105+ subpopulation of AT-MSC has been documented [42]. Endoglin plays a role in TGF-β signaling in chondrogenic differentiation [42]. It has been suggested that coexpression of CD166 and CD105 is an attribute of potential chondroprogenitors [19]. We also observed a high expression of the mentioned surface markers for NSP, which is in consistence with high chondrogenic capacity of NSP. NSP express a subset of MSC-specific antigens, that is, CD166 (ALCAM), CD105, CD106, and CD90 while being negative for early hematopoietic markers CD34 and CD45. Here, we showed that NSP are closely kindred to BM-MSC. However, NSP exhibit some distinctive traits such as inherent expression of S-100, GFAP, and NGFR, knowing that these cells originate from a neural crest-derived tissue [37].
Expression of CD271 (low-affinity NGFR, P75) and S100, neural crest stem cell markers [43], reflects the ectomesenchymal embryonic origin of NSP and emphasizes that nasal septal cartilage contains at least a subpopulation of stem-/progenitor-like cells expressing many features attributed to the mesoectodermal-derived cells. CD133 is a well-known specific stem cell surface marker that is expressed on epithelial cells, HSCs (hematopoietic stem cell), and also neural stem/progenitor cells [44]. The ability to form neurosphere-like floating clusters is reported for HSCs and neural stem cells [45]. Interestingly, NSP express this marker to a high level, which is consistent with their stemness properties and also their ability to form floating bodies [45]. A continual drawback with chondrocytes expansion had been their dedifferentiation accompanied by losing Coll-II expression after in vitro monolayer expansion [46], which is vastly documented by indicating the disappearance of Coll-II and chondromodulin expression in cultivated cells compared with freshly isolated cells [47]. Lack of Coll-II expression in the mixed cell cultures isolated from septal cartilage may show how sensitive septal chondrocytes are to in vitro conditions, causing them to lose their original phenotype after a few doublings. The immediate adhered cells of the digested tissue showed high clonogenecity and high expression of stemness markers, which ruled out the possibility that the expanded clonal population is dedifferentiated chondrocytes, rather than a progenitor subset.
Demonstration of plasticity in differentiation pathway is essential for identification of progenitor populations. Thus, we assessed the in vitro differentiation to different cell types not originally present in cartilage. These cells retain their differentiation potential throughout culture time even after extensive cryopreservation, without any compromise in their differentiation capability, which demonstrates that the process of senescence of NSP is not accompanied with their terminal differentiation. In contrast, the heterogeneous population of BM-MSC loses its plasticity after less than 10 passages [27]. Meanwhile, the capability of these cells to transdifferentiate to endodermal cell lineages has yet to be determined.
In the attempt to induce adipogenic, the very small and rare vesicles stained with Oil Red dye indicated low adipogenic potential, if any, of NSP that along with their high osteogenic and chondrogenic potential can be attributed to their original niche, which is in close vicinity to nasal bone and cartilage. The influence of the original site of cell extraction on cell behavior and differentiation potential has been previously reported for MSC from alveolar and iliac bone, such that less chondrogenic and adipogenic capacity was observed for alveolar bone-derived MSC [46]. The effect of the natural niche of the stem/progenitor populations on their in vitro differentiation plasticity has been vastly studied; for example, higher osteogenic, adipogenic, and chondrogenic potential has been particularly emphasized for periosteum porgenitors, AT-MSC, and synovial membrane progenitors [48], respectively.
The high osteogenic and chondrogenic potential of NSP relative to BM-MSC can be attributed to the high expression level of BMP-2 in these cells. BMP-2 is needed not only for chondrogenesis but also for chondrocyte maturation, with a role in induction of Coll-II and X [26]. Besides, this protein is a potent stimulator of osteogenesis through overrepresentation of RUNX2 and osteocalcin. Rosen et al. reported that in media devoid of BMP-2, progenitors express PPARg2, an important hallmark gene for adipocyte differentiation [49], whereas the expression of this gene was dramatically suppressed when medium was supplemented with BMP-2 [26,50]. In another study, Skillington et al. reported that BMP-2 can lead to an increased proliferation along with adipogenesis suppression [51]. BMP signaling is one of the major pathways in induction, migration, and aggregation of neural crest cells into clusters [52]. This protein is also needed for the formation of cell clusters in vitro [53]. The higher expression of BMP-2 in NSP in comparison with BM-MSC can lead to the observed higher propensity of NSP to give rise to osteocytes and chondrocytes and their disinclination to differentiate into adipocytes.
Recently, there has been increasing concern regarding chromosomal abnormality and immortality of the ex vivo expanded cells [10]. Despite contradictory reports on spontaneous transformation of different stem cells in long-term culture, even the least possibility of such potentially tumorgenic abnormalities should not be ignored [11]. To exploit the delightful proliferative and differentiative features of progenitor cells residing in the septal cartilage, it is critical to examine these cells for their susceptibility to chromosomal abnormalities that can trigger unwanted cancers [11]. Herein, we proved the safety of NSP after prolonged ex vivo expansion by karyotype analysis and in vitro and in vivo tumorgenic assay.
Footnotes
Acknowledgment
This work was supported by funding from Stem Cell Technology Research Center, Tehran, Iran.
Author Disclosure Statement
Authors have nothing to declare.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.