
Abstract
Endothelial cells (ECs) are desired for their therapeutic potential in a variety of areas including gene therapy, cardiac regeneration, development of tissue-engineered vascular grafts, and prevascularized tissue transplants. Pluripotent embryonic stem cells (ESCs) can be induced to differentiate into ECs in vitro using embryoid bodies, monolayer cultures, or by genetic manipulation and immortalization. However, obtaining homogeneous cultures of proliferating ESC-derived ECs without genetic manipulation is a challenging undertaking and often requires optimization of protocols and rigorous purification techniques. Moreover, current differentiation methods that use medium containing fetal calf or bovine serum components introduce additional challenges because of our limited ability to control the differentiation signals and batch-to-batch variations of serum. We have explored the development of new medium formulations for deriving ECs from murine embryonic stem cells (mESCs) using only chemically defined reagents. We present 2 different medium formulations along with the detailed methodologies, including the optimization of extracellular matrix–derived substrates known to play a role in cell attachment and proliferation as well as cell differentiation. Characterization of the ESC-derived ECs indicate that (1) chemically defined medium formulations reproducibly generate superior ECs compared with previous serum-containing formulations, (2) fibronectin, and not collagen type-IV, is the optimal substrate for EC induction in our chemically defined medium formulations, (3) without additional activation of Notch-signaling, ESC-ECs develop predominantly into venous ECs, and (4) using these medium formulations, a second rigorous selection step is not required to generate proliferating ECs from ESCs, but it does enhance the final purity of the ECs.
Introduction
E
Vascular ECs or endothelial progenitor cells derived from stem cells could potentially lead to a variety of clinically relevant therapeutic applications [1]. Endothelial progenitor cell transplantation has been shown to induce new vessel formation in ischemic myocardium and hind limb [2 –4], supporting enthusiasm that these cells could be used in strategies for the repair and revascularization of ischemic tissue in patients exhibiting vascular defects [4,5]. Additionally, because ECs inhibit platelet adhesion and clotting, lining the lumen of a synthetic or tissue-engineered vascular graft may aid in patency of vascular grafts [6,7] or in the development of prevascularized tissue-engineered materials. Moreover, because ECs line the lumen of blood vessels and can directly release proteins into the blood stream, they are ideal candidates to be used as vehicles of gene therapy.
EC differentiation from embryonic stem cells
Human and murine embryonic stem cells (ESCs), isolated from the inner cell mass of a developing blastocyst, are pluripotent cells that are also capable of self-renewal as well as to differentiate into cells from all 3 germ layers [8]. ESCs are an especially attractive cell culture system because they can be easily maintained and expanded in culture. Although it is possible to obtain stem cells from adult sources, such as bone marrow and adipose tissue, adult cells exhibit limited pluripotency compared with ESCs or induced-pluripotent stem cells. Additionally, adult stem cells can be difficult to identify, isolate, and expand in culture. For these reasons, ESCs are an ideal cell culture system for studying stem cell fate and vascular development.
Successful methods for the in vitro differentiation of ECs from ESCs [9 –16] and adult stem cells [17 –19] have been previously described. One common method used in the derivation of several cell types from ESCs, including ECs, involves the formation of a 3-dimensional aggregate called an embryoid body [9,14]. This structure allows the differentiation of ESCs toward various cell types from all 3 germ layers. Unfortunately, it is difficult to control the cells' microenvironment within the embryoid body. Conversely, a 2-dimensional monolayer induction system allows for easier cell visualization real time and better control over the cells' microenvironment [12,13,15]. Endothelial promoting growth factors, such as vascular endothelial growth factor (VEGF), can be also added to the differentiation medium to increase cell differentiation and proliferation of a specific cell phenotype.
Our laboratory and others have published methods for the differentiation of ECs from ESCs [9 –13,15]. These methods incorporate treatment with VEGF to promote EC specification; however, they also rely on fetal bovine serum to further promote differentiation and proliferation of the EC populations. Unfortunately, the presence of serum in the induction medium formulations often leads to problems with reproducibility due to uncontrolled variations from batch-to-batch of serum and also limits one's ability to directly control stem cell fate.
For these reasons, serum-free replacements have been explored and successfully used in the maintenance of ESCs and stem cell differentiation [20 –24], but this has not yet been accomplished for EC induction from mouse ESCs. Although most formulations of serum replacements are proprietary, they are generally free from animal components and do not demonstrate the batch-to-batch variation seen in serum. Using a chemically defined medium, one can more accurately control the cell's microenvironment and, therefore, better evaluate the response of a particular biochemical or physical signal. In addition, the final yield and quality of functionally mature tissue-specific cells derived in vitro from stem cells may be improved by using chemically defined medium formulations that allow directed differentiation, rather than serum-containing formulations.
We set out to explore methodologies for directed differentiation of ESCs toward ECs using only chemically defined medium formulations. Here, we present our novel medium formulations for the induction and culture of these cells at well-defined stages of maturation. Moreover, we explored optimal time points and matrix substrates required for the initial generation of high numbers of Flk-1+vascular progenitor cells and Flk-1+outgrowths. The methods presented in this article allow a more consistent and robust generation of ECs with appropriate expression of endothelial markers as well as an improved low-density lipoprotein (LDL) uptake compared with previous derivation methods that included the use of serum [25].
Materials and Methods
ESC culture
R1 and D3 mESCs (ATTC) and E14 mESCs (gift from Bruce Conklin) were cultured on 0.5% gelatin-coated cell culture dishes in serum-free medium [26]. This medium contains knockout Dulbecco's modified Eagle's medium (DMEM; Invitrogen), 15% knockout serum replacer (Invitrogen), 1×penicillin–streptomycin (Invitrogen), 1×nonessential amino acids (Invitrogen), 2 mM
Induction of Flk-1+cells in a chemically defined medium
Undifferentiated R1 and E14 mESCs were harvested from gelatin-coated dishes using 0.25% trypsin/2.21 mM EDTA (Mediatech) and plated on cell culture plates coated with various commercially available substrates including 0.5% gelatin, 50 μg/mL fibronectin, 50 μg/mL collagen type-I, 50 μg/mL collagen type-IV, and 50 μg/mL laminin as per manufacturer's instructions (BD Biosciences). The initial induction medium optimized by our laboratory was named “NS1D2b.” (Table 1). This consists of alpha-MEM (Cellgro), 20% knockout serum replacer (Invitrogen), 1×penicillin–streptomycin (Invitrogen), 1×nonessential amino acids (Invitrogen), 2 mM
The first induction medium optimized for the generation of Flk-1+ vascular progenitor cells is called “NS1D2b.” The second medium formulation called “LDSk” is optimized for EC specification and expansion.
bFGF, basic fibroblast growth factor; BMP-4, bone morphogenetic protein-4; DMEM, Dulbecco's modified Eagle's medium; EC, endothelial cells; VEGF, vascular endothelial growth factor.
In addition to verifying induction time, we also optimized the substrate for induction of Flk-1+cells by using culture plates coated with either gelatin, fibronectin, collagen type-I, collagen type-IV, or laminin. The adherent cells were dissociated, counted, stained with AlexaFluor 647-conjugated anti-Flk-1 antibodies (Biolegend), and analyzed for Flk-1 expression using a BD LSRII flow cytometer and FlowJo software (TreeStar).
Purification and expansion of ESC-derived ECs in chemically defined conditions
After the initial induction period, the cell population was enriched for Flk-1+vascular progenitor cells using either fluorescent-activated cell sorting (FACS) or a MiniMACS (Miltenyi Biotec). AlexaFluor 647–conjugated anti-mouse Flk-1 antibodies (Biolegend) and anti-AlexaFluor 647 magnetic beads (Miltenyi Biotec) were used to label the Flk-1+expressing cells. Postenrichment, the Flk-1+cells were replated on dishes coated with either fibronectin, laminin, collagen type-I, collage type-IV, or gelatin. These cells were expanded in another chemically defined medium developed in our laboratory that we have named “LDSk.” (Table 1). The medium consists of 70% alpha-MEM (Mediatech) and 30% DMEM (Invitrogen) plus 100 ng/mL VEGF (R&D Systems), 2×Nutridoma CS (Roche), 50 ng/mL basic fibroblast growth factor (Sigma), 2 mM
Second purification of ESC-derived ECs
We have previously published that a second purification is necessary for proliferation of the D3 ESC-derived ECs produced using medium formulations containing serum [10,25,26]. Although we have rigorously examined a variety of purification techniques including magnetic activated cell sorting and FACS, the manual picking of cobblestone colonies consistently results in the most pure EC cultures (>95% purity [10]). Moreover, we found that obtaining relatively homogeneous EC cultures is a critical factor allowing the expansion of the maturing ECs [10]. Here, we also compare ECs generated with and without this second purification step under serum-free conditions.
ECs with cobblestone morphology were manually picked with flame-pulled microtip Pasteur pipettes in a sterile laminar flow hood outfitted with a stereoscope (Zeiss). The 9” Pasteur pipettes (VWR) were flame-pulled to a thin point and attached to a mouth aspirator line (Sigma-Aldrich) with a 0.22-μm filter (Whatman) for performing the sterile manual selection. The culture plates were washed with phosphate-buffered saline followed by 10 min in cell dissociation buffer (Invitrogen) to allow gentle cell scraping and aspiration of the picked cells with the microtip pipette. The manually picked cells were then plated onto fibronectin-coated dishes in LDSk medium.
Flow cytometry analysis
Vascular cells, both with and without the second manual selection, were stained for the following endothelial markers: Flk-1 (Biolegend), vascular endothelial-cadherin (VE-cad; eBioscience), Flt-1 (Santa Cruz), EphB4 (Santa Cruz), ephrin-B2 (Santa Cruz), and Tie-1 (Santa Cruz). Some cells were also stained for calponin (Santa Cruz), a marker indicating smooth muscle cells (SMCs). The secondary antibodies include anti-rat PE (Abcam), donkey anti-rabbit PE (Fitzgerald), and donkey anti-goat FITC (Abcam). All samples were analyzed using a BD LSRII flow cytometer and FlowJo software (TreeStar).
LDL uptake
ESC-derived ECs derived in serum-containing medium and those derived under chemically defined conditions were plated on Permanox microscope slides (Nunc). Commercially available Alexa Fluor 488 Acetylated-LDL (Invitrogen) was diluted to 1:100 in DMEM (Invitrogen) and incubated with the cells for 4 h at 37°C. The slides were then stained with DAPI and fixed with 4% formaldehyde. The slides were imaged with a Leica fluorecent scope.
Results
R1-ESCs were induced toward vascular progenitor cells in chemically defined NS1D2b medium on various substrates over 5 days and then analyzed for the expression of the vascular progenitor cell marker, Flk-1, using flow cytometry analysis (Fig. 1). Unfortunately, the ESCs at day 1 exhibited very low levels of cell adhesion/proliferation and we were not able to consistently obtain enough cells for flow cytometry analysis. The percentage of Flk-1+cells from R1-ESCs cultured for 0, 2, 3, 4, and 5 days on fibronectin under the chemically defined conditions peaked on day 2 (over 70%), continually decreasing over the next 5 days. We also examined the effect of cell-matrix signaling during the 2-day induction of Flk-1+progenitors. We plated these cells on collagen type-I, collagen type-IV, laminin, fibronectin, and gelatin. Figure 2A shows that, contrary to currently accepted reports that collagen type-IV is the optimal substrate for the induction of Flk-1+vascular cells [12,13,15], the actual percentages of Flk-1+cells induced from R1-ESCs under chemically defined conditions did not vary significantly between substrates. We subsequently counted the adherent cells on the various substrates at the end of the 2-day induction period to quantify their ability to promote adhesion and proliferation of the differentiating ESCs (Fig. 2B). Combining Flk-1+cell percentages with total cell numbers, the optimal substrates yielding the greatest total number of Flk-1 cells are gelatin and fibronectin (Fig. 2C). We suspect that these results might not necessarily be due to directed differentiation, but enhanced proliferation of the adherent cells on gelatin and fibronectin. The results of this study shown in Fig. 2 are particularly important to note, as many previous studies, including ours [10,25], use collagen type-IV for differentiation of vascular cells in serum-containing medium. The culture of ESCs on collagen type-IV in a chemically defined medium without serum actually yields the lowest number of Flk-1+cells of all the substrates tested. We hypothesize that the superior results on fibronectin, and not collagen IV, is due to the fact that important adhesion-related proteins, such as fibronectin, found in serum would not be available in our serum-free cultures.
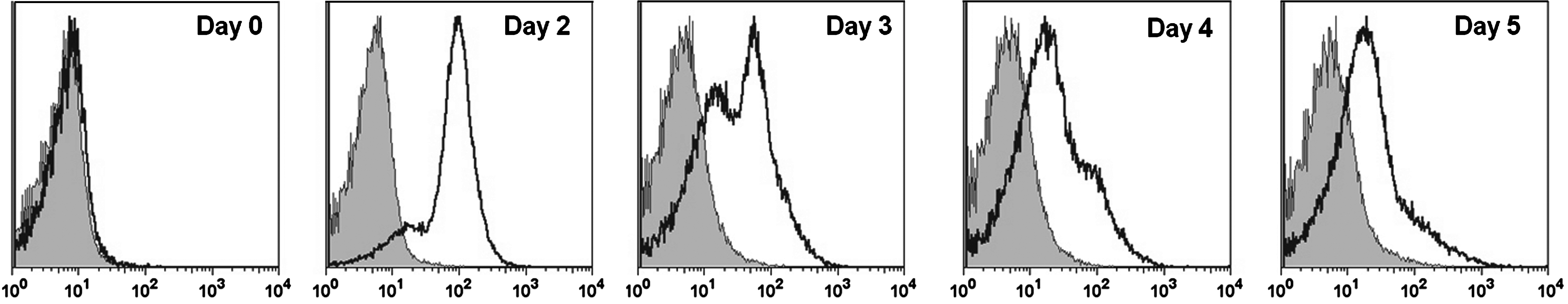
The largest numbers of Flk-1+ vascular progenitor cells are seen at day 2 of induction from embryonic stem cells (ESCs). Histograms of the Flk-1+ expression of R1 mouse ESC on day 0 (prior to induction) and those induced in chemically defined medium on days 2, 3, 4, and 5. The optimal expression of Flk-1+ cells occurs at day 2 for this cell line and subsequently decreases.
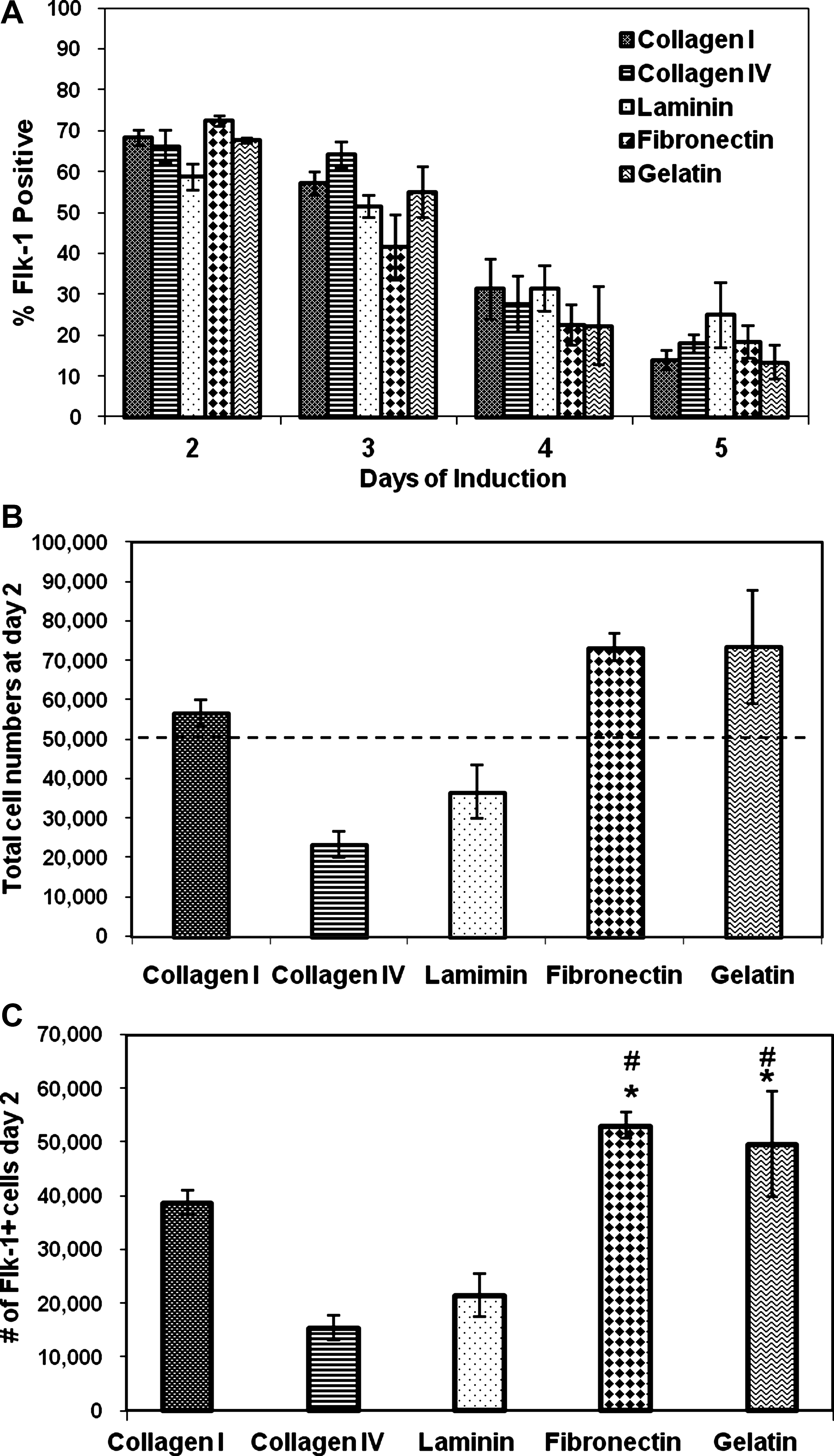
Culture on fibronectin and gelatin yields the largest numbers of Flk-1+ cells at day 2.
The Flk-1+cells were then enriched and plated onto either collagen type-I, collagen type-IV, laminin, gelatin, or fibronectin-coated dishes in LDSk medium. After 2 weeks, the outgrowths from the Flk-1–expressing cells were analyzed for markers of venous endothelium (EphB4), arterial endothelium (ephrin-B2), endothelial VE-cad, and vascular smooth muscle (calponin). Here, we do see marked differences in the induction of vascular cells on different substrates (Fig. 3). Laminin generates the largest number of venous ECs, whereas both collagen IV and fibronectin generate the most number of arterial ECs (Fig. 3D). However, all of these percentages are still quite low at this early stage of endothelial maturation. Looking at an earlier endothelial marker, VE-cad, we see that collagen I, collagen IV, and fibronectin yield the largest percentages of VE-cad+cells (Fig. 3A, D). We then looked closely at the SMC outgrowths and observed that the cells cultured on fibronectin also contained the lowest percentage of contaminating SMCs (Fig. 3B) and that the greatest cell yield was again observed in cells cultured on fibronectin (Fig. 3C). Based on these data, we continued to expand ESC-derived ECs on fibronectin to reduce the percentage of SMCs in the cultures while promoting optimal adhesion and proliferation of our ECs.
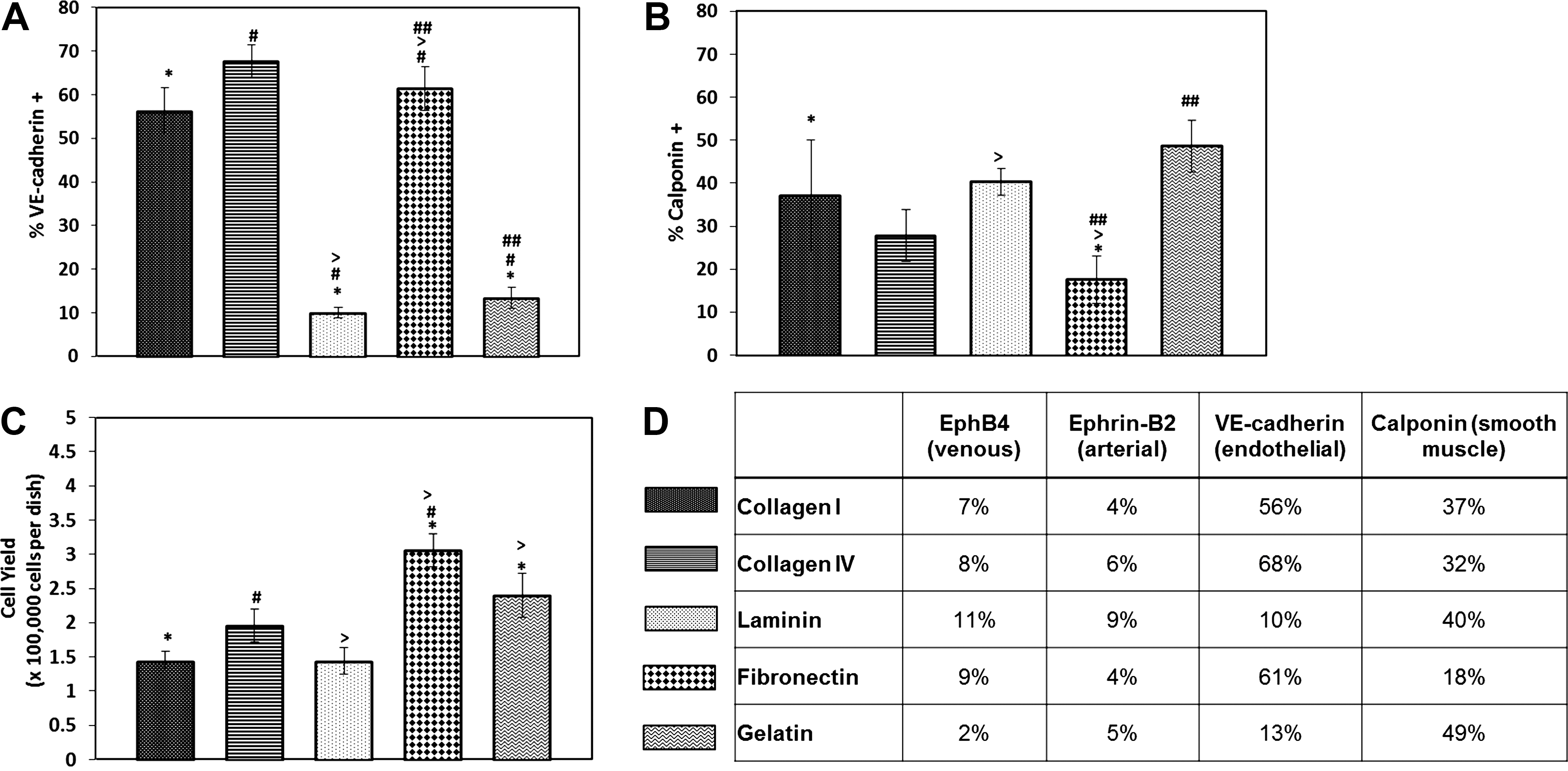
Flk-1+ outgrowths cultured on fibronectin generate the largest number of endothelial cells (ECs) with minimal smooth muscle cell (SMC) contamination.
The expanded Flk-1+ cells subcultured in LDSk medium on fibronectin robustly supported the differentiation and proliferation of ECs, and some of the Flk-1+ exhibited distinct cobblestone-like morphologies (Fig. 4), allowing manual selection to be used for further enrichment of the ECs. The expanded ESC-derived ECs were then analyzed for EC markers Flk-1, VE-cad, Flt-1, Tie-1, venous-specific marker, EphB4, and the arterial-specific marker, ephrin-B2, using FACS analysis (Fig. 5 and Table 2). Here, the differentiation and expansion procedures of the ESC-R1 cells derived in our chemically defined medium formulations, as described here, expressed very high levels of most of the EC markers that were examined, but did not exhibit a high level of ephrin-B2 arterial surface marker, indicating a largely venous phenotype (EphB4) for these cells (Fig. 5, green). We also included characterization of our ESC-ECs derived from serum-containing medium for 2 different ESC lines. Comparing columns II (aqua) and III (red), the serum-containing induction protocol worked well for the ESC-D3 cell line (Fig. 5, red) as previously described [10,25,27], but the same serum-containing medium was not able to generate the same level of quality ECs from the ESC-R1 cell line (Fig. 5, aqua). We expect that this difference is due to the inherent heterogeneity between ESC lines.
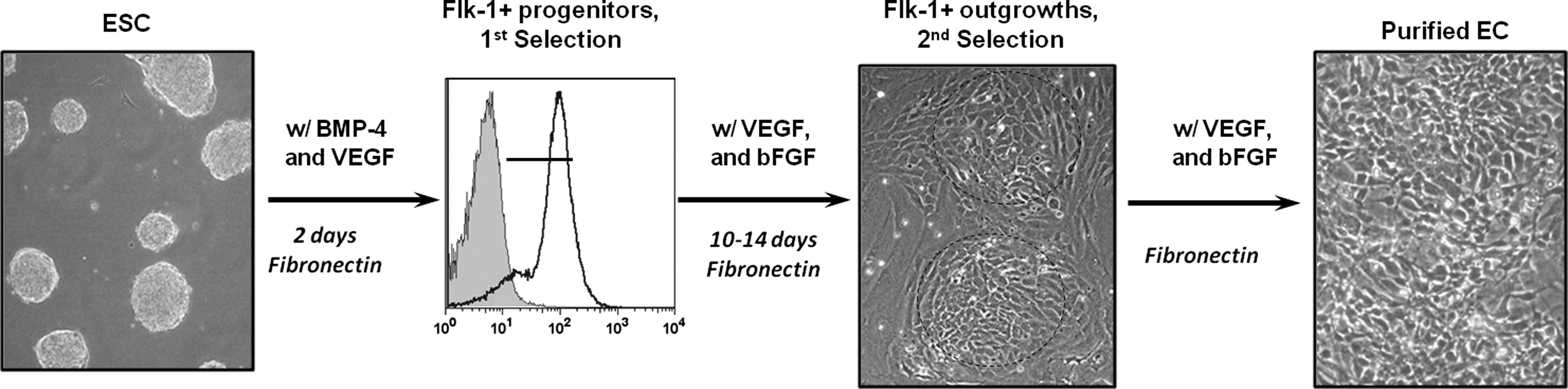
Flowchart of EC differentiation procedure. Undifferentiated ESCs are expanded on gelatin and then transferred to fibronectin for 2 days of induction with bone morphogenetic protein-4 (BMP-4) and vascular endothelial growth factor (VEGF) treatment. The Flk-1+ cells are then isolated and replated on fibronectin in medium containing VEGF and basic fibroblast growth factor (bFGF). The Flk-1+ outgrowths generate cobblestone-like cell sheets. These sheets are manually selected for optimal EC purification.

ECs derived in chemically defined conditions express appropriate endothelial makers. We examined the expression of endothelial markers Flk-1, VE-cadherin, Flt-1, Tie-1, EphB4 (venous), and ephrin-B2 (arterial). The histograms include the ECs derived using R1 ESCs under chemically defined conditions (green). Also included are ECs derived from R1-ESCs (aqua) and D3-ESCs (red) using our previous medium formulation that contained FBS. The last 2 columns include comparisons of the EC marker expression for the ECs from R1-ESCs (gold) and E14-ESCs (purple) derived under the new chemically defined conditions, but these did not include the second EC purification.
The percentage of positive cells expressing the listed EC markers for the ECs derived using our chemically defined medium compared with old serum formulations (quantitative data from Fig. 5). Also included are data comparing the percentage of positive cells with and without a second rigorous manual selection for optimal purification of the ECs.
VE-cad, vascular endothelial-cadherin.
All 3 of the ESC-ECs presented in the first 3 columns (green, aqua, and red) followed a rigorous differentiation and expansion methodology including 2 distinct isolations. These include an early isolation of vascular progenitor cells plus a late isolation that purifies the progenitor cells further into relatively pure (>95%) ESC-ECs [10]. In addition, the second manual selection (or another method of post-Flk-1+ sorting enrichment) of late developing ECs was required for EC proliferation of the ESCs derived using serum-containing medium [10]; we expect, because of the high levels of contaminating SMCs in serum-containing cultures. However, the EC induction methodology presented in this article uses chemically defined mediums that allow the generation of proliferative ECs with minimal contaminating SMCs. Therefore, we sought to characterize the outgrowths of the nonenriched Flk-1+ to explore whether the second late-stage selection is a necessary manipulation under these chemically defined conditions (Fig 5, gold). The data indicate that although these cells do exhibit equivalent expression of EC molecules Flk-1 and VE-cad, the expression levels of Flt-1, Tie-1, and EphB4 were lower compared with the ESC-ECs that did undergo the second isolation, indicating that this second selection process does generate better quality and, potentially, more mature ECs. Figure 6 examined the coexpression of Flk-1 with endothelial markers, EphB4 and Ephrin-B2, as well as the smooth muscle marker, calponin. Interestingly, 45% of the Flk-1+ cells coexpress the marker for venous endothelium, EphB4, but only 8% of these cells express Ephrin-B2. This indicated that the remaining 37%–45% of the Flk-1+ cells have not yet specified an endothelial subphenotype (previous studies indicate that none of the cells are lymphatic; data not shown) and may retain some level of plasticity.
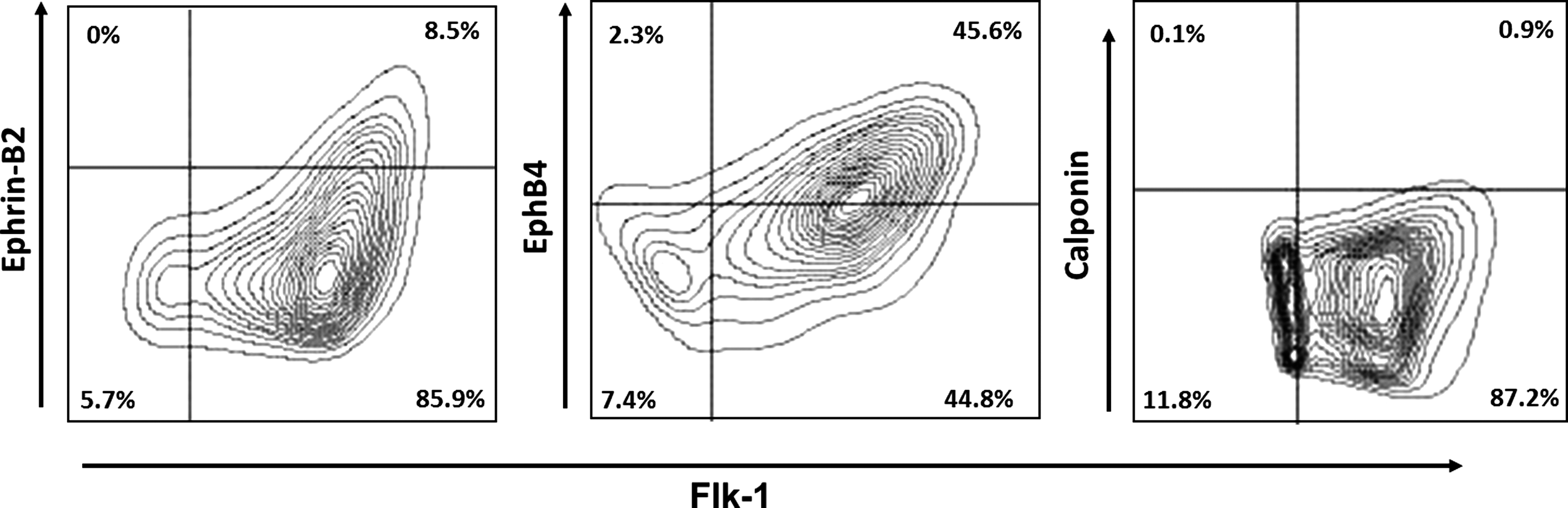
ECs derived in chemically defined conditions coexpress Flk-1 and EphB4 (venous). These cultures contain very few ephrin-B2+ arterial cells and do not contain calponin+ SMCs.
Lastly, the entire derivation procedure was repeated in a third ESC line (E14) to verify that the chemically defined medium formulations presented in this article can be applied toward other mouse ESCs. These ESCs were also able to generate ECs, but require a second manual selection for purification of the ECs (Fig. 5, purple).
Functional assays are also an important indicator of EC quality. The uptake of LDL is considered an important EC function that was not seen in our ESC-ECs derived from D3-ESCs in serum-containing medium [10,25]; however, as seen in Fig. 7A and B, our ESC-ECs derived in chemically defined conditions were able to take up LDL. We also observed that some of the ESC-ECs that did not undergo the second isolation (Fig. 5, gold) could take up LDL (Fig. 7C). However, not all of the cells in the culture could take up LDL (Fig. 7D), indicating that these unpurified cells were also remaining somewhat heterogeneous, even in the chemically defined medium.
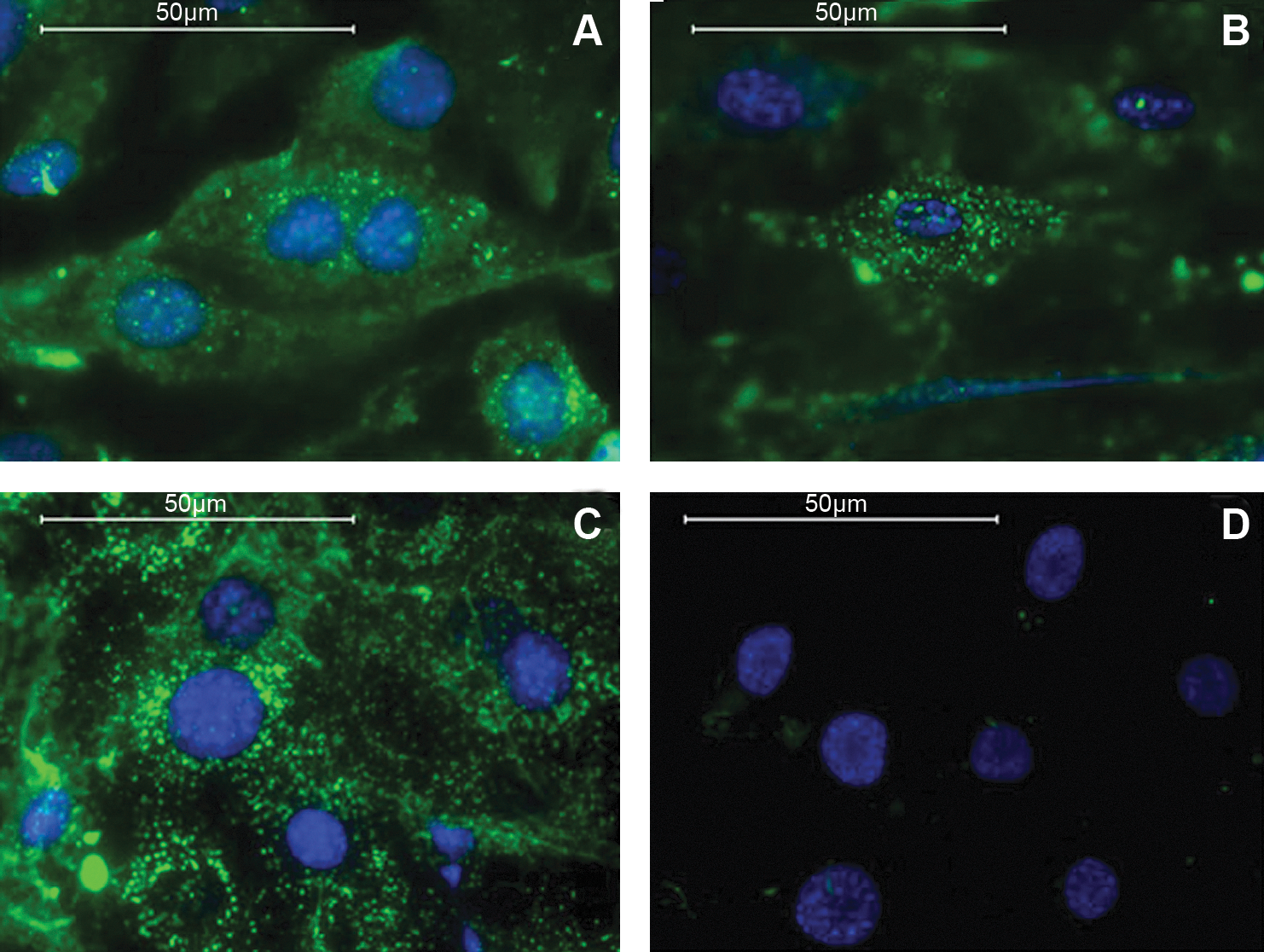
ECs derived in chemically defined conditions take up low-density lipoprotein (LDL). The 2
Discussion
Our initial stage of induction focused on promoting the differentiation of ESCs into Flk-1+ cells similar to studies using serum-containing medium formulations [10 –13,15,16]. Our rationale is supported by the fact that the Flk-1 surface molecule is currently considered to be the first lineage-commitment marker expressed on vascular and hematopoietic progenitors [28 –41]. BMP-4 and VEGF are also known to promote ventral mesoderm and hematopoietic development while inhibiting neuronal development [42 –44]; therefore, these were also incorporated into our serum-free differentiation medium. Conversely, basic fibroblast growth factor, considered to be a pro-angiogenic factor, was not required in our early stage of induction (i.e., generation of Flk-1+ cells) [42] but incorporated with VEGF at the later, more mature, stages of development for enhancing EC proliferation.
We also examined the effect of ECM substrate on the EC inductions. Our results using a chemically defined medium indicate that early-stage EC inductions focused on generating Flk-1+ mesodermal cells can take place on any of the substrates examined, including gelatin. However, the substrates' role in directing the specification of the Flk-1+ cells toward various cell phenotypes was much more enlightening. It seems that fibronectin directs the most EC differentiation, leading to the greatest number of VE-cad+ ECs, whereas culture on gelatin, laminin, and collagen I result in more SMCs. If one wanted to study the codevelopment of ECs and SMCs, collagen type-IV would remain the optimal substrate that allows the simultaneous proliferation and differentiation of both cell types. These data from ESC-EC derivations in a chemically defined medium also challenges the currently accepted belief that collagen IV substrate generates largest number of vascular progenitors and ECs [13,15].
Lastly, it has been proposed that the venous lineage is the default pathway during EC development [16], presumably because of insufficient Notch activation [45]. Based on the high EphB4 and limited ephrin-B2 expression levels of our cells, we found that our ESC-derived ECs also resembled venous endothelium compared with ephrin-B2–expressing arterial endothelium. One study using cells cultured in serum and VEGF found that >90% of the cells became venous ECs, but when Notch signaling was activated by stimulating the cyclic adenosine monophosphate (cAMP) pathway (by adding either 8bromo-cAMP or adrenomedullin—a cAMP-elevating factor—to the serum- and VEGF-containing medium), up to 70% of the cells expressed ephrin-B2 arterial marker [46]. On the basis of this study, one would expect that specifically activating Notch signaling in our chemically defined cultures may also lead to the generation of arterial ECs.
The generation of ECs from ESCs in chemically defined conditions is valuable for potential use of these cells in a variety of therapeutic applications. By eliminating various unknown animal contaminants and removing unspecified and uncontrollable elements found in serum, chemically defined culture conditions allow better controlled studies of the effects of various biological and mechanical signaling variables. Although we can see some developmental variations between different ESC lines, the reproducibility and quality of ESC-derived ECs were significantly increased using the chemically defined conditions described.
Footnotes
Acknowledgment
This work was funded, in part, by a National Institutes of Health-National Service Award (NIH-NRSA) from the National Heart Lung and Blood Institute (NHLBI) (No. F31HL087716).
Author Disclosure Statement
No competing financial interests exist.