
Abstract
In the culture system using human feeder cells, the mechanism through which these cells support undifferentiated growth of embryonic stem cells (ESCs) has not been well investigated. Here, we explored the mechanisms of 3 kinds of human feeder cells, including human placental cells from the chorionic plate, human bone marrow stromal cells, and human foreskin fibroblasts. First, we determined that undifferentiated growth of 2 kinds each of human (H1 and HSF6) and mouse (D3 and CE3) ESCs was possible in all human feeder cell types tested (human placental cells, human bone marrow stromal cells, and human foreskin fibroblasts), without the need for exogenous cytokine supplementation including basic fibroblast growth factor (bFGF) and leukemia inhibitory factor. We then prepared their corresponding endogenous bFGF-knockout feeders using siRNA and tried to maintain human and mouse ESCs in their undifferentiated state; however, neither human nor mouse ESCs could be maintained in bFGF-knockout human feeder cells. The expressions of stemness markers such as Oct-4 and Nanog were significantly decreased in the bFGF-knockout group compared with those in the controls, and differentiation had already occurred, despite the undifferentiated morphologic appearance of the ESCs. In conclusion, human feeder cells are able to support the undifferentiated growth of human and mouse ESCs via bFGF synthesis. Further, a bFGF-dependent pathway might be crucial for maintaining the undifferentiated characteristics of mouse and human ESCs.
Introduction
V
Basic fibroblast growth factor (bFGF) is the key growth factor in the maintenance of undifferentiated growth in hESCs. [4 –6]. Since chemically synthesized serum replacement media are used in place of animal serum in the culture of hESCs, bFGF has to be exogenously supplemented in the culture media when using a mouse feeder cell line such as mouse embryonic fibroblasts (MEF) or when using feeder-free conditions. However, the question of whether bFGF is essential in culture systems using human feeder cells has not been well studied. Recently, we reported that HPC feeders are useful for maintaining undifferentiated hESCs during long-term culture without exogenous bFGF supplementation [7]; we found that HPC feeders express bFGF, suggesting that bFGF synthesized within HPCs might be a key mechanism for supporting hESCs. As a result of these experiments, we wanted to determine whether undifferentiated growth of hESCs is dependent predominantly on bFGF synthesized within HPC. Since the cytokine profile synthesized within HPC has not been completely investigated, it is possible that an alternative pathway which supports undifferentiated hESCs might exist.
We hypothesized that HPCs support the undifferentiated growth of hESCs through bFGF synthesis. Based on this, we want to determine whether the synthesis of bFGF, which supports undifferentiated growth of hESCs, is a characteristic of the placenta itself. Since the placenta is a specialized, transient tissue supporting the growth of the embryo and fetus during pregnancy and is distinguished from other tissues which support the growth of adult human cells, its function to support the undifferentiated growth of hESCs might be different from those of other feeder cells that are derived from human adult tissues.
At present, it is not known whether the properties of HPC that support undifferentiated growth of hESCs exist in other human feeder cells. If undifferentiated growth of hESCs is dependent on endogenous bFGF from HPCs, the question of whether this mechanism is limited to HPC or also exists in other human feeder cells should be explored. Finally, we wondered whether the supporting mechanism of human feeder cells in hESC culture are applicable to mouse ESCs (mESCs) cultured on human feeder cells. We recently reported that mESCs can be maintained in an undifferentiated state on HPC feeders without exogenous leukemia inhibitory factor (LIF) supplementation [8], raising the question of whether mESCs are dependent on LIF or other cytokines from human feeder cells. If bFGF synthesized within feeder cells is useful for supporting undifferentiated growth in both mESCs and hESCs, the feeder cells that synthesize bFGF could be utilized as universal feeders for the culture of hESCs and mESCs. To answer these questions, we investigated the mechanism through which human feeder cells, including HPC, HFF, and HBMSC, support the undifferentiated growth of hESCs and mESCs, focusing on the production of bFGF.
Material and Methods
Preparation of feeder cells
Before the preparation of HPC and HBMSC feeder cells, informed consent of voluntary donors and approval by the Institutional Review Board of Korea University Medical Center were obtained.
For the culture of HPC, we followed the protocol of Kim et al. [3]. Briefly, placental chorionic plates from healthy women who had undergone abortion at 6–8 weeks of gestation were surgically isolated, minced, and incubated in 0.25% trypsin-EDTA (GIBCO®; Invitrogen, Carlsbad, CA) at 37°C for 30 min. The cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM; Invitrogen) with 20% fetal bovine serum (FBS; HyClone Laboratories Inc, Logan, UT), 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C, 5% CO2, and 95% humidity, respectively. The medium was changed every 3 days until the third passage. During culture, cell debris was removed, and fibroblast-like cells were growing adherent to the tissue culture plate. Approximately 2 weeks after inoculation, colonies of fibroblast-like cells were formed. These fibroblast-like cells derived from the placenta chorionic plate (HPC) were cultured to the 12th passage and harvested for human and mouse ESC cultures after trypsinization and irradiation (1,500 cGy). Before being frozen as stock, reverse transcription–polymerase chain reaction (RT-PCR) was used to determine whether the HPCs were contaminated with pathogens that commonly infect the placenta, including cytomegalovirus, herpes simplex virus types 1 and 2, Chlamydia trachomatis, Chlamydia spp., Mycoplasma genitalium, Mycoplasma hominis, and Ureaplasma ureaticum. Previously published primer sequences were used [9,10].
For HBMSC culture, we followed the protocol of Cheng et al. [2]. Briefly, human bone marrow was obtained from healthy human donors during bone marrow harvest for allogeneic stem cell transplantation. Mononuclear cells were isolated from heparinized bone marrow aspirates using standard density (1.077 g/mL) centrifugation with Ficoll (Sigma-Aldrich, St. Louis, MO). Mononuclear cells at the interface were collected and suspended in a medium composed of DMEM with low glucose, 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin. During culture, cell debris was removed, and fibroblast-like cells were seen growing adherent to the tissue culture plate. Approximately 1–2 weeks after inoculation, colonies of fibroblast-like cells appeared. We harvested these bone marrow-derived fibroblast-like cells (HBMSC) using trypsinization. For co-culture with human and mouse ESCs, HBMSCs were irradiated (1,500 cGy) before being transferred to new plates.
For HFF, commercially available cells (ATCC, Manassas, VA; catalog number, CRL-2429) were used. Before being used as feeders, these cells were cultured in DMEM with 20% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C, 5% CO2, and 95% humidity. The medium was changed every 3 days until the second passage. For coculture with human and mouse ESCs, HFFs were irradiated (1,500 cGy) before being transferred to plates.
Transfection of siRNA
The 3 kinds of human feeder cells just described were divided into control and study groups (Fig. 1). For the control group, feeder cells were transfected with control siRNA for bFGF, whereas the study group feeder cells were transfected with bFGF-siRNA. The control and bFGF siRNA were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). Transfection was performed 48 h before transfer of hESCs and mESCs according to protocols provided by the manufacturer. Briefly, in a 6-well tissue culture plate, 2×105 feeder cells per well were seeded in 2 mL antibiotic-free normal growth medium supplemented with FBS and were incubated for 24 h at 37°C, 5% CO2, and 95% humidity. A mixture of 10 μg/mL siRNA and siRNA transfection reagent was added to each well and incubated for 6 h at 37°C, 5% CO2, and 95% humidity, followed by the addition of normal culture medium with antibiotics for 18 h. After that, the cell culture media were replaced and the transfected feeder cells were incubated for 24 h. After the final incubation period, prepared cells were used as feeders in ESC cultures. Before the transfer of human and mouse ESCs, a bFGF-knockout was identified using RT-PCR (Fig. 2A). The sequences for the control and bFGF siRNA are presented in Table 1. The culture medium was replaced with ESC culture medium (see below) immediately before human and mouse ESC transfer.

The overall study scheme. Three kinds of human feeder cells were prepared for each of the control and study groups. Control group feeders were treated with control siRNA for bFGF, whereas study group feeders were transfected with bFGF-siRNA. Two kinds each of human (H1, HSF6) and mouse (D3, CE3) embryonic stem cells were propagated on each type of control and study group feeder, simultaneously. The characterization of ESCs was planned every 10th passage. bFGF, basic fibroblast growth factor; ESC, embryonic stem cell; HPC, human placental cell; HFF, human foreskin fibroblast; HBMSC, human bone marrow-derived stromal cells; RT-PCR, reverse transcription–polymerase chain reaction.

The features of the human feeders.
bFGF, basic fibroblast growth factor.
Culture of human and mouse ESCs
The H1 and HSF6 hESC lines were purchased from the National Stem Cell Bank (Madison, WI) and were initially cocultured with MEFs according to the instructions of the provider. After the hESCs were maintained on MEFs for 10 passages, colonies of hESCs were simultaneously transferred onto either control or bFGF-knockout feeder group cells and were propagated on each of the 3 kinds of human feeder cells. DMEM/F-12 supplemented with 20% Knockout Serum Replacement (GIBCO), 0.1 mM β-mercaptoethanol, and 1% penicillin-streptomycin (Sigma Aldrich) (ESC culture medium) was used for coculture of hESCs and feeder cells. bFGF was not supplemented into the culture media of either the control or study groups. The culture medium was exchanged every 48 h. The colonies of hESCs were passaged weekly using mechanical dissociation. The method of mechanical dissociation was modified based on the published protocol [11]. Briefly, 2 days before transfer, we selected about one-third of the hESCs colonies based on size and differentiation status and excised differentiated and/or raised regions within each selected colony using a 26-G needle under a stereomicroscope. On the day of transfer, we cut each colony into a grid motif containing approximately 3–4 pieces per colony and dislodged the cells with a 26-G needle. After replacing the ESC culture medium in each well in which prepared feeder cells were plated, the whole prepared pieces of hESC colonies were transferred into the new well and then cultured at 37°C, 5% CO2, and 95% humidity. During culture, undifferentiated hESC colonies were counted before mechanical dissociation during each of the first 5 passages and then at every fifth passage thereafter. The D3 and CE3 mESCs that were purchased from ATCC were initially cultured on MEF feeder cells. After maintenance on MEF for 5 passages, colonies of mESCs were simultaneously transferred onto either control or bFGF-knockout feeder group cells. The culture conditions and protocols were the same as those of the hESC culture.
For ESC characterization, additional preparation of 6-well plates was planned at 5, 10, 20, 30, 40, and 50 passages. However, the planned ESC characterization was possible only in the control group, as the ESC colony from the study group could not be maintained. The comparison of ESC features between the groups was possible only at the fifth passage. At the designated passages, we cut each ESC colony into approximately 4–6 pieces per colony, and the additional pieces were used for ESC characterization.
Histochemical staining for alkaline phosphatase
To identify human and mouse ESCs in their undifferentiated states, morphology, expressions of stem cell markers, and differentiation capacity were examined in both control and study groups. Cell morphology was observed each day using an inverted microscope, and expression of stem cell markers was examined via histochemical staining for alkaline phosphatase (ALP) using a commercial kit (Chemicon International Inc., Temecula, CA) every 10th passage (Fig. 1). Briefly, human and mouse ESCs were cultured for 1 week in 6-well culture dishes and were fixed with 4% paraformaldehyde and 90% methanol/10% formaldehyde for 2 min and then washed with rinse buffer (20 mM Tris-HCL, 0.15 mM NaCl, 0.05% Tween-20). The substrate solution was prepared by mixing fast red violet with naphthol AS-BI solution and water in a 2:1:1 ratio. The substrate solution was added to each well and incubated in the dark at room temperature for 15 min. After incubation, plates were washed with rinse buffer and then stained with hematoxylin before being examined using a bright field microscope.
Real-time RT-PCR for stemness markers
To compare the degrees of expression of stemness markers, real-time RT-PCR for human and mouse Oct-4 and Nanog was performed. Total RNA was isolated from cells in colonies with an undifferentiated morphology using Trizol® total RNA isolation reagent (Invitrogen). The extracted RNA was quantified using a NanoDrop Spectrophotometer® (Thermo Fisher Scientific Inc., Wilmington, DE). First-strand cDNA was synthesized from 1 μg of total RNA using the RETROscript cDNA synthesis kit® (Ambion, Austin, TX). Prepared cDNA was amplified using the Bio-Rad iCycler iQ system (Bio-Rad Laboratories, Hercules, CA) with iQ SYBR Green Supermix®(Bio-Rad Laboratories), which included each dNTP, iTaq DNA polymerase, and SYBR Green I. The primers for Oct-4 and Nanog are described in Table 1. Cycle threshold values were normalized for amplication of glyceraldehyde-3-phosphate dehydrogenase. Data were analyzed using the PfaffI method [12].
RT-PCR for germ layer markers
We evaluated the differentiation of hESCs and mESCs with an undifferentiated morphology. For this purpose, the germ layer markers Sox-1, Brachyury, and AFP were analyzed in ESCs using RT-PCR. Total RNA was isolated using Trizol total RNA isolation reagent (Invitrogen). First-strand cDNA was synthesized as just described. Prepared cDNA was amplified with Taq polymerase (Qiagen, Hilden, Germany) using a TaKaRa PCR thermal Cycler Dice TP600® (TAKARA BIO, Shiga, Japan). The primers are described in Table 1. After amplification, products were analyzed on a 1.5% agarose gel and were visualized with ethidium bromide.
Karyotype analysis
Karyotype analysis was initially planned every 10th passage in both the control and study groups (Fig. 1). Since the colonies of hESCs and mESCs could not be maintained in the study group, karyotyping was performed only in the control group. For this analysis, ESCs were incubated with 0.1 μg/mL colcemid for 3–4 h, trypsinized, and then incubated in 0.075 M KCL for 20 min at 37°C. After fixation with 3:1 methanol/acetic acid, the karyotypes of ESCs were analyzed at the 550-band level of resolution.
Statistical analysis
Each ESC lines (H1, HSF6, D3, and CE3) for each feeder cell type in both the control and study groups consisted of 6-well culture dishes, for a total of 36 wells per group (Fig. 1). The experiments were repeated 5 times, and the values for result of each ESC line is presented as the mean of these repetitive experiments (N=30). The results were verified by 2 other specialists (Seung Jin Lee and In Young Choi). The error bars in the graphs indicate the standard deviation. Comparison of the means was performed using the Mann-Whitney U test. Results were considered significant when the P value was less than 0.05.
Results
Undifferentiated growth of hESCs was predominantly dependent on bFGF synthesized in human feeders cells
We recently identified bFGF expression in HPCs [7]. Therefore, in order to investigate the role of synthesized bFGF, 3 kinds of human feeder cells (HPC, HFF, and HBMSC) were prepared for control and study groups (Fig. 1). In the study group, the feeders were treated by bFGF-siRNA for blockage of feeder-synthesized bFGF, whereas control group feeders were treated with control bFGF siRNA. First, we investigated whether or not HFF and HBMSC expressed bFGF and determined that bFGF was well expressed in these feeders (Fig. 2A). Next, the hESCs were propagated in the control and study groups. Since there was no exogenous bFGF supplementation in the culture media of either group, hESCs in the study group were totally deprived of bFGF.
Endogenous bFGF synthesis in all feeder types was effectively blocked by bFGF-siRNA (Fig. 2A). H1 and HSF6 hESCs colonies were transferred to control group feeder cells or bFGF-knockout feeders, simultaneously. Morphologic changes in the colonies were evident in both H1 and HSF6 hESCs. Most of the hESC colonies transferred onto bFGF-knockout feeders became scattered and were dispersed after the first passage compared with those in the control group, and the ALP staining for hESC colonies was consistent with these morphologic changes (Fig. 3A). We counted the numbers of colonies during the first 5 passages and then for every fifth passage and compared the number of hESCs transferred to the study group with that transferred to the control group and found that the number of hESC colonies with undifferentiated morphology that were transferred onto the bFGF-knockout feeders decreased with increasing passages. This result was highly reproducible, and we could not find undifferentiated hESC colonies of either H1 or HSF6 on bFGF-knockout feeders beyond 10–15 passages. Indeed, the undifferentiated growth of hESC colonies was dependent on the bFGF synthesized in the human feeder cells. The mean colony counts at each passage are depicted in Fig. 4A, B.
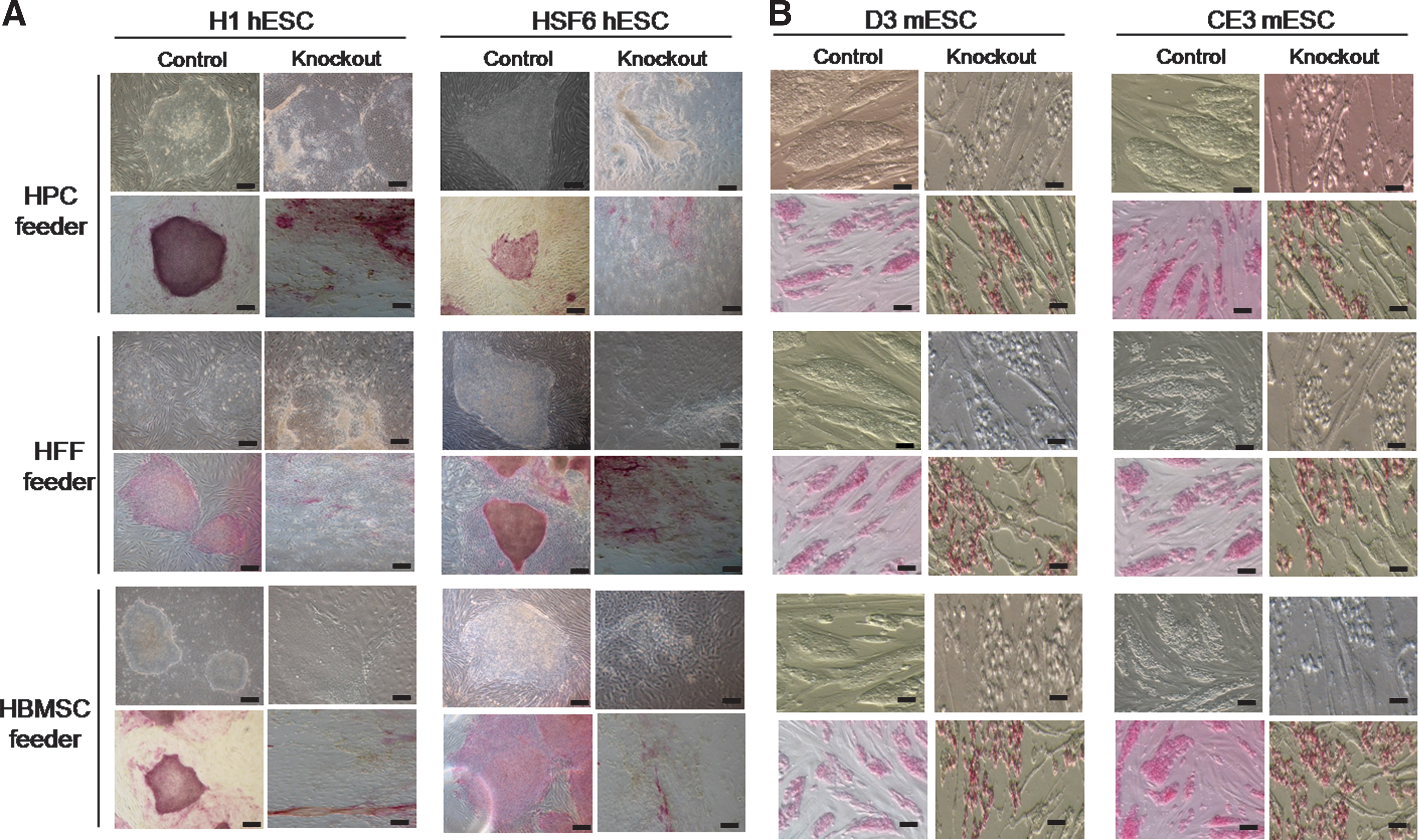
Morphologic changes in ESC colonies on bFGF-knockout feeders according to bright field microscopy.

Mean colony counts for human and mouse ESCs in both control and bFGF-knockout feeders. The results of control and study groups are depicted as solid (•, ▴, ▪) and blank (○, Δ, □) figures, respectively. The circles (•, ○), triangles (▴, Δ), and squares (▪, □) indicate HPC, HFF, and HBMSC feeder groups, respectively.
Endogenous bFGF in human feeder cells also supports the undifferentiated growth of mESCs
Since undifferentiated growth of mESCs is known to be dependent on LIF, we investigated whether human feeder cells express LIF. Our results showed that LIF was not expressed in any of the prepared human feeder cells (Fig. 2B). Therefore, we postulated that the maintenance of an undifferentiated state in mESCs might be dependent on bFGF as well as LIF, because of the following reasons: ESCs derived from larger animals than the mouse, i.e., primates and rabbit, are dependent on bFGF but not on LIF to maintain an undifferentiated state [13], and the role of bFGF in undifferentiated mESC culture has been scarcely investigated. The same protocol used in hESCs was repeated using D3 and CE3 mESCs (Fig. 1), and the results were very similar to those of hESCs: most of the mESCs colonies transferred onto any tested variety of bFGF-knockout feeder became scattered and dispersed after the first passage compared with those in the control group, and alkaline phosphatase staining for mESC colonies was also consistent with these morphologic changes (Fig. 3B). The number of mESC colonies with an undifferentiated morphology and that was transferred onto the bFGF-knockout feeders decreased with increasing passage, and we could not find any D3 and CE3 undifferentiated mESC colonies on bFGF-knockout feeders beyond 10–15 passages (Fig. 4C, D). Therefore, the undifferentiated growth of mESCs could be maintained on human feeder cells by bFGF synthesized within the human feeders cells themselves, as is true in hESCs.
bFGF-knockouts in human feeder cells have a negative impact on stemness in human and mouse ESCs
To investigate the impact of bFGF-knockouts on stemness marker expression, the degree of stemness marker (Oct-4 and Nanog) expression was compared using real-time RT-PCR analysis, which was initially planned for every 10 passages (Fig. 1). However, because since both hESC and mESC colonies on bFGF-knockout human feeder cells could not be maintained for more than 10–15 passages, this analysis was performed after 5 passages. Only colonies with undifferentiated morphology were used in this analysis. The degrees of expression of stemness markers were significantly decreased in all bFGF-knockout feeder groups compared with those in the control groups for both H1 and HSF6 hESCs (Fig. 5, P<0.0001, respectively). Because Since the long-term culture of hESCs was possible in the control group of each type of feeder, we could evaluate the expressions of stemness markers Oct-4 and Nanog in hESCs using RT-PCR and performed karyotype analysis every 10 passages. Our results showed that the expressions of stemness markers and normal karyotype were well- conserved over the passages (data not shown).
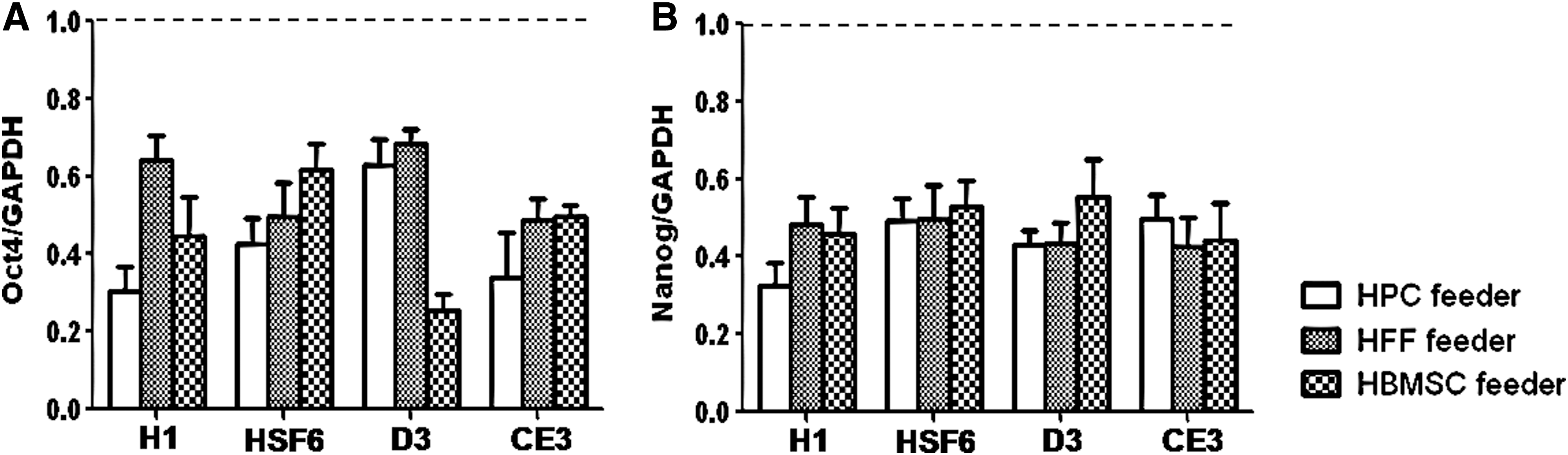
The impact of bFGF-deprivation on stemness marker expressions in human and mouse ESCs on human feeder cells analyzed using real-time RT-PCR. At each real-time RT PCR analysis, cycle threshold values were normalized relative to amplification of GAPDH. Data were analyzed using the PfaffI method. For comparison of the relative strengths of gene transcription between the control and study groups, the value of the bFGF-knockout feeder group was adjusted after setting the value of the control group to 1. The presenting data are the means of these adjusted values of the bFGF-knockout feeder group (n=30: the experiment was repeated 5 times). Error bars represent standard deviation.
Similar results were also observed in mESC cultures. The degrees of expression of stemness markers were significantly decreased in all bFGF-knockout feeder groups compared with those of the control groups in both D3 and CE3 mESCs (P<0.0001, respectively) (Fig. 5). In the control group, both stemness markers and normal karyotype were well conserved over all passages, as in the case of hESCs (data not shown). We concluded that bFGF-knockouts in human feeder cells have a negative impact on stemness in human and mouse ESCs and that transfection of control siRNA for bFGF did not influence stemness in human or mouse ESCs.
bFGF-knockouts in human feeder cells cause differentiation of human and mouse ESCs even though they are morphologically undifferentiated
We evaluated the impacts of bFGF-knockouts on the differentiation of human and mouse ESCs. To investigate whether differentiation had already occurred in the ESCs colonies with undifferentiated morphology, we initially planned RT-PCR analysis for germ layer markers such as Sox-1, Brachyury, and AFP every 10 passages in both control and bFGF-knockout groups. However, due to aforementioned issues, we could only compare the study group with the control group after 5 passages.
No expression of any germ layer marker was observed in the control feeders in either H1 or HSF6 hESCs. However, hESCs of the bFGF-knockout feeder group expressed all of these germ layer markers (Fig. 6A), indicating that differentiation had already occurred under bFGF-deprived conditions, even though the hESCs appeared undifferentiated. In D3 and CE3 mESCs, similar results were obtained. In bFGF-knockout feeders, mESCs were also already differentiated, despite their undifferentiated appearance (Fig. 6B).
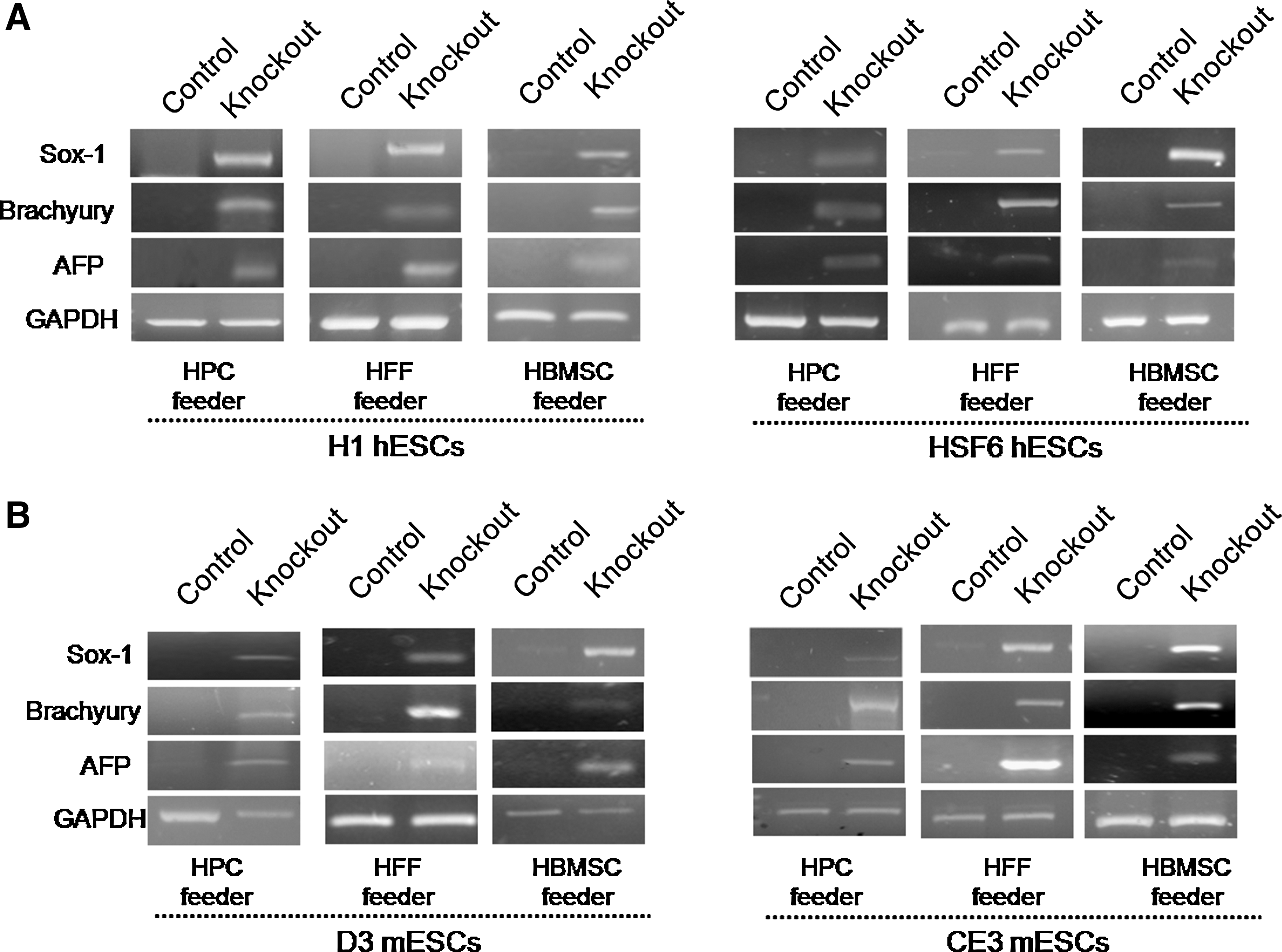
The impact of bFGF deprivation on maintenance of an undifferentiated state in human and mouse ESCs with human feeder cells.
These findings once more suggest that undifferentiated growth of hESCs on human feeder cells was dependent on bFGF synthesized within the feeder cells and that bFGF plays a crucial role in the undifferentiated growth of mESCs, similar to what we observed for hESCs.
Discussion
The present results can be summarized as follows. Undifferentiated growth of H1 and HSF6 hESCs could not be maintained in our culture conditions on the 3 kinds of human feeder cells, HPC, HFF, and HBMSC, when endogenous bFGF synthesis was blocked without an exogenous bFGF supply. Second, the expressions of stemness markers, such as Oct-4 and Nanog, in H1 and HSF6 hESCs of bFGF-knockout groups were significantly decreased compared with those in the hESCs of control groups. Further, evidence indicated that differentiation had already occurred in H1 and HSF6 hESCs of the bFGF-knockout groups, although the morphologies of the hESCs colonies appeared to be undifferentiated. Lastly, when the experiment was repeated in 2 different mESCs cell varieties, D3 and CE3, on human feeder cells, the results were reproducible.
The role of bFGF in the culture of hESCs has been well demonstrated in many studies. In a culture system using animal feeders such as MEF or under feeder-free conditions, the undifferentiated growth of hESCs is totally dependent on the exogenous supplementation of bFGF. In our culture system presented here using human feeder cells such as HPC, HFF, and HBMSC, however, we showed that the need for exogenous bFGF could be eliminated by directly supplying bFGF from the feeder cells to adjacent hESCs. This could be a unique biological feature of human feeder cells, because MEF, which is the most widely used animal feeder cell, does not express bFGF [7]. Even when supplied by human feeder cells, bFGF did not lose its key support role in the undifferentiated growth of hESCs. Undifferentiated growth of hESCs seems to always be dependent on bFGF, no matter the source. The findings that hESCs began to express stemness markers at a low level and to differentiate under bFGF-deprived conditions even though they visually appeared to be undifferentiated indicate that the impact of bFGF on the undifferentiated state of hESC is strong and decisive. Of course, it is possible that there are cytokines as effective as bFGF; however, based on the currently available data on the conditions necessary to support undifferentiated growth in hESCs, it seems unlikely that there exists an alternative bFGF-independent pathway which supports this undifferentiated growth.
It is well known that undifferentiated growth of mESCs is mainly dependent on the presence of LIF [14,15]. In the present study, mESCs were found to be well maintained and propagated in an undifferentiated state on human feeder cells when no exogenous cytokines including LIF were supplemented. Human feeder cells do not express LIF, indicating that the undifferentiated growth of mESCs on human feeder cells was not dependent on LIF. Instead, it was dependent on the bFGF synthesized within the human feeder cells themselves. These data suggest that there is an LIF-independent, bFGF-dependent pathway supporting the undifferentiated growth of mESCs. Although the signaling pathway of LIF is not yet clearly understood, some pathways have been identified. LIF could activate the Janus kinase-signal transducer and activator (JAK/STAT) pathway [16] and/or the PI3-knase/Akt pathway [17] in mESCs. PI3K/Akt signaling is noteworthy in association with bFGF, because PI3/Akt signaling is known to be activated by exogenous bFGF in hESCs culture, which is crucial for maintaining pluripotency [18]. Therefore, a possible mechanism for supporting undifferentiated mESCs growth on human feeder cells might be the activation of the PI3K/Akt pathway by bFGF synthesized within these feeder cells. However, this needs to be confirmed in further studies. To the best of our knowledge, studies on the role of bFGF in undifferentiated growth of mESCs culture have been very rare.
bFGF is known to be an essential cytokine in vitro cultures of large mammalian ESCs such as rabbit and primate (monkey and human), and LIF and the activation of the downstream pathway JAK/STAT3 are dispensable for maintaining ESCs in an undifferentiated state [13,19,20]. Our present data indicated that bFGF supported undifferentiated mESCs proliferation. Thus, we suggest the possibility that bFGF might be a prime regulator of undifferentiated growth in mammalian ESCs.
Exogenous bFGF in the culture media has been shown to have bidirectional impacts on the proliferation of stem cells. In pluripotent stem cells such as ESCs, bFGF exerts antidifferentiating activity on cultured ESCs. However, in multipotent stem cells such as lineage-specific stem cells, the exogenous bFGF drives stem cells into differentiation. In particular, adding bFGF to the culture media of this stage of stem cells has been shown to produce a neural inductive activity [21]. The blockage of bFGF activity in mESCs cultured for neural differentiation was reported to inhibit the neural differentiation [22]. Since the microenvironment of feeder-synthesized bFGF is different from that of exogenously supplied bFGF, we could not initially be sure that bFGF synthesized within feeder cells was responsible for the antidifferentiation activity in the ESCs. Nevertheless, the overall results of this study suggest that bFGF synthesized in feeder cells during mouse and human ESC culture has antidifferentiation activity for ESCs, similar to that of exogenously supplied bFGF; blockage of feeder-synthesized bFGF induced differentiation in all germ-layers without the propensity for a specific lineage.
In conclusion, human feeder cells (HPC, HFF, and HBMSC) support undifferentiated growth of hESCs and mESCs via a bFGF-dependent mechanism, based on bFGF produced by the feeder cells. bFGF might be crucial for maintaining the characteristics of mammalian ESCs. There have been many studies regarding feeder-free culturing of ESCs; however, the efficiency and cost of a feeder-free culture are not satisfactory. ESC culture systems should have both high cloning efficiency and safety to ensure that the derivatives of hESCs can be used for clinical purposes. Considering the possible clinical application, our current culture system using human feeder cells appears to be the best of the current hESC culture methods.
Our present and earlier results [3,7,8] suggest that bFGF-dependent support of human feeder cells for undifferentiated growth of hESCs as well as mESCs seems to be a unique activity of human feeder cells. To the best of our knowledge, this is the first report on the mechanism of human feeder cells. However, in order to generally apply our results, there are some limitations to be solved. First, since our results are the first observations, our data should be validated by other investigators. Second, the more long-term impact of exogenous bFGF deprivation on ESCs cocultured with human feeder cells should be investigated. Third, the role of endogenous bFGF in other types of human feeder cells should be elucidated.
Footnotes
Acknowledgments
This research was supported by a grant (SC-2240) from the Stem Cell Research Center of the 21st Century Frontier Research Program, funded by the Ministry of Science and Technology, Republic of Korea.
Author Disclosure Statement
No authors indicated a potential conflict of interest, and no competing financial interests exist in connection with this manuscript.