
Abstract
The purpose of this study was to test the hypothesis that the SOX trio genes (SOX-5, SOX-6, and SOX-9) have a lower level of expression during the chondrogenic differentiation of mesenchymal stem cells (MSCs) compared with chondrocytes and that the electroporation-mediated gene transfer of SOX trio promotes chondrogenesis from human MSCs. An in vitro pellet culture was carried out using MSCs or chondrocytes at passage 3 and analyzed after 7 and 21 days. Then, MSCs were transfected with SOX trio genes and analyzed for the expression of chondrogenic markers after 21 days of in vitro culture. Without transforming growth factor-β1, the untransfected MSCs had a lower level of SOX trio gene and protein expression than chondrocytes. However, the level of SOX-9 gene expression increased in MSCs when treated with transforming growth factor-β1. GAG level significantly increased 7-fold in MSCs co-transfected with SOX trio, which was corroborated by Safranin-O staining. SOX trio co-transfection significantly increased COL2A1 gene and protein and decreased COL10A1 protein in MSCs. It is concluded that the SOX trio have a significantly lower expression in human MSCs than in chondrocytes and that the electroporation-mediated co-transfection of SOX trio enhances chondrogenesis and suppresses hypertrophy of human MSCs.
Introduction
T
Mesenchymal stem cells (MSCs) from adults are capable of self-regeneration and differentiation into a range of cell types [6,7]. Several studies have examined the potential of MSCs as an alternative to chondrocytes for cartilage repair [8,9]. Since MSCs are undifferentiated cells, they need to undergo chondrogenic differentiation when intended for use in cartilage tissue engineering. However, the quality of engineered cartilaginous tissues from MSCs has not outperformed those from chondrocytes [10]. Therefore, to achieve chondrogenesis of acceptable quality from MSCs, our understanding on the differentiation of MSCs should be expanded.
Several lines of evidence point toward the importance of SOX transcription factors for the maintenance of chondrocytic phenotypes [11,12]. These factors belong to a family of regulatory molecules related to the sex-determining factor SRY (sex-determining region Y; 15). SOX-9 is expressed in all chondrogenic cells, except hypertrophic chondrocytes [12], binding to and activating chondrocyte-specific enhancer-elements in COL2A1, COL9A1, COL11A2, and aggrecan in vitro [11 –13]. Two other members of the SOX family (SOX-5 and SOX-6) are needed for perfect chondrogenesis [14,15]. These factors are also co-expressed with SOX-9 in all chondroprogenitors and differentiated chondrocytes and cooperate with SOX-9 to activate the COL2A1 gene [12]. SOX transcription factors also play an important role during mesenchymal condensation and chondrogenesis in the developmental processes [12]. Considering that a large number of embryogenic molecular events are recapitulated in the in vitro chondrogenesis from MSCs, SOX transcriptional factors merit attention as possible candidates for gene transfer to enhance chondrogenesis from human MSCs. If genetically modified MSCs that have enhanced chondrogenic potential comparable to chondrocytes are safely provided, they will hopefully replace autologous chondrocyte as the cell source for cartilage tissue engineering. Thus, the purpose of this study was to test the hypothesis that the SOX trio (SOX-5, SOX-6, and SOX-9) have a lower level of expression during the chondrogenic differentiation of human MSCs compared with chondrocytes and that the electroporation-mediated gene transfer of these transcription factors promotes chondrogenesis of human MSCs.
Materials and Methods
Procurement of samples, cell isolation, and cultivation
The bone marrow samples used to isolate the MSCs were obtained from 3 patients (mean age, 75 years; range, 68–78 years) undergoing hemiarthroplasty from femoral neck fractures. Chondrocytes were obtained from 3 patients (mean age, 72 years; range, 68–75 years) undergoing total knee arthroplasties. Minimally osteoarthritic areas (Mankin score, 0–3) in the articular cartilage of the lateral femoral condyles were used to obtain chondrocytes. This study was approved by the Institutional Review Board at our university, and informed consent was obtained from all of the individuals included in the study. MSCs were isolated from fresh bone marrow samples and expanded, as described in previous studies [8,16]. For chondrocyte cultures, fragments of articular cartilage were chopped with a scalpel, washed twice with phosphate-buffered saline (PBS), and digested briefly with 0.25% trypsin-EDTA (Gibco BRL) for 15 min at 37°C, then incubated for 16–18 h with 0.1% collagenase in F-12/DMEM at 37°C with shaking. Tissue debris was removed by filtration through a 100-μm cell strainer (BD Bioscience), and the cells were collected by centrifugation at 500×g for 5 min. Isolated chondrocytes were cultured and expanded in a monolayer in DMEM/F-12 containing 10% FBS and 1% antibiotics at 37°C in a humidified atmosphere with 5% CO2. The viability of the MSCs and chondrocytes were always >95%.
In vitro pellet culture
For the induction of chondrogenesis, the in vitro pellet culture was carried out using 2.5×105 MSCs or chondrocytes at passage 3 in chondrogenic medium (CM; DMEM/F-12 supplemented with 1% insulin-transferrin-selenium, 10–7 M L-dexamethasone, 50 μM L-ascorbate-2-phosphate, 50 μM L-proline, and 1 mM sodium pyruvate). The MSC pellets were cultured either with or without 10 ng/mL of transforming growth factor (TGF)-β1 (R&D Systems Inc.).
For a pellet culture, 1 mL of the cell suspension was aliquoted into 15-mL polypropylene centrifuge tubes and spun in a benchtop centrifuge (Hanil) at 500 ×g for 5 min. The tubes were placed in an incubator at 37°C in a humidified atmosphere containing 95% air and 5% CO2 for <21 days. The caps of the tubes were loosened to allow air exchange. The medium was changed every 3rd day. Pellets were taken for analysis at 7 and 21 days after culture.
Reverse transcription and real-time polymerase chain reaction analysis
RNA was isolated using the standard guanidine isothiocyanate Tri-Reagent® (Sigma Chemical) according to the manufacturer's recommendations. The isolated RNA samples were converted to cDNA using reverse transcriptase (SuperScript III®; Invitrogen) and oligo (dT) primers. All of the polymerase chain reaction (PCR) were performed on the LightCycler 480 system® (Roche Diagnostics) in standard 20 μL reactions. The reactions were performed for SOX-5, SOX-6, SOX-9, type I collagen (COL1A1), type II collagen (COL2A1), type X collagen (COL10A1), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a housekeeping gene. The primers and reaction conditions used for amplification are listed in Table 1. After polymerase activation (95°C for 10 min), 45 cycles were run with 10 s denaturation at 95°C, 10 s annealing at 65°C, and 10 s extension at 72°C. Melt-curve analysis was performed immediately after the amplification protocol under the following conditions: 5 s at 95°C (hold time on reaching temperature), 1 min at 65°C, and 1 s at 97°C. The temperature change rates were 20°C per sec (except for the final step, which had a temperature change rate of 0.1°C per sec). The peak melting temperature obtained represented the specific amplified product. To guarantee the reliability of the results, all samples were processed in triplicate. The test was considered positive if the signal from the amplified product was clearly visible in both samples. Each assay was performed using the positive and negative controls. The data were standardized to the housekeeping GAPDH values for all samples using the delta Ct method (User bulletin #2 Applied Biosystems; Roche Molecular System). Seven pellets were used for 7 genes per donor.
Histology and immunohistochemistry
Pellets were fixed and prepared for tissue section using a routine method. For Safranin-O staining, the sections were deparaffinized with xylene and ethanol, treated with aqueous Safranin-O (0.1%) for 30 min, and washed with distilled water. Immunohistochemistry for SOX-5, SOX-6, and SOX-9 was performed using a Dakocytomation LSAB2 System HRP kit (DAKO). Primary antibodies were rabbit SOX-5 polyclonal antibody (Abcam), rabbit SOX-6 polyclonal antibody (Abcam), rabbit SOX-9 polyclonal antibody (Santa Cruz Biotechnology, Inc.), mouse collagen type I polyclonal antibody (Millipore), rabbit collagen type II monoclonal antibody (Millipore), mouse collagen type X monoclonal antibody (Sigma-Aldrich), and mouse Ki-67 monoclonal antibody (Santa Cruz Biotechnology, Inc.) diluted 1:250, 1:2,000, 1:500, 1:200, 1:200, 1:200, and 1:200 in goat serum, respectively. Primary antibodies were reacted overnight at 4°C. After 3 washes in 1× wash buffer (0.05% Tween 20 in PBS: DAKO), the sections were incubated with horseradish peroxidase-labeled goat anti-mouse secondary antibodies (DAKO) for 30 min. After extensive washing, the sections were reacted with a substrate buffer and diaminobenzidine chromogen (50:1; DAKO) for 5 min and mounted. A semi-quantitative assessment of histological grading was performed based on the scale of intensity, which was precisely defined as high (3), moderate (2), low (1), faint, or no reaction (0). Negative controls were specimens on which all the processes just described were performed except for the application of the primary antibody. The score was obtained from each sample from the agreements of 3 microscopists who were blinded to the samples.
Construction of plasmid for the transfection of SOX genes
The coding regions of human SOX-5, SOX-6, and SOX-9 from NIH Mammalian Gene Collection cDNA Clones (Invitrogen Corporation) were amplified by PCR and cloned into the pECFP-C1 expression vectors (Fig. 1A). To increase the cloning efficiency, the 5′ end was made sticky, and the 3′ was made end blunt. After PCR, the insert was prepared using the enzymes, SmaI and BglII (Takara Bio Inc.). SmaI was applied to digest the 3′ end, followed by T7 polymerase (Takara Bio Inc.), which induced the blunt end. BglII was used to produce a sticky 5′ end. For the vector, SmaI and BglII were used for the same purpose. The prepared vector and insert underwent ligation using Ligation Mix (Takara Bio Inc.) and underwent a transformation to obtain their respective clones. PCR products were verified by DNA sequencing (Bioneer).
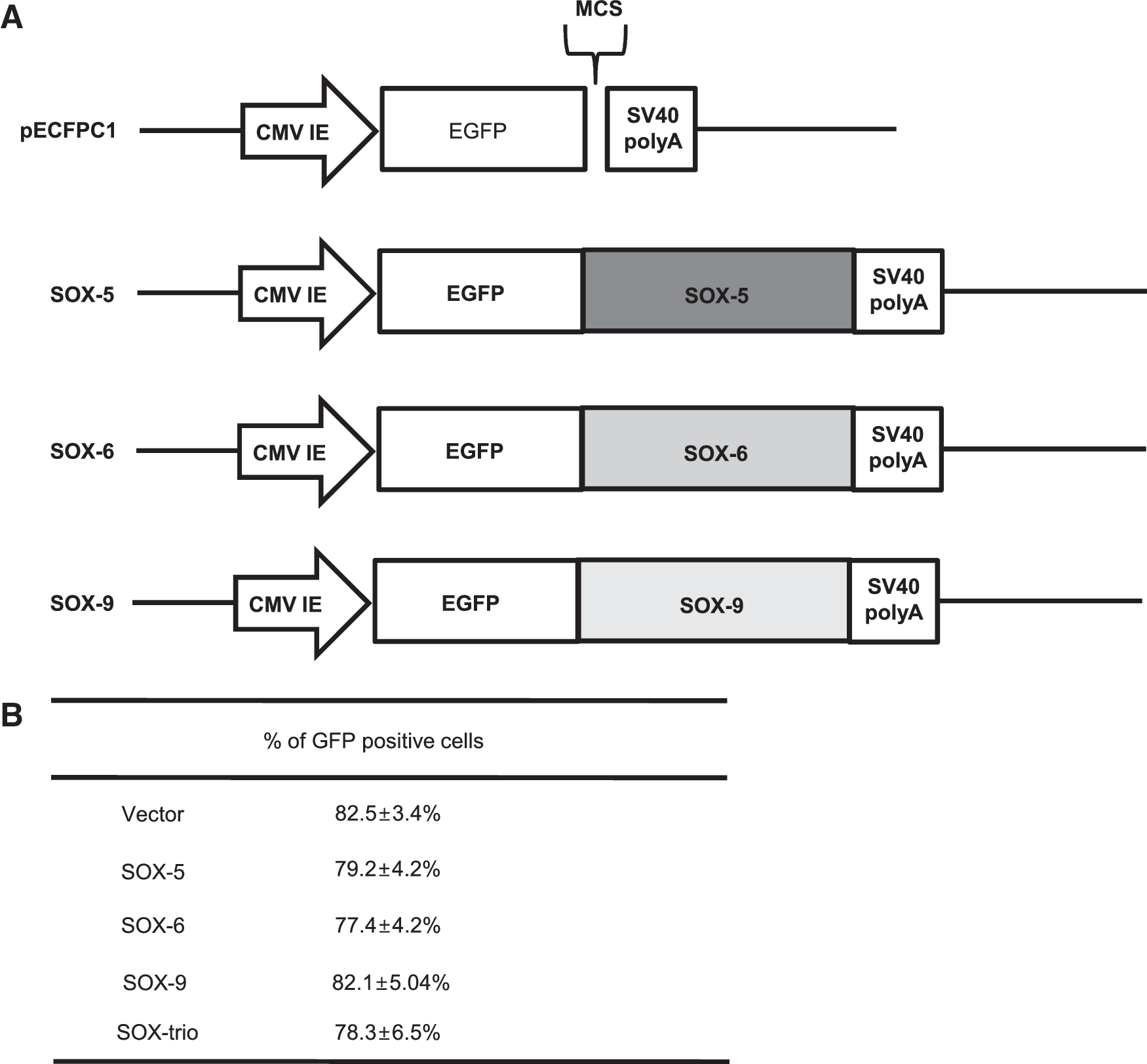
Construct of the plasmid for SOX gene transfer
Transfection of MSCs by electroporation
Subconfluent MSCs were harvested and resuspended in a buffer provided by the manufacturer at a density of 3×105 cells/mL and mixed with 0.5 μg of the constructed plasmids. Then, electroporation was performed with the Microporator (Invitrogen) using the programs recommended by the manufacturer, as follows: 1,400 voltage; 20 ms; and 2 pulses. After electroporation, cells were plated on a 12-well plate and placed at 37°C in 5% CO2. Five subsets were prepared, as follows: MSCs transfected with pEGFP-C1 without interposed genes of transcription factors (negative control); MSCs transfected with pEGFP-C1 interposed with SOX-5; MSCs transfected with pEGFP-C1 interposed with SOX-6; MSCs transfected with pEGFP-C1 interposed with SOX-9; and MSCs transfected with one-third doses (0.17 μg) of each plasmid (SOX-5, SOX-6, SOX-9). Positive controls were MSCs that underwent chondrogenic induction with CM containing 5 ng/mL of TGF-ß2, as based on our previous study [17] and chondrocytes which were cultured in the CM without TGF-ß2. Transfected MSCs were cultured in the pellets in the same way as described in the previous section in CM without growth factors. The pellets were analyzed after 21 days of culture.
Confirmation of transfection efficiency using flow cytometry
After performing gene transfer using the microporator, the transfection efficiency of each gene was confirmed using flow cytometry (Beckman Coulter, Inc.) 24 h after transfection. For flow cytometry, the cells underwent washing thrice and were suspended in a media consisting of Hank's buffered salt solution (Welgene) with 2% FBS (Invitrogen/GIBCO). Thereafter, the cells were analyzed using CXP software (Beckman Coulter, Inc). MSCs that did not undergo gene transfer were used as negative controls. The gene transfection efficiency was high for all subsets: ∼80% of cells were transfected (Fig. 1B).
Confirmation of GFP expression from in vitro cultured pellets
After 7, 14, and 21 days of in vitro culture, the pellets were embedded in an optimal cutting temperature aqueous embedding medium (Tissue-Tek®; Sakura) within a mold, frozen in a metal pan over a bath of liquid nitrogen, and stored at –70°C until used for cryomicrotomy. All frozen tissue blocks were cryo-sectioned to a thickness of 5 μm. Lastly, sections were mounted using a DAPI mounting solution (Vector Laboratories). GFP expression was confirmed using fluorescence microscopy.
DNA quantification and GAG analysis
The pellets of each group were digested overnight in papain buffer at 60°C. DNA content was determined using a Quant-I T™ dsDNA assay kit and the Qubit Fluorometer system (Invitrogen). GAG production was determined using a Blyscan kit (Biocolor), according to the manufacturer's instructions. GAG content was determined using a standard curve drawn using standard solutions containing chondroitin 4-sulfate from bovine trachea. Absorbance was measured at 656 nm on the Spectra max plus 384 (Molecular Devices). GAG levels were expressed as μg of GAG per μg of DNA.
Characterization of apoptosis
To determine the degree of apoptosis in the MSCs cultured in pellets, the cleavage of genomic DNA during apoptosis was assessed using the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) method [18] according to the protocol provided by the manufacturer (DeadEnd™ Colorimetric Apoptosis DetectionSystem: Promega). Total cell counts and cell morphology were determined by optical microscopy, and only those cells exhibiting both TUNEL-positive staining and the characteristic pattern of apoptosis (blebbing of plasma membrane and nuclear condensation) were counted as apoptotic. The apoptotic rate was defined as the percentage of apoptotic cells (TUNEL-positive cells with apoptotic morphology) to the total cells counted. Five spots were investigated from the pellets and counted. The tissue sections stained in the absence of terminal deoxynucleotidyl transferase were employed as a negative control for nonspecific TUNEL labeling.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis and Western blotting
Protein extracts were analyzed by Western blotting for SOX-5, SOX-6, SOX-9, type I collagen, type II collagen, type X collagen, Runx-2, and osterix after 21 days of in vitro culture. Briefly, cells were washed twice with cold PBS and suspended in 50 μL of RIPA lysis solution (Pierce) according to the manufacturer's instruction. Protein concentrations were determined using a Qubit assay kit (Qubitfluorometer; Invitrogen), and equal amounts of protein extracts were fractionated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (Bio-Rad) and transferred onto a BioTrace™ NT nitrocellulose transfer membrane. After blocking with TBS-T (10 mM Tris, 150 mM NaCl, and 0.05% Tween-20) containing 3% non-fat powdered milk (Bio-rad Laboratories, Inc.), the blots were incubated with the specific primary antibody for 2 h at room temperature, washed, incubated with secondary antibody for 1 h at room temperature, and washed again. The blots were developed using the SuperSignal® West Pico Chemiluminescent Substrate (Pierce) following the manufacturer's protocols. After 3 washes, the protein bands were visualized with an enhanced chemiluminesence Western blot analysis system (Amersham Biosciences). Primary antibodies were rabbit SOX-5 polyclonal antibody (Santa Cruz Biotechnology, Inc.), rabbit SOX-6 polyclonal antibody (Santa Cruz Biotechnology, Inc.), rabbit SOX-9 polyclonal antibody (Santa Cruz Biotechnology, Inc.), mouse collagen type I polyclonal antibody (Millipore), rabbit collagen type II monoclonal antibody (Millipore), mouse collagen type X monoclonal antibody (Sigma-Aldrich), goat Runx-2 polyclonal antibody (Santa Cruz Biotechnology, Inc.), and rabbit Osterix polyclonal antibody (Santa Cruz Biotechnology, Inc.) diluted 1:200 in TBS-T/2.5% non-fat dry milk. Secondary antibody was horseradish peroxidase-conjugated goat anti-rabbit, goat anti-mouse, and rabbit anti-goat IgG antibody (Santa Cruz Biotechnology, Inc.) diluted 1:2,000 in TBS-T/2.5% powdered non-fat dry milk. This experiment was repeated thrice per sample, and the samples were from 3 different people.
Statistical analysis
Descriptive statistics were used to determine the group means and standard deviations in the numerical data. Statistical analysis was performed using analysis of variance, followed by Bonferroni's correction for multiple comparisons. The level of significance was set at a P<0.05.
Results
SOX-5, SOX-6, and SOX-9 gene and protein expression from MSCs and chondrocytes at passage 3
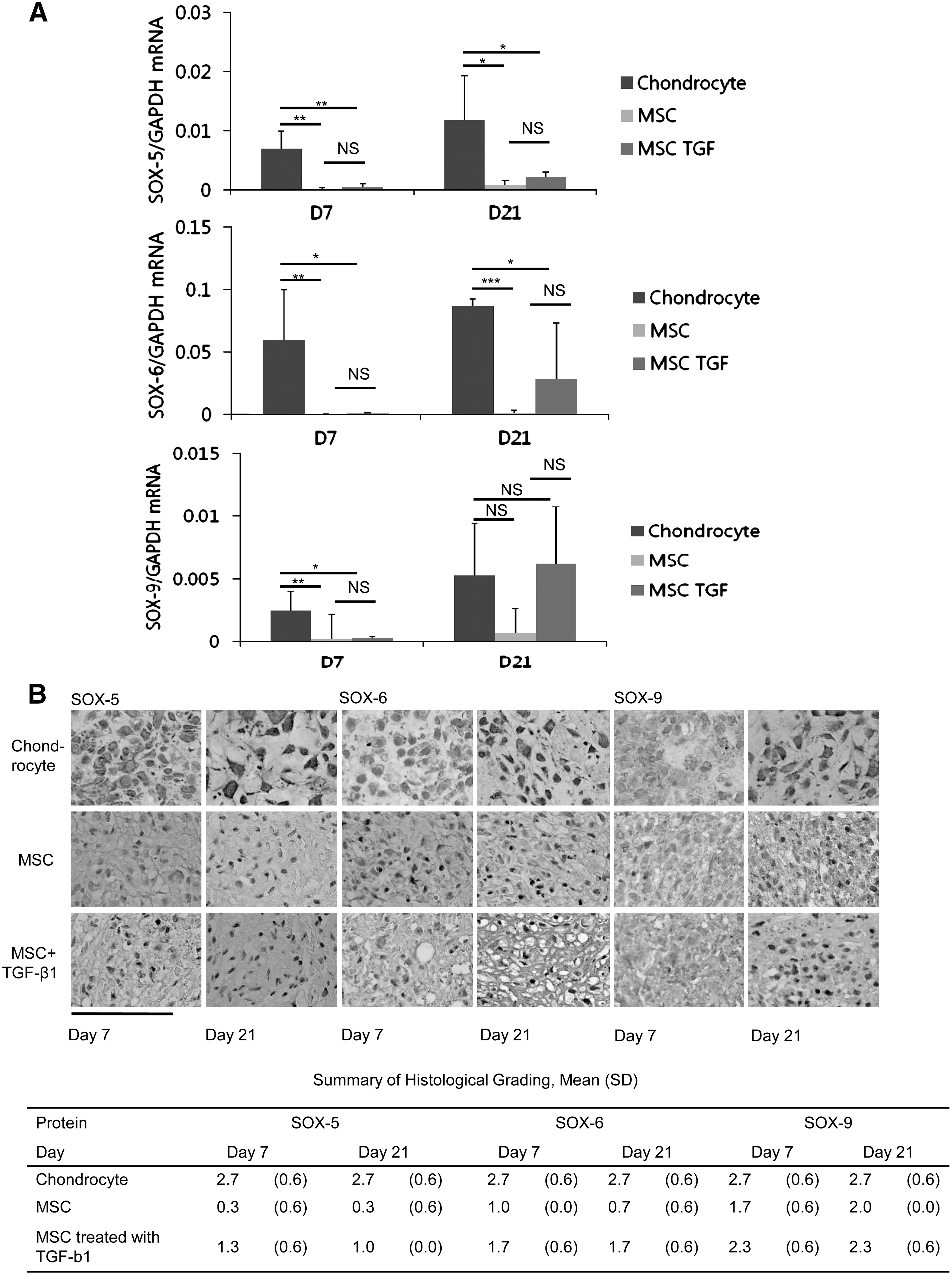
To examine the expression of the SOX trio gene and protein during the in vitro chondrogenesis from MSCs, MSCs were cultured either with or without TGF-β1 treatment and compared with the chondrocytes cultured without TGF-β1.
SOX-5 gene expression was much lower in the untreated MSCs than in the chondrocytes on days 7 (P<0.01) and 21 (P<0.05). The addition of TGF-β1 increased the level of the SOX-5 gene expression in MSCs but not to a level comparable to that in chondrocytes (P<0.05). The level of the SOX-6 gene expression was also much lower in the untreated MSCs than in chondrocytes on days 7 (P<0.01) and 21 (P<0.001). The addition of TGF-β1 increased the expression of the SOX-6 gene in MSCs on day 21, but the level of expression was still significantly lower than in chondrocytes (P<0.05). The level of the SOX-9 gene expression was significantly lower in MSCs than in chondrocytes on day 7, either in the presence (P<0.05) or absence (P<0.01) of TGF-β1. However, on day 21, the addition of TGF-β1 notably increased the level of the SOX-9 gene expression to a level comparable to that in chondrocytes (Fig. 2A).
Results of real-time polymerase chain reaction
The findings based on immunohistochemistry showed that SOX trio had robust expression from chondrocytes. SOX-5 and SOX-6 was faintly expressed in MSCs at the early (day 7) and later time periods (day 21). The presence of TGF-β1 elevated the expression of SOX-5 and SOX-6 protein in MSCs, but the expression was still much weaker than that in chondrocytes. SOX-9 expression was evident in MSCs, although weaker than in chondrocytes. SOX-9 expression was increased with TGF-β1 treatment (Fig. 2B).
Expression of GFP and SOX trio in MSCs transfected with pECFP-C1-SOX genes
The transfected MSCs were cultured in pellets and analyzed by fluorescent microscopy after 7, 14, and 21 days to examine the expression of GFP. Pellets from all transfected groups expressed green fluorescence throughout the periods, whereas untransfected MSCs (TGF-β2–treated groups) did not (Fig. 3).
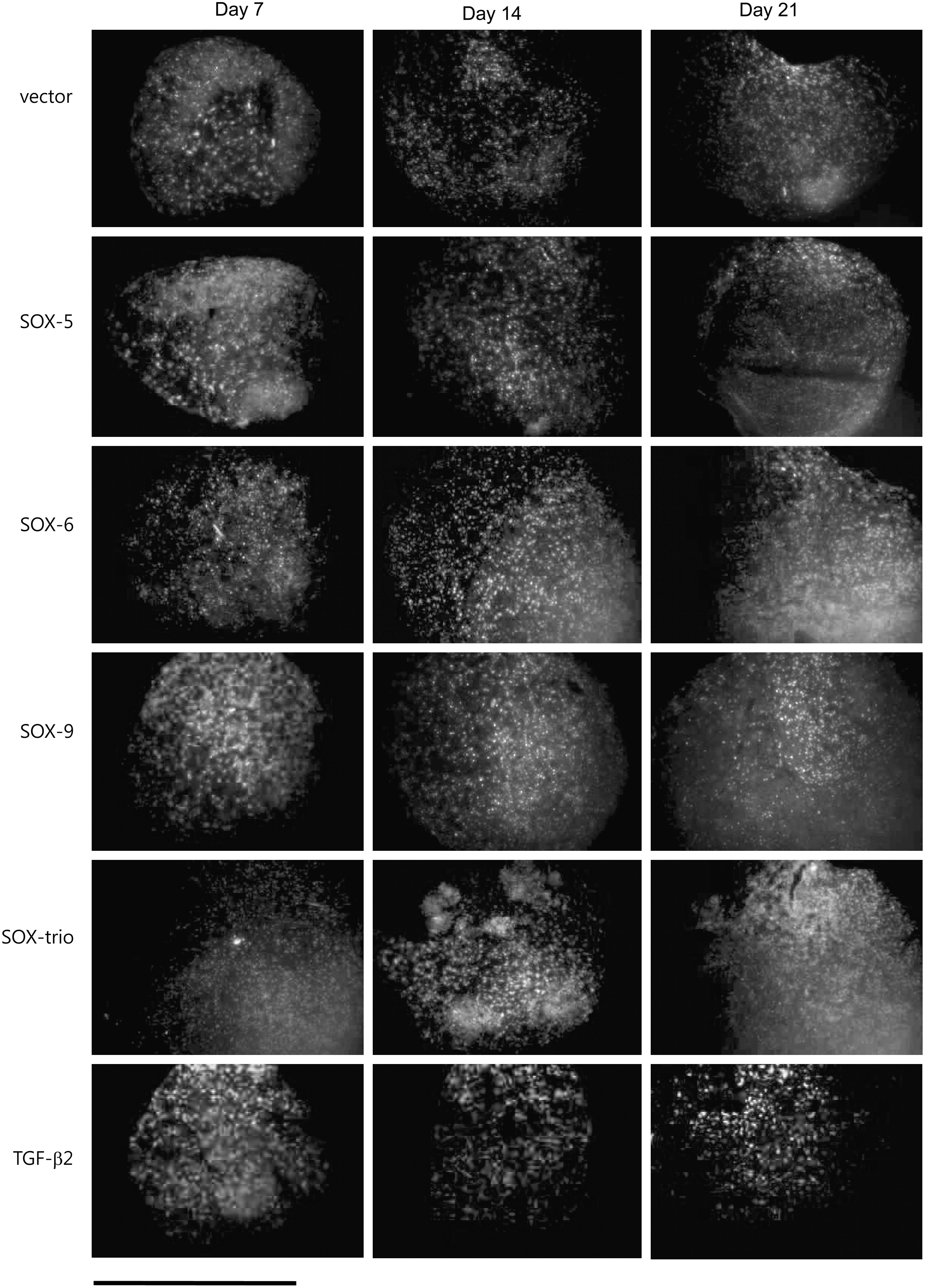
Expression of GFP (bright) in fluorescent microscopy in MSCs transfected with pEGFP-C1 without interposed gene (vector), SOX-5, SOX-6, and SOX-9, all 3 genes (SOX-trio), and also in untransfected MSCs treated with TGF-β2 (TGF-β2) after 7, 14, and 21 days of in vitro culture. The images were merged with DAPI staining, which indicates the nucleus. Scale bar is equal to 2 mm.
The transfected MSCs were also analyzed for the gene and protein expression of SOX trio after 21 days of pellet culture. Even after 21 days, the MSCs transfected with a single gene of SOX-5, SOX-6, or SOX-9 had a100–500-times greater gene expression of the corresponding genes (P<0.01). When the SOX trio were co-transfected, each gene was expressed in a greater amount than the control (P<0.01), although the level of expression was lower than in single gene transfer (Fig. 4A). Western blotting also demonstrated the strong protein expression of transfected gene in single gene transfer and moderate expression of SOX trio proteins in SOX trio co-transfection (Fig. 4B). The findings from immunohistochemistry generally mirrored the findings from Western blotting, although SOX-9 could also be detected to a degree in vector-transfected, SOX-6- transfected, and TGF-β2-treated MSCs (Fig. 4C). These findings together evidenced the effective transcription and translation of transfected genes in the transfected MSCs.

Gene expression by real-time polymerase chain reaction
The effect of SOX gene transfection on the cell number, proliferation, apoptosis, and GAG synthesis
Transfected MSCs were cultured in pellets for 21 days and analyzed. The amount of DNA and GAG was measured to see whether there was a change in the cell number and the production of chondroid extracellular matrix. There was no significant difference in the DNA content among the subsets of MSCs, whereas chondrocytes had significant lower DNA contents than the control MSCs (Fig. 5A). These results showed that transfection of SOX genes did not have a significant effect on cell number. The level of GAG normalized to DNA significantly increased (7-fold) when all 3 SOX trio genes were co-transfected, which was greater than those from the MSCs treated with growth factor or chondrocytes. Subsets with a single gene transfer of SOX-9 also had a significantly increased level of GAG compared with the control (P<0.05) (Fig. 5B). Safranin-O staining, which detects proteoglycan synthesis, showed the strongest metachromasia with co-transfection of the SOX trio, corroborating the results of GAG analysis (Fig. 5C). A high-magnification view from HE staining demonstrated a more differentiated morphology of the chondrogenic cells with abundant extracelluar matrix in SOX-9 transfected or SOX-trio co-transfected MSCs compared with other groups (Fig. 5D). The cell proliferation was also measured by counting the number of cells positive for Ki-67 antigen, which is a cellular marker for proliferation [19]. Apoptosis was measured by counting the number of TUNEL-positive cells. There was no significant difference in the percentage of Ki-67 positive cells (Fig. 5E) or TUNEL positive cells (Fig. 5F) among all groups.
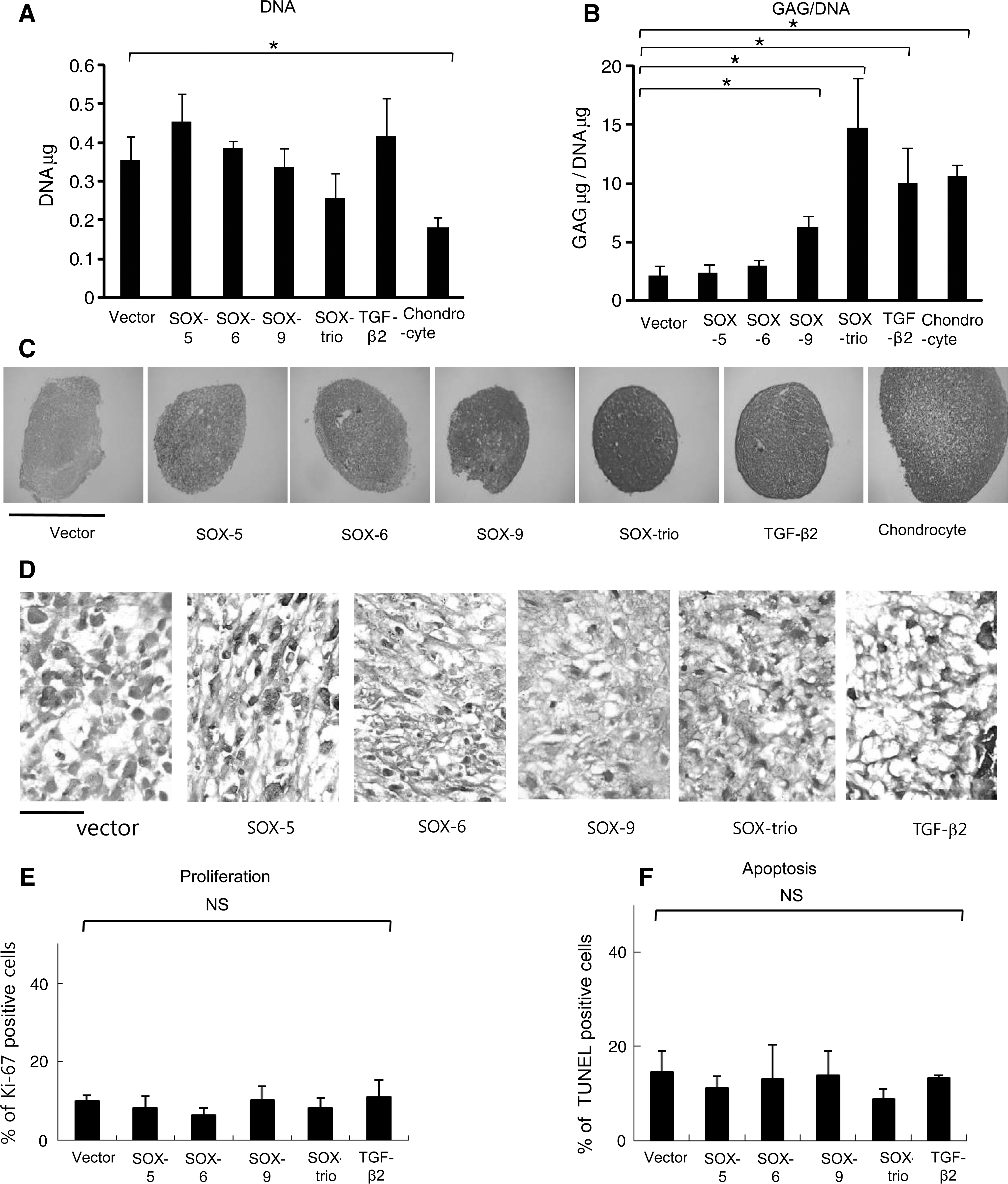
DNA levels
Enhancement of chondrogenic markers in MSCs transfected with SOX genes
Transfected MSCs were cultured in pellets for 21 days and analyzed for the markers of chondrogenic differentiation. The gene expression of COL2A1, a chondrogenic marker, significantly increased (6-fold) when all 3 SOX genes were co-transfected (P<0.05), similar to the MSCs treated with growth factors or chondrocytes (Fig. 6A). COL1A1, which signifies inadequate differentiation, or COL10A1, which indicates hypertrophy, did not show a significant increase with single gene transfer or co-transfection of the SOX trio, whereas both genes markedly increased with TGF-β2 treatment (Fig. 6B, C).

Gene expression of COL2A1
Western blotting demonstrated that type II collagen dramatically increased with the co-transfection of the SOX trio, which was greater than that induced with TGF-β2 treatment. Type I collagen did not notably increase as a result of single or co-transfection of SOX genes, whereas it greatly increased with TGF-β2 treatment. Type X collagen markedly decreased with co-transfection of SOX trio. Runx-2, the master transcription factor of osteogenic differentiation and an additional marker of hypertrophy [20], also decreased in the SOX-trio transfected MSCs. Osterix, a marker of osteogenic differentiation downstream of Runx-2 [21], decreased in SOX-9 transfected, SOX-trio co-transfected, and TGF-β2-treated MSCs (Fig. 4D).
The findings from immunochemistry generally, if not completely, paralleled the results from Western blotting. The SOX-trio co-transfected MSCs had a high level of type II collagen expression in the cell and matrix, whereas type I and X collagen expression was definitely lower than other groups (Fig. 5E). These results suggested that SOX-trio co-transfection enhanced chondrogenesis while suppressing hypertrophy of MSCs.
Discussion
Although MSCs are known to differentiate into a whole lineage of musculoskeletal tissues, individual cells have different abilities to differentiate into chondrocytes [22]. Since the SOX trio were sufficient to induce in vitro neochondrogenesis from fibroblasts when expressed together [23], it was reasoned that if these genes had a lower level of expressions in MSCs than in chondrocytes, the transfection of these genes would overcome the intrinsic limitations of MSCs and enhance chondrogenesis from MSCs.
Although previous studies have used viral gene delivery methods [23 –25], these methods are associated with the risk of immunologic reactions and mutagenesis [26,27] and are, therefore, not indicated for the treatment of nonlethal disease. Therefore, nonviral methods should be used if considered for cartilage tissue engineering. We used a microporation method, which has a capillary tip and pipette-based gene transfer technique [28]. This technique circumvents a number of harmful effects of a cuvette-based electroporation method and provides an easier and more efficient method for hard-to-transfect cells [29]. Our results also demonstrated high transfection efficiency and persistent expression of GFP and SOX proteins until 21 days after transfection.
Transfection of all SOX trio genes was needed to enhance chondrogenesis from MSCs, whereas the individual SOX genes were not effective. Even when SOX-9 was transfected, the evidence of chondrogenesis as shown by the expression of type II collagen and proteoglycan synthesis was much less than when the SOX trio were co-transfected. Our results are generally in agreement with the results from Ikeda et al. [23], who reported that the SOX trio were needed to induce chondrogenic differentiation from embryonic stem cells and fibroblasts. Although the retroviral transfection of SOX-9 enhanced the re-expression of the chondrocytic phenotype in passaged chondrocytes and adeno-associated virus-mediated over-expression of SOX-9 restored the extracellular matrix in human osteoarthritic articular cartilage [24,25], our results indicated that co-transfection of the SOX trio gene is required to induce significant chondrogenic phenotypes from MSCs. This can be explained by the intrinsically low expression of SOX-5 and SOX-6 in MSCs compared with chondrocytes. These overall results suggest that a low level of SOX-5 and SOX-6 is also important in explaining the inferior chondrogenic capacity of MSCs and that these genes also constitute key targets for gene transfer to induce successful chondrogensis from MSCs. It is also of note that co-transfection of the SOX trio was not associated with an increase of type I or type X collagen as observed in MSCs treated with TGF-β. Rather, it induced a decrease in type X collagen, indicating the suppression of hypertrophy.
Although nonviral gene transfers are safe methods for patients, low transfection efficiency poses a barrier for clinical application. Our data, however, demonstrated a remarkably high transfection efficiency. This high efficiency raises the potential for successes when used for tissue engineering applications. The half life of a transgene is known to be brief in electroporation. In addition, translated protein is supposed to be rapidly destroyed, because the SOX trio are transcription factors. Nevertheless, a greater amount of proteoglycan synthesis and a still higher expression of the chondrogenic gene 21 days after transfection show the effectiveness of the SOX trio gene transfection. Although in vivo testing is necessary to determine whether the positive in vitro findings of this study could be translated into a good clinical outcome, our results shed light on the possible use of MSCs with enhanced chondrogeneic potential to replace the current use of chondrocytes.
There are limitations in this study. Since MSCs and chondrocytes were obtained from only 3 older patients, it would be difficult to confidently generalize that the same result could be obtained in samples from younger individuals. In addition, although electroporation is much safer than viral approaches, plasmid DNA also can also be inflammatory to the transduced cells and the phenomenon of insertional mutagenesis cannot be completely excluded by transfection approaches. Nevertheless, although the use of growth factors such as TGF-β is associated with increased expression of type I and X collagen in treated MSCs, it is a remarkable finding that transfection of SOX trio reduces hypertrophic markers such as type X collagen and Runx-2.
In conclusion, SOX trio have a significantly lower level of expression in human MSCs than in chondrocytes, and the electroporation-mediated co-transfection of SOX trio enhances chondrogenesis and suppresses hypertrophy of human MSCs. The result of this study can be used for planning gene transfer strategies to induce successful chondrogenesis in cartilage tissue engineering.
Footnotes
Acknowledgments
This study was supported by a grant from the National Research Foundation of Korea (2010-0028762).
Author Disclosure Statement
No competing financial interests exist.