
Abstract
Hemophilia A (HA) is caused by mutation in factor VIII (FVIII) gene in humans; it leads to inadequate synthesis of active protein. Liver is the primary site of FVIII synthesis; however, the specific cell types responsible for its synthesis remain controversial. We propose that the severity of the bleeding disorder could be ameliorated by partial replacement of mutated liver cells by healthy cells in HA mice. The aim of this investigation was to study the cellular origin of FVIII by examining bone marrow cell therapy for treatment of HA in mice. Recipient liver was perturbed with either acetaminophen or monocrotaline to facilitate the engraftment and differentiation of lineage-depleted (Lin−) enhanced green fluorescent protein-expressing bone marrow cells. Immunohistochemical analysis of liver tissue was conducted to identify the donor-derived cells that expressed FVIII. This identification was confirmed by transmission electron microscopy and quantitative gene expression analysis. The phenotypic correction in HA mice was determined by tail-clip challenge and FVIII level in plasma by Chromogenix and activated partial thromboplastin time assays. Immunohistochemical analysis showed that von Willebrand factor and cytokeratin-18-expressing endothelial cells and hepatocytes, respectively, were obtained from BM-derived cells. Both cell types expressed FVIII light chain mRNA and protein, which was further confirmed by transmission electron microscopy. The transplanted HA mice showed FVIII activity in plasma (P<0.01) and survived tail-clip challenge (P<0.001). Thus, we conclude that BM-derived hepatocytes and endothelial cells can synthesize FVIII in liver and correct bleeding phenotype in HA mice.
Introduction
H
The FVIII gene has been identified in different tissues of mammals. Using RNase protection analysis, Wion et al. [2] showed that FVIII mRNA is expressed in the liver, purified hepatocytes, the spleen, and lymph nodes. Pancreas, kidney, and muscle also showed low-level expression of FVIII mRNA [2]. Liver perfusion [3,4] and orthotopic liver transplantation studies in both animals and humans [5 –7] have suggested that the primary site of FVIII synthesis is the liver. The site of the cellular biosynthesis of FVIII remains controversial despite studies that date back nearly 50 years [8,9]. Independent studies involving transplantation of hepatocytes [3,4,10,11] and liver sinusoidal endothelial cells (LSECs) [12,13] showed that, in either case, FVIII is synthesized in the recipients. Further, in another investigation, it was reported that only endothelial cells of the liver are responsible for phenotypic correction in HA mice [14]. From these studies, it can be concluded that both hepatocytes and LSECs are capable of synthesizing functional FVIII protein in the liver.
Recently, our laboratory showed that phenotypic correction is possible by transplanting bone marrow cells (BMCs) in HA mice following the induction of acute liver injury with acetaminophen [15]. Acetaminophen causes necrosis of hepatocytes. The BM-derived cells transdifferentiated into hepatocytes and synthesized FVIII protein [15]. We also found transdifferentiation of BMCs into LSECs in the same liver injury model. Monocrotaline (MCT), a pyrrolizidine alkaloid, is reported to cause injury of LSECs and hepatic parenchymal cells depending upon the administered dose [16,17]. In an earlier study, MCT was specifically used to damage LSECs and subsequently liver regeneration by transplanting donor LSECs [13]. It was claimed that FVIII was only produced by the donor LSECs [13]. In this investigation, we have chosen 2 liver-damaged models; in case of acetaminophen, hepatocytes are typically damaged, whereas MCT primarily affects LSECs. These 2 models will allow to compare the relative contribution of BM-derived cells in differentiation of hepatocytes and endothelial cells and finally to evaluate FVIII synthesis by these cells. Acetaminophen and MCT are supposed to induce acute liver injury to provide a suitable microenvironment for hepatic and endothelial differentiation of the donor cells. Our study indicates that FVIII mRNA and protein are expressed by hepatocytes and LSECs in both liver injury models.
Materials and Methods
Animals
Six- to 8-week-old male HA mice [B6;129S4-F8tm1Kaz /J] and enhanced green fluorescent protein (eGFP)-expressing wild-type (WT) mice [C57BL/6-Tg(UBC-GFP)30Scha/J] were used in this investigation. Mice were obtained from Jackson Laboratories and maintained in an experimental animal facility in our institute. Mice were kept in an isolator and fed with autoclaved acidified water and γ-irradiated food ad libitum. All experiments were conducted as per procedures approved by the Institutional Animal Ethics Committee.
Acute liver injury model
Acute liver injury was induced by a single intraperitoneal injection of acetaminophen (500 mg/kg b.w.) or MCT (300 mg/kg b.w.) in male HA mice. Acetaminophen was administered as described in our earlier report [15]. In the case of MCT, mice were kept fasting for 18 h prior to treatment, at which point feeding resumed until the end of the experiments. Mice were sacrificed at days 1, 2, and 3 after administration of acetaminophen or MCT, and sera samples were collected for the estimation of alanine aminotransferase (ALT) levels. The liver was dissected, fixed in 4% paraformaldehyde, equilibrated with 30% sucrose, embedded in optimal cutting temperature resin, and frozen at −80°C. Five-micrometer cryostat sections were stained with hematoxylin and eosin for histologic examination. The ALT assay was carried out using a standard kit (Transasia Bio-Medicals).
Liver reconstitution
Lineage-depleted (Lin−) (CD5, CD11b, CD45R, 7-4, Gr-1, Ter119-depleted) BMCs were isolated from eGFP-transgenic female mice by negative selection using a magnetic cell sorter (Miltenyi Biotech.). Two hundred fifty thousand cells were transplanted to acetaminophen- or MCT-treated (day 1) and control HA male mice by tail vein injection.
Immunohistochemistry
Liver tissue was fixed and frozen as described in our earlier report [15]. Five-micrometer serial sections were permeabilized with buffer containing 1% bovine serum albumin and 0.15% Triton X-100 in phosphate-buffered saline (PBS) for 30 min at room temperature. The sections were then stained with mouse anti-GFP (Stressgen) or rabbit anti-GFP, mouse anti-cytokeratin-18 (CK-18; sc-32329), and goat anti-von Willebrand factor (vWF) antibodies (sc-8086; Santa Cruz Biotechnology) for 1 h at room temperature. The secondary antibodies were conjugated with Alexa Fluor 488, 594, or 546 (Molecular Probes, Inc.). For FVIII staining, tissue sections were blocked in 2% goat serum and stained with rabbit anti-FVIII light chain (lc)-specific antibody (sc-33584; Santa Cruz) or the corresponding isotype control serum (S-5000; Vector Laboratories). Sections were incubated with Alexa Fluor 546-conjugated goat anti-rabbit IgG, and the nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). Sections were imaged with an Olympus fluorescence microscope using LCPlanFl 20× and 60× objectives and with a Zeiss LSM 510 META confocal laser-scanning microscope using a Plan-Apochromat 63×/1.4 oil objective. Olysia BioReport and LSM 510 software were used for acquisition of images by the Olympus and Zeiss microscopes, respectively. For confocal microscopy, the images were processed by Zeiss LSM Image browser, version 4.2.0.121. Finally, the images were composed and edited in Photoshop 6.0 (Adobe).
Isolation of hepatocytes and LSECs
Single-cell suspension of hepatocytes and LSECs were prepared according to methods reported in the literature [18
–20]. Details of the cell isolation method and subsequent characterization are provided in Supplementary Methods (Supplementary Data are available online at
Phenotype correction analysis
Phenotype correction was assessed by tail-clip challenge, as described in the literature [21]. The tails of transplanted mice were clipped at a length of 1.5 cm, without subsequent cauterization. Three groups of mice were subjected to tail-clip challenge: WT (positive control), HA (negative control), and HA transplanted (HAT) test mice. Clotting of blood and subsequent survival of mice were considered indicative of normal/correction of the HA phenotype. In another assay, whole blood clotting times of the 3 mouse groups were determined [22]. In brief, blood was withdrawn in a capillary tube (nonheparinized) from the eye. One end of the loaded capillary tube was gently broken at regular intervals and slowly pulled apart to view insoluble fibrin strands.
FVIII assays
Levels of FVIII protein were measured using an enzyme-linked immunosorbent assay (ELISA) (Supplementary Methods). The amount of FVIII antigen in HAT plasma was determined relative to plasma from WT and HA mice, which were analyzed simultaneously. The FVIII activity was determined using the COATEST SP4 FVIII kit (Instrumentation Laboratory Company).
Coagulation assay
The FVIII assay was performed as described in the literature [23], with minor modifications. Blood samples were collected in microfuge tubes containing sodium citrate. The samples were centrifuged, and the plasma fractions were immediately stored at −70°C. The activated partial thromboplastin time (aPTT) assay was performed by incubating 100 μL of aPTT reagent (Diagnostica Stago) with 100 μL of plasma at 37°C for 5 min. Then, 100 μL of 25 mM CaCl2 was added, and the clotting time was determined. To establish a standard curve, pooled mouse plasma at dilutions of 1:10, 1:50, and 1:250 in Owren-Koller buffer was subjected to an aPTT assay. The clotting times of the diluted plasma were considered equivalent to that of 100%, 20%, and 4% normal plasma, respectively. For the test mice, 100 μL of diluted (1:10) mouse plasma, 100 μL of hFVIII-deficient plasma, and 100 μL of aPTT reagent were used, which was followed by 100 μL of CaCl2. The plasma FVIII activity with respect to normal pooled plasma was determined by comparing clotting times from the standard curve.
Reverse transcriptase–polymerase chain reaction and real-time RT–polymerase chain reaction
Details of the total RNA extraction from whole liver tissue and isolated cells and cDNA synthesis are provided in Supplementary Methods. cDNA synthesized from the whole liver or hepatocytes/LSECs of WT mice were considered as positive controls, and cDNA synthesized from the whole liver of HA mice or Lin− WT BMCs was considered as a negative control. Primer pairs were designed using Primer 3 software [for RT–PCR] and Primer Express® software version 2.0 (Applied Biosystems) for real-time reverse transcriptase–polymerase chain reaction (RT–PCR). The sequences of the primers and amplification conditions of the reactions are shown in Supplementary Table S1. Real-time RT-PCR was performed with the Master Cycler ep Realplex 4 (Eppendorf AG) according to the manufacturer's instructions. Real-time RT-PCR was carried out in quadruplicates of each 10 μL reaction volume containing 5 μL of 2×Power SYBR® Green PCR Master Mix (Applied Biosystems), 0.5 μM of each primer, and 1 μL of cDNA. For normalization, the expression level of the housekeeping gene (GAPDH) was measured as an endogenous control. For quantification of each PCR result, we calculated the ΔCt value between the target gene of each cell type and its endogenous control. ΔCt values of hepatocytes or endothelial cells were subtracted from the ΔCt value of HA whole liver or Lin− BMCs to obtain ΔΔCt. The 2−ΔΔCt value (relative) was calculated using Realplex 2.2 software (Eppendorf) and expressed as log fold change.
Transmission electron microscopy
Immunogold labeling of tissue sections was performed according to methods published in the literature [24]. In brief, ultrathin (70 nm) tissue sections were taken on nickel grids. The sections were initially blocked with 2% skimmed milk and then incubated separately on droplets of a 1:50 dilution anti-albumin or anti-vWF antibodies for 4–6 h at 6°C–8°C. After washing the sections with PBS containing 1% fish gelatin (6×2 min), we further incubated the sections for 2 h with anti-goat IgG conjugated to 10-nm colloidal gold particles. The sections were thoroughly rinsed with PBS containing 1% fish gelatin (6×2 min). In the second step of immunolabeling, all sections were incubated on droplets of anti-FVIII antibody (1:50) for 4 h at 6°C–8°C. Washed sections were incubated with anti-rabbit IgG conjugated to 40-nm colloidal gold particles (1:20) for 2 h. The washed sections were stained with uranyl acetate and lead citrate before microscopic analysis (Morgagni 268D; FEI). Except for primary antibodies, all other reagents were procured from TAAB Laboratories Equipment Ltd.
Statistical values
The results of multiple experiments were reported as mean±standard error of the mean (SEM). Tukey–Kramer multiple comparison tests were then performed to compare the mean values.
Results
Lin−GFP+ BMCs differentiate into hepatic and endothelial cells in damaged HA mouse liver
Previously, we showed that acetaminophen causes damage to hepatic tissue [15]. In this study, we examined the pathology of liver sections and determined serum ALT levels in MCT-treated mice. It is known that MCT causes injury to hepatic parenchymal and endothelial cells in rodents [16,17]. The extent of liver injury was determined by serum ALT levels and histopathology of liver tissue (Supplementary Fig. S1A, B). We observed that in MCT-treated mice, hepatic injury was comparatively less than in acetaminophen-treated mice, and the damage was mostly in the endothelial cells of the centrilobular region (Supplementary Fig. S1B). This was supported by the low ALT levels of the mice in case of MCT treatment (Supplementary Fig. S1A, right panel). In either of 2 liver injury models, the mortality was restricted to 20% of the mice that received the drug (Supplementary Fig. S1C).
Twenty-four hours after liver injury, 2.5×105 Lin−GFP+ BMCs were transplanted into acetaminophen- and MCT-treated groups of HA mice. To monitor the differentiation of engrafted cells into target cells, we analyzed serial liver sections of transplanted mice at different times by immunohistochemistry (IHC). Representative micrographs taken at 6 months after transplantation showed that eGFP+ cells had engrafted in MCT-treated mouse liver and expressed both endothelial (vWF) and hepatic (CK-18) markers (Fig. 1A). At the end of 6 months of transplantation, 4.15%–7.3% BM-derived cells were detected in the recipient liver, irrespective of the injury model. These BM-derived cells expressed either vWF or CK-18. These results suggested that in damaged liver microenvironment, donor-derived cells tend to change into endothelial and hepatocyte lineages. We have not shown the results for acetaminophen-treated mice here, because such results have been reported by our group [15]. Further, to support in favor of the formation of donor-derived hepatocytes, we stained HAT mice liver cells with albumin antibody. Results showed that mononucleated and binucleated GFP+ cells express albumin, suggesting that these hepatocyte-like cells were derived from BMCs (Supplementary Fig. S2). On the basis of above and our earlier [15] results, it has been proposed that in the present liver-damaged model, fusion may not be the principle mechanism for the formation of hepatocyte-like cells from BMCs.
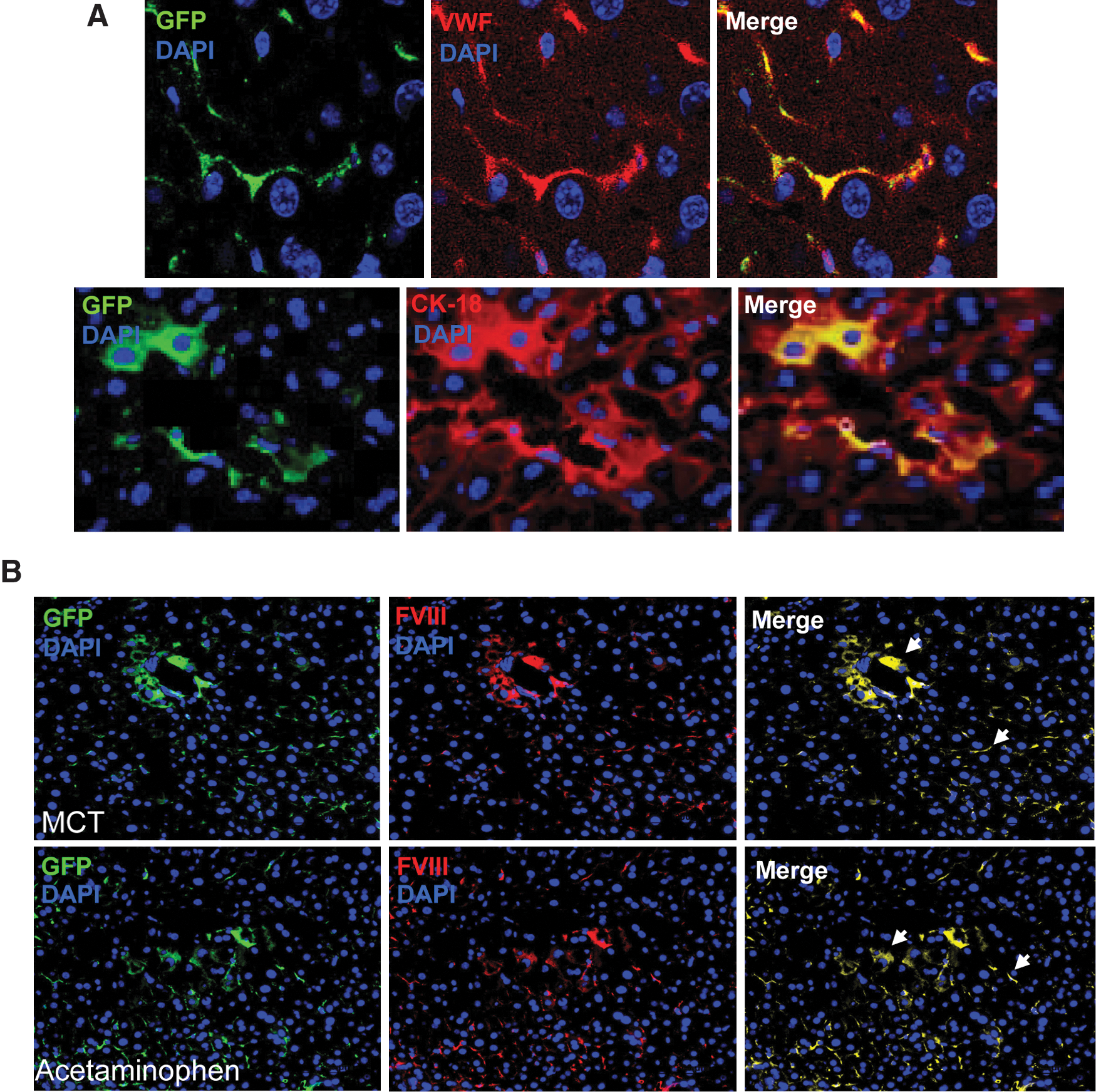
IHC of liver sections.
Liver engrafted cells express FVIII protein
The expression of FVIII in liver tissue was examined by IHC. The mutant mouse used in this investigation has a disrupted exon 16 within the A3 domain of the FVIII lc gene. Thus, the intact light chain protein is not expressed [25,26] in this mouse, although the heavy chain protein is normally expressed. To determine whether HAT mice express the FVIII lc protein, serial liver sections of control and transplanted mice were stained with either isotype control or specific antibodies. The FVIII (lc) protein was not detected in the liver section of HA mice costained with either vWF or CK-18 (Supplementary Fig. S3), indicating that FVIII is not synthesized in HA mice. Interestingly, the FVIII (lc) protein was expressed in both MCT- and acetaminophen-treated HAT mice (Fig. 1B). No background staining of FVIII was observed with the isotype control antibody (Supplementary Fig. S4A) and the secondary antibody conjugates (Supplementary Fig. S4B), indicating that the aforementioned staining was specific. Careful examination of these images revealed that 2 morphologically distinct cell types expressed FVIII protein (Fig. 1). It was proposed that these cell types are hepatocytes and endothelial cells. Such expression would not be possible in HA mice unless BMCs differentiated into these 2 cell types. The expression of FVIII (lc) protein was observed near the central vein as well as in other regions of the liver sections (Fig. 1B). Further, confocal analysis of liver sections also confirmed that, in the MCT model, engrafted BMCs express FVIII protein (Supplementary Fig. S5). To ensure that the FVIII (lc) antibody does not bind with eGFP protein we stained eGFP-BMCs with the same antibody. It was found that FVIII antibody did not recognize eGFP protein (Supplementary Fig. S6), indicating that the staining of FVIII-expressing cells was specific. As each donor cell expressed FVIII (lc) protein and they were either hepatocyte- or endothelial-like cells, we have determined the relative contribution of these 2 cell types in overall FVIII synthesis. On the basis of scoring on serial IHC sections in acetaminophen-treated mice, about 61%±10.5% of BM-derived hepatocyte-like cells contributed in the synthesis of clotting factor; these values were 39%±6.9% in case of endothelial-like cells. In case of MCT-treated mice, these values were 25%±6.5% and 75%±12.2% for hepatocyte- and endothelial-like cells, respectively. These results suggested that the contribution of hepatocytes in synthesizing FVIII was significantly (P<0.05) higher than that of LSECs in acetaminophen-treated transplanted mice. In contrast, in case of MCT-treated mice, endothelial cells synthesized major fraction (P<0.05) of the FVIII protein.
To confirm that FVIII is expressed in hepatocytes and LSECs, we stained liver sections of WT mice with the respective antibodies. It was found that FVIII (lc) protein was expressed by CK-18-expressing hepatocytes (Fig. 2A, top panel) and vWF-expressing LSECs (Fig. 2A, bottom panel). Further, simultaneous 2-channel fluorescence and differential interference contrast images of WT mice liver sections morphologically illustrate 2 distinct structures of cells that expressed FVIII (lc) protein (Fig. 2B). One cell type was polygonal shaped and binucleated and appears to be hepatocytes (Fig. 2B, left panel), and another elongated cell was known to be as LSECs (Fig. 2B, right panel). These results suggest that, in WT mice, FVIII protein was expressed by both hepatocytes and endothelial cells of the liver, whereas in HAT mice, FVIII was produced by BMC-derived hepatocytes and endothelial cells.
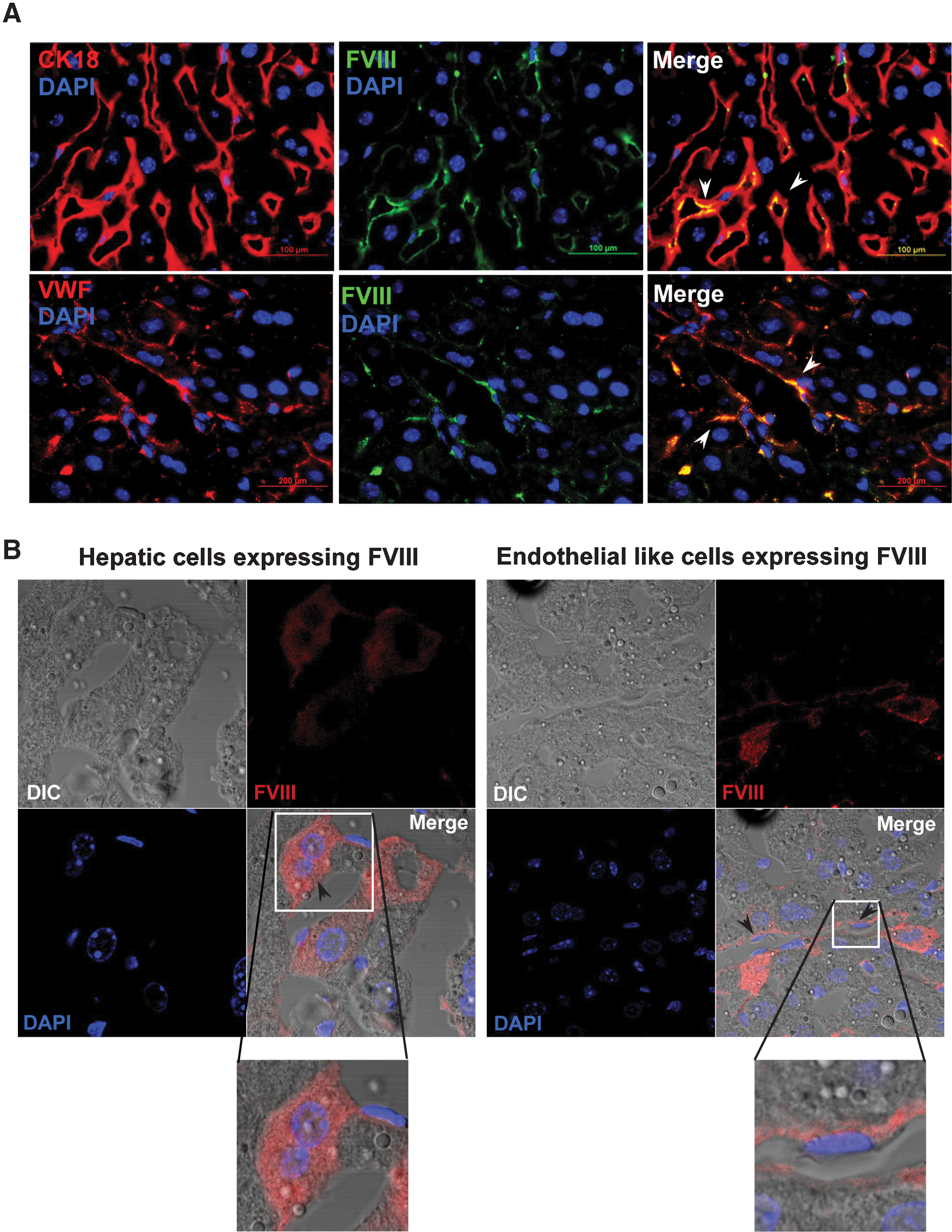
Expression of FVIII (lc) protein in WT mice.
Plasma FVIII activity and bleeding
The analysis of IHC results demonstrated the potential therapeutic effects of transplanted BMCs in both damaged liver models. To confirm this therapeutic potential, we analyzed the plasma of HAT mice using different assays, and mice were also subjected to tail-clip challenge. Results of an ELISA test showed that FVIII antigen levels in the plasma of HAT mice were significantly higher (P<0.05) compared with HA mice (Fig. 3A). The average antigen levels in the 2 liver-damaged models were comparable and more than 60% of the normal FVIII level in the WT mice. To evaluate the therapeutic effect of BMCs, the same plasma samples were analyzed by COATEST assay (Fig. 3B). The average FVIII activities in plasma collected from MCT- and acetaminophen-treated HAT mice were 28.5%±6.1% (n=5) and 39%±6.7% (n=5) of normal plasma, respectively (Fig. 3B). The phenotypic rescue in HAT mice was further examined by an aPTT assay. In WT mice, the plasma FVIII activity was 115%±18% (n=6) of that in normal pooled mouse plasma. The FVIII activity in HA mice was 0.8%±0.4%, which increased to 42.1%±5.6% and 53.8%±8.1%, respectively, in MCT- and acetaminophen-treated HAT mice (Fig. 3C). Tukey-Kramer multiple comparison analysis concluded that the plasma FVIII activity in HAT mice was significantly (HA to HATMCT: P<0.01; HA to HATAceta: P<0.01) higher in the 2 test groups compared with the HA mice. These results suggest that the average FVIII activity estimated by COATEST assay was lower than that observed by aPTT assay. However, an ELISA-based assay showed that FVIII-like protein expression was 1.5-fold higher than that measured by these 2 functional assays.
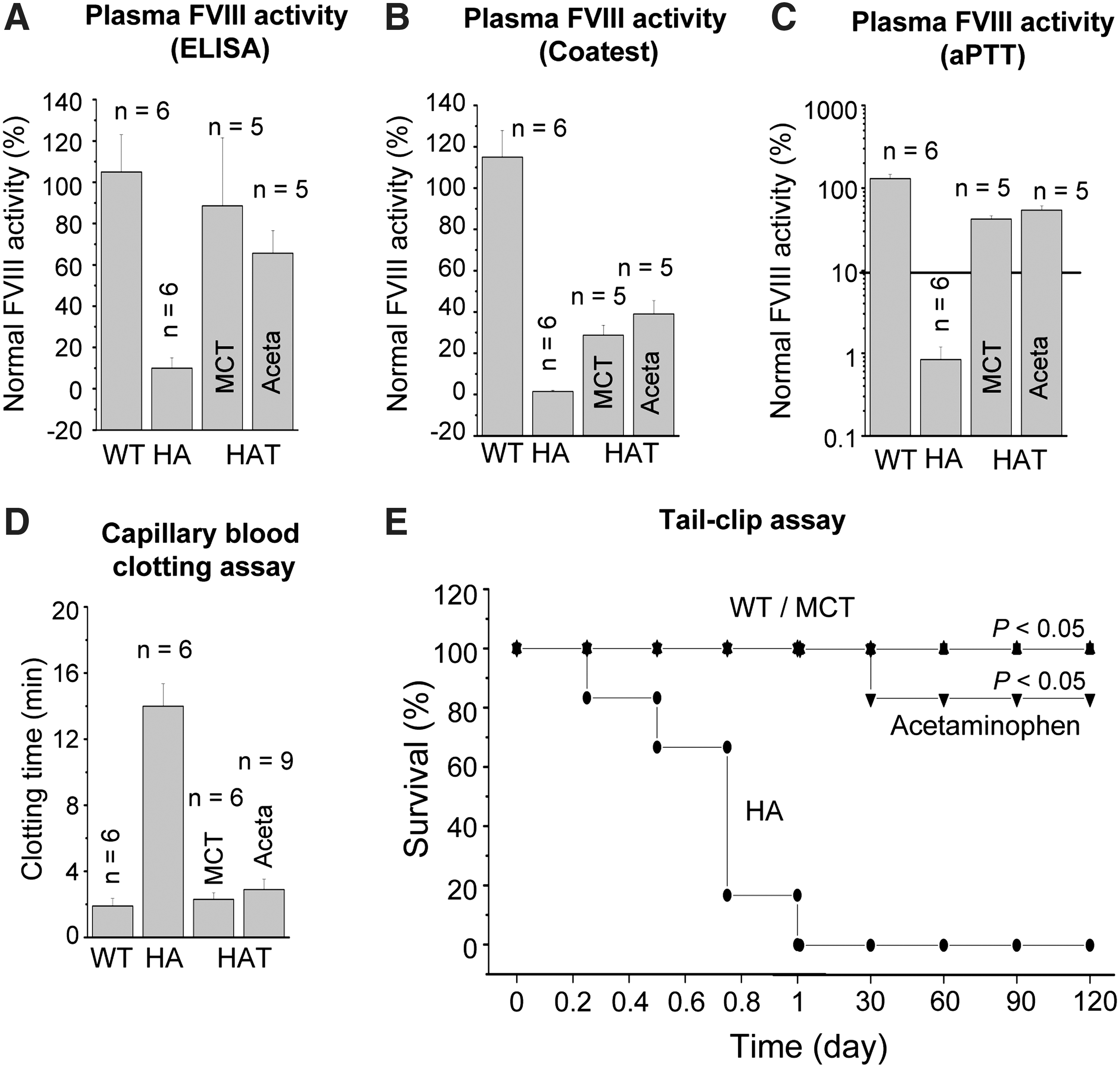
Relative FVIII activity and functional recovery.
Further, phenotypic correction in HAT mice was determined by a blood clotting and tail-clip challenge experiment. The whole blood clotting time in WT mice was 1.9±0.5 min (n=6), which was increased to 14±1.5 min (n=6) in HA mice (Fig. 3D). The whole blood clotting time was significantly (P<0.01, n=15) reduced at 6 months after transplantation (Fig. 3D). Similarly, Lin− cells therapy did exhibit a significant (P<0.05) survival benefit compared with the control HA mice (Fig. 3E). The survival rate in the tail-clip challenge experiment was increased from 0% to 100% and 0% to 87%, respectively, for MCT- and acetaminophen-treated HAT mice. The death of lone mouse in the transplantation group was due to some unknown reasons. All the mice that received Lin− BMCs achieved hemostasis within 4 h of tail-clip. The mice that did not stop bleeding died within 10–20 h of the tail-clip. It was found that none of the HA mice survived more than 20 h after the tail-clip. These results suggest that BMC-derived hepatocytes and endothelial cells produced FVIII protein in the liver of HA mice and also that FVIII was secreted into the blood circulation.
FVIII lc mRNA is expressed in both hepatocytes and LSECs
To confirm the result that FVIII protein is expressed by hepatocytes and endothelial cells, we conducted RT-PCR and real-time RT-PCR analysis of light chain mRNA in both cell fractions. Hepatocytes and LSECs were separated from the perfused liver cells of WT and HAT mice using standard protocols [18 –20]. The purity of isolated cells was determined by immunocytochemistry using hepatocyte- and endothelial-specific markers. LSECs are much smaller in size than hepatocytes. Morphological and immunocytochemical analyses of the cells suggest that the isolated endothelial cell fractions were about 93% pure, whereas no contaminating LSEC was detected in the hepatocyte fractions (Supplementary Fig. S7). These cells were used to isolate mRNA for RT-PCR and real-time RT-PCR analyses. The rationale for designing primers has been previously described [15,25]. The PCR results showed that a 637-bp gene fragment was amplified in WT mouse whole-liver tissue as well as in the purified fractions of hepatocytes and endothelial cells. The same gene fragment was not amplified in mutant (HA) mouse liver (Supplementary Fig. S8).
Because in RT-PCR analysis the difference in the expression of the 637-bp FVIII (lc) gene fragment by hepatocytes and LSECs was not appreciable, we conducted real-time RT-PCR to determine the relative expression of mRNA in these cells. Hepatocytes and endothelial cells were isolated from WT and HAT mice, and the mRNA was subjected to RT-PCR using primers designed for real-time RT-PCR. As negative controls, HA mice whole liver and Lin− BMCs mRNA were used. The results suggest that both hepatocyte and endothelial cell fractions expressed FVIII (lc) gene, because a 101-bp fragment was amplified (Fig. 4A). Further, the expression of same gene was discernible in the Lin− BMCs. In real-time RT-PCR experiments, relative signals of the test samples were determined with respect to the signal of the HA (whole liver) and Lin− BMCs. Hepatocytes and LSECs isolated from WT mice showed extremely high relative expression of the target amplicon as gene expression by HA mice was negligible (Fig. 4B). Among these 2 cell types, LSECs expressed about 5-fold more FVIII (lc) mRNA (Fig. 4B). In MCT-HAT mice, the expression of the gene product in hepatocytes and LSECs was comparable, despite variability from mouse to mouse. Similarly, in the case of acetaminophen-HAT mice, FVIII (lc) mRNA was expressed by both cell types, although the fold expression was lower than that for MCT-HAT mice (Fig. 4B). In case of Lin− BMCs serving as control, the relative expressions of the gene were much lower when compared with the HA (Fig. 4B). Taken together, the RT-PCR and real-time RT-PCR data suggest that FVIII (lc) mRNA is expressed by both hepatocytes and LSECs of WT mice. Further, in both MCT- and acetaminophen-induced liver injury models, BMC-derived hepatocytes and LSECs expressed the FVIII (lc) gene.
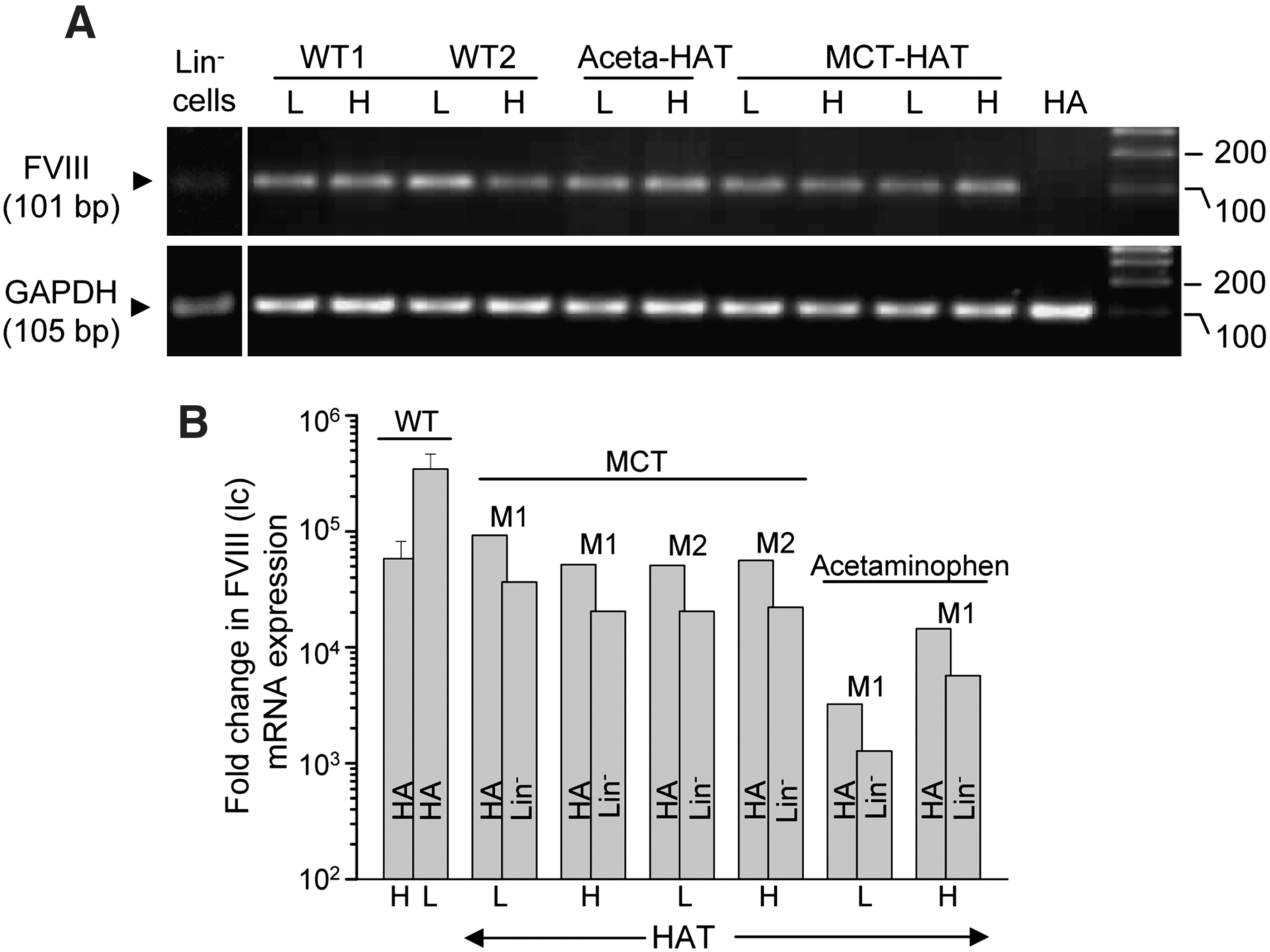
mRNA expression of FVIII (lc) gene in hepatocytes and endothelial cells.
Transmission electron microscopy confirms that both hepatocytes and LSECs express FVIII (lc) protein
Postembedded immunogold-labeled transmission electron micrographs also confirmed the finding that FVIII (lc) protein is expressed by hepatocytes as well as by endothelial cells. WT, HA, and HAT mouse liver sections were either stained with anti-albumin and anti-FVIII (lc) or anti-vWF and anti-FVIII (lc) antibodies. It may be noted that HA mouse liver sections showed albumin and vWF staining, whereas FVIII (lc) protein was not expressed (Fig. 5, middle panel). We observed that 1 to 2 colloidal gold particles, targeted to FVIII (lc) protein, were nonspecifically attached to the HA mouse liver sections. In WT and HAT mice, coexpression of albumin and FVIII (lc) proteins was observed in the cytoplasm of the target cells (Fig. 5, left upper and lower panels). These results indicated that FVIII protein is expressed by hepatocytes. Similarly, vWF-expressing cells also synthesized FVIII protein in WT and HAT mice, confirming that endothelial cells are also a source of FVIII (Fig. 5, right upper and lower panels).
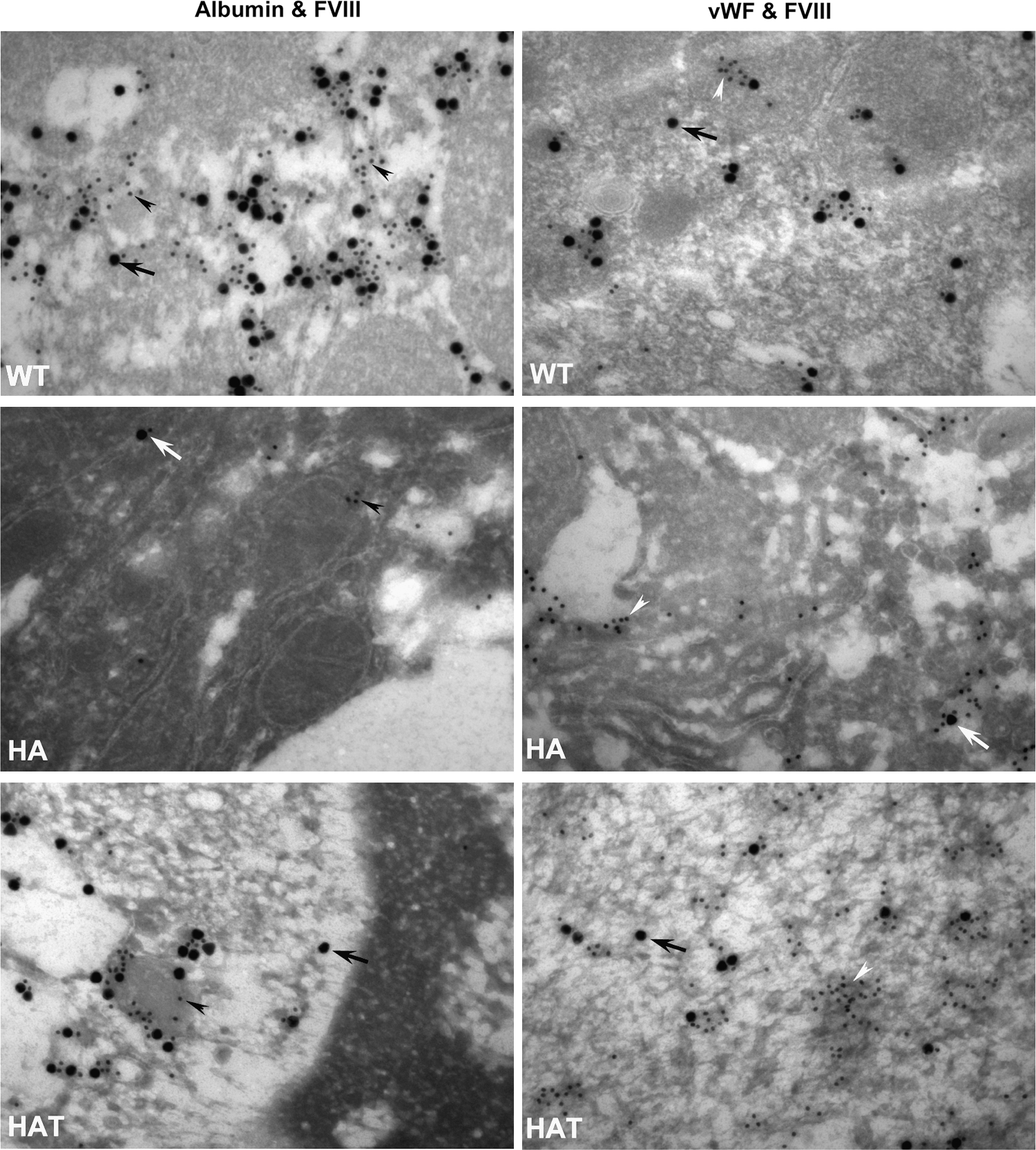
Transmission electron microscopy analysis for the expression of FVIII (lc) protein in hepatocytes and endothelial cells. Ultrathin sections of liver were labeled with
Discussion
Cell-based therapy using isolated primary hepatocytes [27,28] or LSECs [12 –14] has been suggested to treat clotting disorders. Coagulation factors VIII and IX are synthesized in the liver before entering the circulation. We hypothesized that the partial replacement of mutated liver cells by healthy cells in HA mice could ameliorate the severity of the bleeding disorder. In the past few years, a novel and exciting option to regenerate damaged liver from BM-derived cells has been proposed by many investigators [29 –34]. Recently, we have shown that Lin−BMCs can correct the bleeding phenotype in HA mice by producing active FVIII protein [15].
In the present study, we provide evidence that the FVIII gene and protein are expressed by both hepatocytes and endothelial cells of the liver sinusoids in WT mice. In acetaminophen- or MCT-treated HA mice, BMCs converted into hepatocytes and endothelial cells, leading to correction of the bleeding phenotype of the mouse. We propose that it is a case of direct differentiation as our earlier results [15,34] did not show a significant level of cell fusion in the models of damaged liver. The cellular origin of FVIII remains elusive. In 2 separate studies [13,14], it was shown that LSECs, but not hepatocytes, are responsible for phenotypic correction in HA mice. These reports contradicted an elegant study showing that human hepatocytes transplanted under the kidney capsules of mice render therapeutic benefit [27]. Kumaran et al. [14] transplanted hepatocytes mixed with Cytodex-3 microcarriers in the peritoneal cavity and showed that the transplanted hepatocytes engrafted in the cavity remained viable and functional for 1 week. However, hepatocytes did not produce phenotypic correction in HA mice, whereas LSECs did. Although earlier reports [35,36] showed that the expression of some proteins was maintained by hepatocytes for more than a week in the peritoneal cavity, the same may not be true in the case of the FVIII-like complex protein. We believe that the ability of hepatocytes to produce FVIII was lost in the peritoneal cavity, because they were removed from their natural hepatic environment. This loss of FVIII production may not be the case with LSECs, because endothelial cells occur naturally in all tissues. Also, the failure to express FVIII by hepatocytes [14] may be due to the short duration of experiments. Further, the tolerogenic properties of the liver may have an important role in the expression of FVIII by hepatocytes in HA mice; these properties are lacking in the peritoneal cavity [37 –39].
In an earlier study, we proposed that FVIII is synthesized by hepatocytes, primarily by direct differentiation [15]. In the present investigation, we confirmed that both hepatocytes and endothelial cells are able to synthesize FVIII. We used 2 different liver injury models: acetaminophen is known to be toxic to hepatocytes [40] and MCT to endothelial cells [17]. It has been found that acetaminophen and MCT are also toxic to endothelial cells and hepatocytes, respectively. Histology of liver sections and serum ALT levels confirmed that MCT is less toxic to hepatocytes than endothelial cells. However, in both cases of liver injury, we found that BMCs converted into hepatocytes and endothelial cells. Interestingly, the FVIII (lc) gene was expressed in hepatocytes as well as in endothelial cells. In HAT mice, we did not find much difference between the gene expressions in these cells, but, in WT mice, FVIII (lc) gene expression was much higher in LSECs than in hepatocytes. Further, IHC results confirmed that HAT mice synthesized the lc FVIII protein in LSECs and hepatocytes; such synthesis was absent in HA mice. Finally, these results have been validated by transmission electron microscopy, which demonstrated that both hepatocytes and endothelial cells express FVIII (lc) protein. Overall, the above results make us believe that FVIII synthesis is not specific to endothelial cells, as shown by others [12 –14]; FVIII is also synthesized by hepatocytes.
FVIII was not only expressed by the hepatocytes and endothelial cells of HAT mice but also secreted in the active form, as supported by coagulation and tail-clip challenge assays. Our results do not demarcate the FVIII-synthesizing ability of these 2 cell types; for that purpose, separate transplantation of hepatocytes and LSECs in different groups of mice would be necessary, which is beyond the scope of this investigation. However, we have speculated that FVIII-synthesizing ability by hepatocytes and endothelial cells in HAT mice may change with the nature of the hepatotoxic drugs used to perturb liver. In acetaminophen model, about two-thirds of secreted FVIII was probably synthesized by hepatocytes, whereas in MCT model about three-fourths of the circulating factor might have been synthesized by LSECs. Phenotype correction of HA mice was observed as early as 2 months following BMC transplantation (data not shown). This study was conducted in 1 fixed dose of Lin− BMCs, although early recovery may be possible at higher cell dose because of better engraftment. This is a subject of further investigation. Most interestingly, our previous study [15] did not show the formation of FVIII inhibitors during 18 months of experiments, which was thought to be due to the tolerogenic properties of the liver. Therefore, BMC therapy appears to be an efficient alternate method for treating HA. However, inflicting acute liver injury by acetaminophen or MCT before BMC therapy could be potentially hazardous in HA patients. The extent of risk due to hemorrhage may be contained by following alternate strategies: (1) inflecting minimum liver injury by acetaminophen, which can reduce hemorrhage but allow engraftment of enough competent BMCs undergoing differentiation and synthesizing adequate quantity of FVIII; (2) conditioning patients with therapeutic dose of purified FVIII prior to inflecting minimum liver injury and BMC therapy; and (3) transplanting in vitro differentiated BMCs (hepatocytes/LSECs) in the undamaged liver of the patient.
Earlier studies showed BMCs have a tendency to fuse with primary hepatocytes [41 –44] and Purkinje neurons and cardiomyocytes [44]. Mice with fetal metabolic liver disease, tyrosinemia type I, were rescued by transplantation of purified hematopoietic stem cells [41,42]. The donor partners were believed to deliver their genome, which includes the fumarylacetoacetate hydrolase (Fah) gene, into preexisting liver cells. It was proposed that in resultant hybrids, liver gene expression was activated and donor gene expression was suppressed [45]. Similarly, cell fusion can be promoted between donor-derived Lin− cells and hepatocytes of HA mice to obtain therapeutic benefit of transplantation. Thus, cell fusion may avoid inflicting liver injury by acetaminophen; hence, the potential threat due to hemorrhage can be significantly contained. Spontaneous cell fusion is a very rare event; therefore, therapeutic effects can be achieved only by selective growth of few fused hepatocytes. The second approach to improve the efficiency of fusion between host hepatocytes and donor cells could be using specific chemicals or proteins, which enhances fusion between 2 adjacent cells. There is a need to further study on improving fusion efficiency and selective proliferation of donor-derived cells in the present model to obtain optimal therapeutic benefit.
We conclude that BMC therapy might control the bleeding disorder in HA mice. This therapy may provide a cost-effective, constant, and prophylactic dosage of FVIII in the diseased animals. In the damaged liver microenvironment, engrafted cells can change themselves in the lineage of hepatocyte- and endothelial-like cells. Both cell types are capable of synthesizing active FVIII in HA mice. Thus, BMC therapy is a potential alternative approach for managing HA.
Footnotes
Acknowledgments
The last author is thankful to Department of Biotechnology, Government of India, for providing grants to Centre for Molecular Medicine for pursuing the work. The last author is also indebted to Prof. Avadhesha Surolia, director, NII for his special support to complete the work.
Author Disclosure Statement
None of the authors or their immediate family members has any actual or potential commercial association that might create a conflict of interest in connection with this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.