
Abstract
Human parthenogenetic embryonic stem cells (hpESCs) established from artificially activated oocytes have a wider immune-matching ability because of their homozygosity in the major histocompatibility complex alleles. Whether these cells possess the differentiation capacity similar to regular human embryonic stem cells (hESCs) derived from fertilized eggs is unclear. The aims of this study were to determine whether hpESCs could be differentiated into multipotent mesenchymal stem cell (MSC)-like cells in vitro and then compare these cells with those derived from hESCs. MSC-like cells were obtained from both hpESCs and hESCs, which exhibited similar cell surface marker expression profiles. Further analyses revealed that cells derived from hpESCs possessed stronger osteogenic but weaker adipogenic potentials compared with cells derived from hESCs. This is the first work that demonstrates the differentiation of hpESCs into multipotent MSC-like cells. These hpESCs could be a potential source for cell-based therapies.
Introduction
H
Mesenchymal stem cells (MSCs), which are able to give rise to multiple mesenchymal tissues including osteoblasts, chondrocytes, and adipocytes, have been observed in many adult tissues such as bone marrow, fat, and muscle [11,12]. MSCs derived from adult tissues have become a useful cell source for cell-based therapy [13]. Unfortunately, these cells have a limited proliferation capacity in culture [14,15]. By sequential differentiation of hESCs, a multipotent population that possesses adipogenic, osteogenic, and myelogenic characteristics has been successfully established by several groups [16 –21]. These cells could potentially be an unlimited source for cell-based therapy. The purposes of this study were to investigate whether multipotent MSCs could be generated from hpESCs and then compare those MSCs with those derived from regular hESCs.
Materials and Methods
Cell culture
The hpESC line, chHES-32, was kindly provided by Dr. Lu from the Institute of Reproductive and Stem Cell Engineering, Central South University [2]. The regular hESC line, hES1, was established in our laboratory [22]. Cells were cultured on a feeder layer of mitomycin C-inactivated (Sigma) mouse embryonic fibroblasts in hESC culture medium. The medium consisted of knockout Dulbecco's modified Eagle's medium (DMEM; Invitrogen) supplemented with 20% knockout serum replacement (KSR; Invitrogen), 1% nonessential amino acid, 50 U/mL penicillin, 50 μg/mL streptomycin, 2 mM
Cell differentiation
Differentiation of ESCs into MSCs was performed as described by Hwang et al. [16]. Briefly, chHES-32 and hES1 cells were differentiated into embryoid bodies (EBs) for 10 days on a bacteriological Petri dish (Fisher Scientific) in the same medium as for hESC culture but without bFGF. EBs were then transferred onto a 0.1% (w/v) gelatin-coated plate and cultured for 4 days in DMEM medium supplemented with 10% fetal bovine serum (FBS; HyClone), 50 U/mL penicillin, and 50 μg/mL streptomycin. Cells that migrated from EBs were then collected by physically removing the core of the EBs and passaged every 4–5 days in the same medium using enzymatic digestion with 0.25% trypsin/EDTA (Invitrogen). For cell proliferation assays, cells at passage 5 (P5) and P15 were seeded at a density of 2×103 cells/well in a 96-well plate. Viable cells at the indicated time points were measured using a Cell Counting Kit-8 (Dojindo). The absorbance at 450 nm reflects the relative number of viable cells in each well. Each sample was measured in triplicate.
Immunofluorescence staining
ESC-derived cells at P5 were fixed with 4% cold paraformaldehyde (Sigma) in phosphate-buffered saline (PBS) for 15 min and then permeabilized with 0.25% Triton X-100 (Sigma) in PBS for 10 min. Fixed cells were blocked for 30 min with blocking solution comprised of PBS, 1% bovine serum albumin, and 1% goat serum (Sigma) followed by incubation with rabbit anti-human vimentin (1:100) or goat anti-human nestin (1:100; all from Santa Cruz Biotechnology) antibodies at 4°C overnight. Anti-goat immunoglobulin G (IgG) or anti-rabbit IgG secondary antibodies labeled with Alexa Fluor 488 or Alexa Fluor 555 (1:1,000; Invitrogen), respectively, were added and allowed to incubate. Nuclei were counterstained with 4′,6′-diamidino-2-phenylindole (DAPI; Invitrogen).
Flow cytometry analysis
ESC-derived cells at P5 were harvested, and aliquots of 500,000 cells resuspended in 200 μL washing buffer (PBS supplemented with 4% FBS) were prepared. Freshly diluted fluorescein isothiocyanate (FITC)–conjugated anti-CD31 (1:200; BD Pharmingen) and -CD44 (1:200; eBioscience) and phycoerythrin (PE)-conjugated anti-CD34, -CD105, -CD166, -CD105 (1:200; BD Pharmingen), -CD13, -CD29, and -CD90 (1:200, eBioscience) monoclonal antibodies were added and incubated on ice for 30 min. After 3 washes, the cells were resuspended in washing buffer for analysis. The cells incubated with rat anti-Stro-1 antibody (1:200; R&D Systems) were then treated with FITC-conjugated goat anti-rat IgM (1:1,000; eBioscience). FITC-conjugated isotype-matching Igs were used to determine nonspecific staining. For HLA-A, -B, -C, and -DR staining, the cells were incubated with biotin-conjugated anti-human HLA-A, -B, -C, or -DR antibodies (1:200; eBioscience), followed by incubation with PE-conjugated streptavidin (1:1,000; eBioscience). The cells were analyzed on a flow cytometer (Epics Altra; Beckman Coulter), and data were analyzed with CXP software (Beckman Coulter).
Multilineage differentiation
Adipogenic differentiation
Adipogenic differentiation was induced by culturing cells on 22-mm glass cover slips at a density of 3,000 cells/cm2 in adipogenic medium (DMEM supplemented with 10% FBS, 1% antibiotics, 0.5 mM isobutylmethylxanthine, 1 μM dexamethasone, 10 μM insulin, and 200 μM indomethacin; all from Sigma) for 3 weeks. Cells cultured in the regular culture medium were used as controls. The medium was changed every 3 days. Adipogenic differentiation was measured by Oil red O staining as previously described [23]. The proportion of adipogenic differentiation was determined by counting Oil red O-positive and -negative cells with the assistance of a microscope. At least 5 fields of view were counted for each sample.
Osteogenic differentiation
For osteogenic differentiation, cells were seeded at a density of 3,000 cells/cm2 and cultured in osteogenic medium (DMEM supplemented with 10% FBS, 1% antibiotics, 100 nM dexamethasone, 50 μM ascorbate-2-phosphate, and 10 mM β-glycerophosphate; all from Sigma) for a maximum period of 3 weeks. The medium was changed every 3 days. As a control, cells were cultured in the regular culture medium. Osteogenic differentiation was measured by alkaline phosphatase (ALP) staining with an ALP analysis kit (Renbao). ALP activity was evaluated as previously described [16,24] and normalized to the total protein content, which was determined with a BCA protein assay kit (Pierce). All experiments were conducted in triplicate.
Chondrogenic differentiation
Chondrogenic differentiation was conducted as previously described [23]. Briefly, cells were grown in monolayer culture with chondrogenic medium [DMEM supplemented with 10% FBS, 1% antibiotics, 50 μM ascorbate-2-phosphate (Sigma), 10 ng/mL TGF-β1 (R&D Systems), and 500 ng/mL IGF (R&D Systems)] for 4 weeks. Medium change was performed every 3 days. Cells maintained in the regular culture medium were used as a control. Chondrogenic differentiation was measured by immunohistochemical staining of type II collagen [23]. The chondrogenic differentiation percentage was determined by counting the type II collagen-positive area. At least 5 fields of view were counted for each sample.
Myogenic potential
For myogenic differentiation, cells were seeded at a density of 3,000 cells/cm2 and induced in a myogenic medium (DMEM supplemented with 10% FBS, 1% antibiotics, and 10 ng/mL TGF-β1) for 3 weeks as previously reported [25,26]. The medium was changed every 3 days. Cells maintained with regular culture medium were used as a control. After myogenic induction, the cells were stained with mouse anti-human α-smooth muscle actin (SMA), myosin heavy chain (MHC), and calponin (1:200; Dako) antibodies at 4°C overnight, followed by incubation with anti-goat IgG secondary antibodies labeled with Alexa Fluor 488 (1:1,000). Nuclei were counterstained with DAPI.
Reverse transcription–polymerase chain reaction
Total RNA was extracted with TRIzol® (Invitrogen) and reverse transcribed into cDNA with an RT-PCR Kit (TaKaRa). The primer sequences (all from Sangon Technology Co.), reaction conditions, and the sizes of each product are listed in Table 1. The amplified products were separated on 1.2% (w/v) agarose gels and visualized with the assistance of ethidium bromide (Sigma) and ultraviolet light. Levels of expression were analyzed and compared with β-actin, which was used as the reference gene.
LPL, lipoprotein lipase; ALP, alkaline phosphatase; COMP, cartilage oligomeric matrix protein; ColII, collagen type-II; SMA, smooth muscle actin; AFP, α-fetoprotein; PDX1, pancreatic and duodenal homeobox 1.
Statistical analysis
The differences between cells derived from hpESCs and hESCs, including cell proliferation, cell surface marker expressions, Oil red O staining, ALP staining, as well as gene expressions analyzed by reverse transcription–polymerase chain reaction (RT-PCR), were compared using unpaired Student's t-test (SPSS software, version 16.0). A p-value less than 0.05 was considered to be statistically significant.
Results
Generation of fibroblastic cells from hpESCs and hESCs
After 10 days of suspended culture in the bacteriological Petri dishes, EBs had formed from hpESCs (chHES-32) and regular hESCs (hES1) (Fig. 1A). The EBs were then plated on gelatin-coated tissue culture plates. Fibroblastic cells migrated from EBs and were observed after 5–7 days. The cells were collected by physically removing clumps of EBs, digested with trypsin/EDTA, and subcultured and passaged every 4–5 days. Both types of cells derived from hpESCs and hESCs showed fibroblastic morphology. The cells were cultured for over 30 passages without obvious morphological changes. However, the cells derived from hpESCs grew slower (P<0.05) than the cells derived from hESCs. In addition, both types of cells at later passage (P15) grew slower than those at an earlier passage (P5; Fig. 1B).

Generation of mesenchymal-like stem cells from human parthenogenetic embryonic stem cells (hpESCs) and human embryonic stem cells (hESCs).
Characterization of ESC-derived cells
Cells derived from hpESCs and hESCs were characterized by their cell surface marker expression profiles. The hpESC-derived cells expressed mesenchymal cell surface markers, including CD13, CD29, CD44, CD90, CD105, and CD166, but did not express CD31, CD34, CD45, and Stro-1 (Fig. 2A). Further, HLA-ABC was expressed, whereas HLA-DR was not (Fig. 2A). Similar expression patterns were observed in cells derived from hESCs (Fig. 2B and Supplementary Fig. S1; Supplementary Data are available online at

Characterization of mesenchymal-like stem cells derived from hpESCs and hESCs.
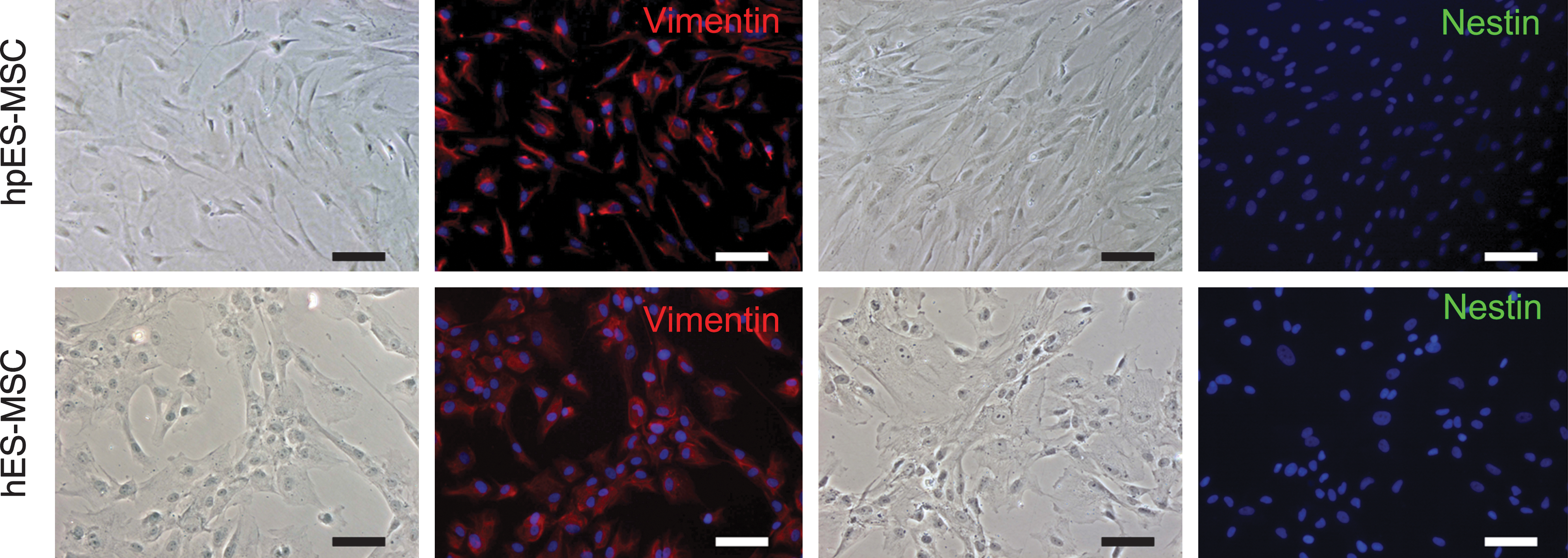
Immunofluorescence staining for vimentin (red) and nestin (green) in cells derived from hpESCs and hESCs. Cell nuclei (blue) were counterstained with 4′,6′-diamidino-2-phenylindole (DAPI) (scale bars indicate 100 μm).
Differentiation potential of ESC-derived cells
The strong proliferation capacity and expression profile of the ESC-derived cells suggest that they were likely MSCs. The adipogenic, osteogenic, chondrogenic, and myogenic differentiation potentials were analyzed and compared between cells derived from hpESCs and hESCs.
After 3 weeks of adipogenic induction, about 10% of Oil red O-positive cells were observed among the hESC-derived cells, with less than 1% positive cells observed in the hpESC-derived cells (Fig. 4A, B). No Oil red O-positive cells were observed in the control groups. Adipogenic differentiation was confirmed by RT-PCR analysis. Expression of adipogenic-related genes, including leptin and LPL, was high in hESC-derived cells following induction but low in hpESC-derived cells that were either induced or uninduced (Fig. 4C, D), indicating that hpESC-derived cells possess lower adipogenic potential.

Adipogenic differentiation of mesenchymal-like stem cells derived from hpESCs and hESCs.
The osteogenic differentiation potential was detected by ALP staining after 2 weeks of osteogenic induction. Cells that were positive for ALP were observed in both types of ESCs after induction, but in very few of the cells that had not been subjected to induction (Fig. 5A). Quantitative analysis of ALP activity showed that hpESC-derived cells displayed higher ALP activity than hESC-derived cells after induction (Fig. 5B), indicating that hpESC-derived cells possessed greater osteogenic potential. This was confirmed by RT-PCR analysis. Osteogenic differentiation–related genes, including ALP and Runx2, were highly expressed in hpESC-derived cells compared with hESC-derived cells that had been subjected to induction. Both genes were also expressed in hpESC-derived cells even though they had not been subjected to osteogenic induction, indicating that cells spontaneously progressed down the osteogenic pathway in culture.
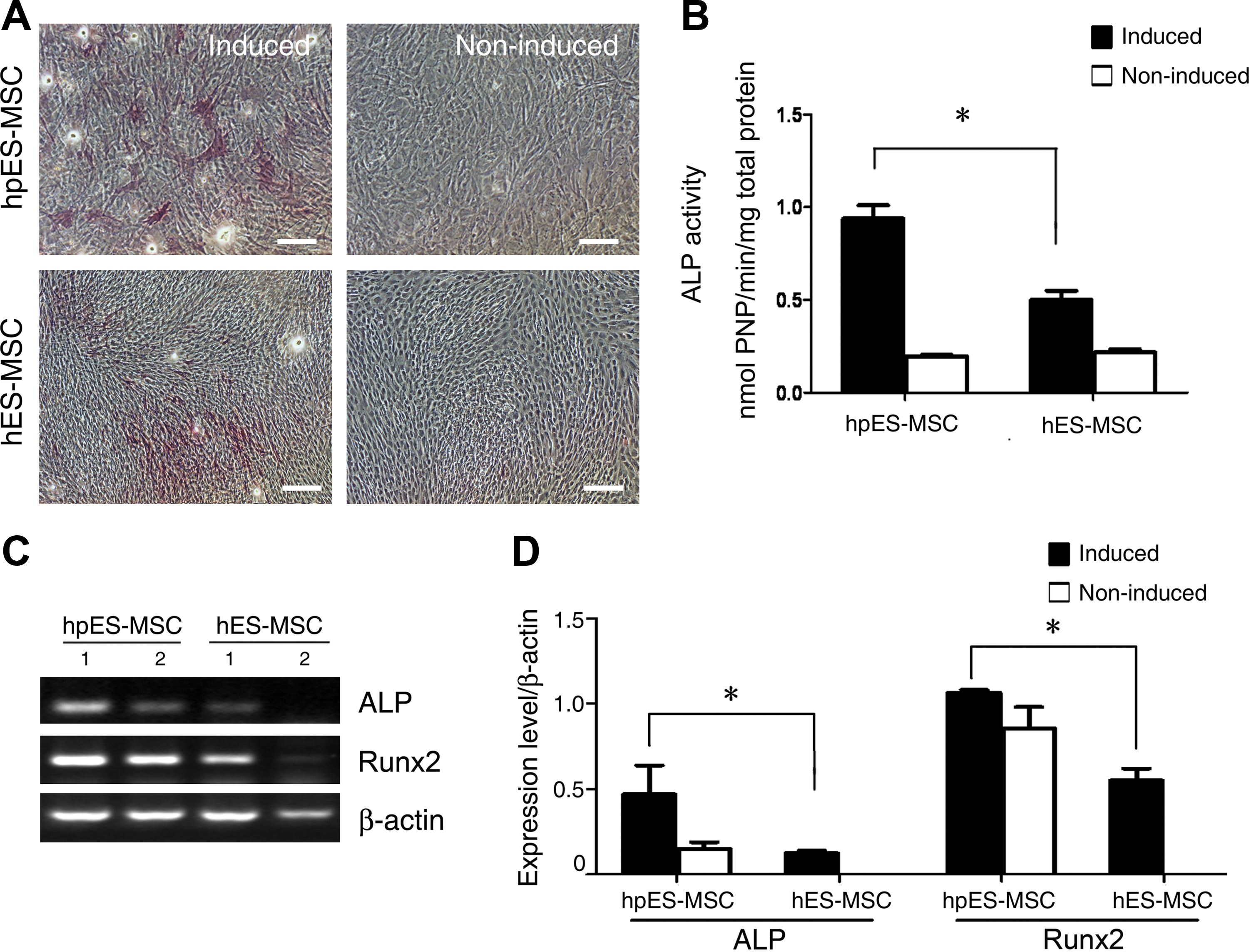
Osteogenic differentiation of mesenchymal-like stem cells derived from hpESCs and hESCs.
The chondrogenic potential of ESC-derived cells was determined by immunohistochemical staining for type II collagen. Around 1% and 1.5% of type II collagen-positive cells were observed in hpESC- and hESC-derived cells, respectively, postinduction. No significant difference was observed between these 2 types of cells (Fig. 6A, B). RT-PCR analysis showed that type II collagen was expressed in uninduced hpESC- and hESC-derived cells. Expression was upregulated following chondrogenic induction in both types of cells (Fig. 6C, D). The cartilage oligomeric matrix protein (COMP) transcript was only detected in the groups that had been subjected to induction, with no significant difference observed between the 2 types of cells (Fig. 6C, D).

Chondrogenic differentiation of mesenchymal-like stem cells derived from hpESCs and hESCs.
Following myogenic induction, cells were incubated with anti-MHC, anti-SMA, and anti-calponin antibodies to assess their myogenic potential. As shown in Fig. 7A, SMA- and calponin-positive cells were observed in both types of cells with or without induction. MHC-positive cells were observed in the induced groups, and a much lower number was observed in the uninduced groups. RT-PCR analysis showed that SMA and caldesmon were expressed in induced and uninduced cells from both types of cells (Fig. 7B, C), indicating that ESC-derived cells can spontaneously differentiate into muscle cells in vitro.

Myogenic differentiation of mesenchymal-like stem cells derived from hpESCs and hESCs.
Discussion
ESCs are a potential source for cell-based therapy because of their unlimited proliferation capacity and pluripotent characteristics. However, differentiation of ESCs toward a certain lineage remains a significant challenge. Several induction and isolation methods have been established for generating MSCs from hESCs [16,27 –29]. In the present study, we utilized the EB differentiation method, which involves hpESCs and hESCs forming EBs, followed by adherent culture of the EBs in the presence of FBS. After several rounds of passaging, a fibroblastic population was achieved, which expressed mesenchymal markers including CD29, CD44, CD105, and CD166, but not the hematopoietic marker CD45 and the endothelial cell markers CD31 and CD34. Additionally, these cells could be further differentiated into adipocytes, osteoblasts, chondrocytes, and myocytes, suggesting that these are mesenchymal-like stem cells.
Sequential differentiation steps are critical for generating MSCs from hpESCs and hESCs. When cells were directly cultured on plates in the presence of FBS but without the EB formation step, cells differentiated but eventually died (data not shown), indicating that the EB formation step is necessary. Generating cells of ectoderm, mesoderm, and endoderm from hpESCs during EB differentiation has been previously demonstrated [2]. The presence of ectoderm, mesoderm, and endoderm markers in early passages confirmed that cells of all 3 germ layers were achieved initially (Supplementary Fig. S2). Only the expression of mesoderm markers were maintained at high levels after P5 (Supplementary Fig. S2), suggesting that mesodermal cells were selectively expanded in culture. This is likely due to the presence of some growth factors in the FBS that favor mesodermal cell growth, such as bone morphogenetic proteins [30,31].
Interestingly, the expression of AFP was sustained in hpESC-derived cells but not in hESC-derived cells at passage 5 (Supplementary Fig. S2). One explanation could be that hpESC-derived cells might maintain at mesendoderm stage, whereas hESC-derived cells have already committed to mesodermal progenitors. Another explanation could be that endodermal cells were contaminated in hpESC-derived population. Further single-cell–based analyses are required to address this question. Compared with the cells derived from hESCs, hpESC-derived MSCs showed similar fibroblastic morphology and cell surface marker expression patterns. However, hpESC-derived MSCs exhibited a lower proliferation capacity at the same passages. Additionally, hpESC-derived MSCs showed higher osteogenic but lower adipogenic differentiation capacity compared with the hESC-derived MSCs in vitro. Whether these phenomena are dependent on the cell line or are a true characteristic of hpESCs remains unclear. It has been shown that epigenetic modifications exist between hESC lines, which could be the reason for the differences between these cell lines [32]. The differences between hpESC-derived MSCs and hESC-derived MSCs need to be further confirmed with other parthenogenetic ESC lines. Few studies have quantitatively analyzed the differentiation efficiency of hES-derived MSCs into adipocytes, osteocytes, chondrocytes, and myocytes. Comparing our data with other reports, adiopgenic, osteogenic, and myogenic differentiations were similar [16,21], but percentages were lower than those reported by Barberi et al. [27]. The major difference is that Barberi et al. conducted cell sorting utilizing CD73, which was able to purify the progenitors. In our study, and in other studies, a pooled population was used, which was possibly contaminated with differentiated fibroblasts. The existence of endoderm marker (in hpESC-derived MSCs) and ectoderm marker (in hESC-derived MSCs) at later passages also indicated that these cells were likely a quasi-pure population of mesenchymal progenitors that contaminated with cells of other germ layers.
Similar to cells derived from hESCs, hpESC-derived MSCs also exhibited high expression levels of HLA-ABC, but not HLA-DR. This pattern is analogous to that observed in MSCs of the bone marrow [33,34]. As parthenogenetic ESCs are homozygous for major histocompatibility alleles, theoretically they have greater immune matching capability [7]. Further studies are being conducted with hpESC-derived MSCs to determine the validity of this hypothesis. The risk of unbalanced expression of the imprinting genes or their possible loss, and false expression profiles in parthenogenetic cells, which may eventually lead to diseases or tumor formation, should be investigated.
Undifferentiated ESCs require growth factors such as bFGF to maintain their pluripotent state. Expansion of ESCs is labor intensive and time consuming. However, hpESC- and hESC-derived MSCs can be expanded for over 30 passages in the presence of serum without specific growth factors. The cells could be dissociated into a single cell suspension by enzymatic digestion during passaging, making it much easier for cell expansion. Further, the cells maintained their multipotent differentiation capacity even after P15. The characteristics of the ESC-derived MSCs determined in this study show their massive potential for tissue engineering and regenerative medicine applications.
In conclusion, this study demonstrated that MSC-like cells could be efficiently generated from hpESCs and hESCs. A large number of multipotent cells could be easily achieved after sequential steps of differentiation. This would provide a stable source of cells for cell-based therapy and could also allow for the investigation of gene imprinting and X chromosome inactivation in the future.
Footnotes
Acknowledgments
This work was supported by the Major State Basic Research Development Program of China (2005CB522700, 2007CB948004, 2011CB964704, 2010CB944804), National Basic Research Program of China (30800231), Shanghai Pu Jiang Foundation (to W.J.Z.), Shanghai Shu Guang Foundation (to W.J.Z.), and Shanghai-Northeast England Science and Technology Cooperation Program (075407072). The authors also appreciate the technical support from Deli Liu, Demin Ying, Lijuan Zong, and Juanjuan Wu.
Author Disclosure Statement
The authors have nothing to disclose and no competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.