
Abstract
Mesenchymal stem cells (MSCs) from adult and neonatal tissues are intensively investigated for their use in regenerative medicine. The purpose of this study was to compare the onset of replicative senescence in MSCs isolated from equine bone marrow (BMSC), adipose tissue (ASC), and umbilical cord tissue (UCMSC). MSC proliferation (cell doubling), senescence-associated β-galactosidase staining, telomere length, Sox-2, and lineage-specific marker expression were assessed for MSCs harvested from tissues of 4 different donors. The results show that before senescence ensued, all cell types proliferated at ∼1 day/cell doubling. BMSCs significantly increased population doubling rate by passage 10 and ceased proliferation after a little >30 total population doublings, whereas UCMSCs and ASCs achieved about 60 to 80 total population doublings. UCMSC and ASCs showed marked β-galactosidase staining after ∼70 population doublings, whereas BMSCs stained positive by ∼30 population doublings. The onset of senescence was associated with a significant reduction in telomere length averaging 10.2 kbp at passage 3 and 4.5 kbp in senescent cultures. MSCs stained intensively for osteonectin at senescence compared with earlier passages, whereas vimentin and low levels of smooth muscle actin were consistently expressed. Sox-2 gene expression was consistently noted in all 3 MSC types. In conclusion, equine BMSCs appear to senesce much earlier than ASCs and UCMSCs. These results demonstrate the limited passage numbers of subcultured BMSCs available for use in research and tissue engineering and suggest that adipose tissue and umbilical cord tissue may be preferable for tissue banking purposes.
Introduction
I
MSCs from different tissue sources appear to have variable differentiation potential in vitro [4,5,11,12] and probable advantages in their clinical applications. BMSCs or adipose-derived nucleated cell fractions cause histological improvement in experimental tendon lesions [13,14] and improved clinical outcome for tendon and ligament injuries [15,16]. Therefore, MSCs derived from each tissue source need to be individually evaluated and targeted for potential future therapeutic strategies in tissue repair. A common challenge is that cell-based approaches, and especially cell seeding of scaffolding materials, require large numbers of MSCs [17]. MSC expansion is associated with extensive subculturing and cell passage. Depending on the tissue source, primary MSC numbers may be limited to only a few thousand cells, such as in bone marrow (BM), compared with several hundreds of thousands of cells such as in adipose (AT) or muscle tissue [18,19].
The effects of long-term in vitro expansion on MSC molecular changes are unknown. However, subculturing will eventually lead to cellular aging and increase the potential of genetic and epigenetic cellular alterations and the risk of spontaneous cell transformation and cancer [20]. Early MSC senescence during subculturing may limit MSC expansion or require alternative methods of enhancing in vitro cell proliferation.
MSCs are pluripotent cells capable of extensive replication and maintenance of their pluripotentiality in cell culture [21]. However, compared with embryonic stem cells, adult MSCs have a finite ability to self renew. Cultured human fibroblasts enter senescence after ∼50 cell doublings [22], coined the Hayflick limit [23]. Similarly, human BMSCs cease growth at ∼40 to 50 cell doublings [24]. Equine BMSCs have been cultured to 30 population doublings without apparent changes in cell doubling time [18]. Similarly, equine ASCs have been cultured by the same investigators to 28 total population doublings (passage 10) [19] but were recently reported to have undergone replicative senescence by passage 5 [25]. Little is known about equine UCMSCs. However, they have been reported to proliferate up to passage 12 without apparent signs of senescence [1]. The long-term in vitro replicative potential and senescence characteristics of equine MSCs require further investigation.
Cellular senescence is associated with a number of characteristic markers. Changes in cellular morphology to a flat and enlarged appearance with granular deposits from intracellular debris, followed by permanent growth arrest, are the most striking signs of cellular aging [26]. Cell replication is achieved through mitosis, a process during which the double-stranded DNA is replicated by DNA polymerase. This enzyme is not able to fully replicate newly synthesized DNA at the chromosomal termini or telomeres. A marker of senescence is progressive telomere shortening during each mitotic phase. In highly proliferative cells, such as embryonic stem cells, the enzyme telomerase maintains telomere length [27]. However, telomerase is not expressed in the majority of somatic cells due to transcriptional silencing. Telomerase activity will also eventually cease in the aging cell.
During replicative senescence in mammalian cells, an endogenous lysosomal β-galactosidase (SA-β-Gal) is over-expressed and will accumulate within the aging cell. The expression of p16Ink4a in senescent cultures correlates positively with the presence of SA-β-Gal and negatively with proliferation marker Ki67 [28]. The presence of SA-β-gal activity allows visualization of aging cell populations in situ as well as in vitro, similarly to the lipid peroxidation product, lipofuscin [26]. Cellular aging has also been associated with changes in gene expression of markers associated with extracellular matrix such as osteonectin, fibronectin, and a1-procollagen and cell protection [29,30].
Cellular pluripotency and self-renewal are regulated by endogenous embryonic genes such as SRY-related HMG box protein (Sox-2), Nanog, and OCT4 [31,32]. Adult MSCs also appear to express these genes [33,34], but only little is known about their expression in equine MSCs. Equine BMSCs and umbilical cord blood MSCs appear to express Sox-2, but it is unknown whether MSCs lose their expression during the onset of replicative senescence [3,35].
To date, there are no comparative data on the senescence characteristics of equine MSCs derived from adult and neonatal tissues. The importance of understanding the onset of senescence in cultured MSCs is apparent when considering the extensive use of these cells for regenerative medicine techniques requiring millions of MSCs for intra-lesional therapies and seeding of scaffolding materials. The purpose of this study was to compare the onset of senescence in cultured MSCs from equine BM, AT, and umbilical cord tissue (UCT).
Materials and Methods
Research animals and materials
MSCs used for this study were collected from adult horses (BM and AT, n=4) and foals (UCT, n=4) according to approved animal care and use protocols of the University of California, Davis. All chemical reagents were obtained from Sigma Chemical Co. (St. Louis, MO), Invitrogen (Grand Island, NY), or Fisher Scientific International Inc. (Hampton, NH) unless otherwise noted.
Tissue collection
Similarly to a previous description [18], BM was collected from 4 adult horses, ranging in age from 2 to 6 years (3.5±1.7 years), using a BM aspiration needle (Tyco Healthcare, Mansfield, MA) and heparinized syringes (APP Pharmaceuticals, Schaumburg, IL) at 1,000 IU heparin/20 mL BM aspirate. AT was harvested from 4 horses ranging in age from 2 to 9 years (6±2.9 years) exactly as previously described [19]. BM and AT were transported to the laboratory for immediate processing.
UCT was collected at foaling and processed within 24 h as previously described [7]. Briefly, small sections of UCT were collected at the time of birth and rinsed with tap water to remove gross contamination. The UCT was further rinsed with 0.05% chlorhexidine (Nolvasan, Fort Dodge Animal Health, Fort Dodge, IA). The samples were shipped to the Regenerative Medicine Laboratory (William R. Pritchard Veterinary Medical Teaching Hospital, University of California, Davis) in a 4°C preconditioned temperature-controlled Thermal Isolation Chamber (Minnesota Thermal Science LLC, Plymouth, MN), which contained a temperature-tracking device (Global Sensor Log Tag, Mt. Holly, NC).
Isolation and culture of MSCs
BM was diluted at a 1:1 ratio in calcium and magnesium-free Dulbecco's phosphate buffered saline (DPBS), and the BM nucleated cells were obtained using a commercial centrifugation device AXP™ AutoXpress™ (Thermogenesis, Rancho Cordova, CA). Nucleated cells were removed and washed in DPBS. The pellet was resuspended in stromal culture medium containing α-MEM (Invitrogen, Carlsbad, CA) supplemented with 10% characterized fetal bovine serum (Hyclone, Logan, UT) and 1% penicillin/streptomycin. Primary nucleated cells were incubated at 37°C with 5% CO2 and 21% O2, and BMSCs were passaged at 70% confluence, by incubating for ≤5 min with 0.05% trypsin-EDTA and cryopreserved for future experiments.
ASCs were isolated from AT via collagenase digestion as previously described [19]. The stromal–vascular fraction pellet containing the nucleated cell portion of the AT harvest was cultured in stromal medium, and ASCs were passaged at 70% confluence for cryopreservation.
For the isolation of UCMSCs, amnion and large blood vessels were removed from the UCT; and the tissues were processed for UCMSC isolation exactly as described [4]. The harvested primary nucleated cells were cultured in stromal medium, and UCMSCs were passaged at 70% confluence for cryopreservation.
Cryopreservation
All cells were cryopreserved at ∼1×106 cells/mL in α-MEM, 10% fetal bovine serum, and 10% DMSO (Sigma-Aldrich Corp., St. Louis, MO). Cryovials containing the MSCs were placed in a 5100 Cryo 1°C Freezing Container (Wessington Cryogenics, Tyne and Wear, United Kingdom) for 24 h at −80°C before transfer to liquid nitrogen.
Cell doubling assays
MSCs were thawed from early passage cells (P2 for ASC and BMSC, P3 for UCMSC). Thawed cells were allowed to grow for one passage before they were subcultured and incorporated into the study. ASC and BMSC cell lines were studied from P3 to senescence, whereas UCMSC lines were studied from P4 to senescence. All passages and total population doublings are listed in Tables 1 –3. Cells were plated at a density of 5,000 cells/cm2 in 2-T25 flasks for cell proliferation studies, and in multiple larger flasks for MSC collections at various passages. MSCs were passed when they reached 70% confluence, counted at each passage using a Coulter AcT-Diff Cell Counter (Coulter, Hialeah, FL), and cell doubling times were calculated as previously reported [18].
For P11 only 2 donors remained. Population doubling time (PDT, days/PD) increased significantly after about 27 PDs (P<0.01). The data represent the arithmetic mean±standard error of the mean (SEM).
Cum., cumulative.
For P21-22, only 2 donors remained. Population doubling time (days/PD) increased significantly after about 54 PDs (P<0.01). The data represent the arithmetic mean±SEM.
For P23, only 2 donors remained. Population doubling time (days/PD) increased significantly after about 78 PDs (P<0.0001). The data represent the arithmetic mean±SEM.
Telomere length assay
MSCs were collected at multiple passages; and 5×106 cells/sample were washed, pelleted, and frozen at −80°C. Genomic DNA was isolated from frozen cell pellets (Qiagen DNeasy Kit, Valencia, CA) and quantitated on a NanoDrop spectrophotometer (NanoDrop products, Wilmington, DE). Telomere length was measured using the TeloTAGGG Telomere Length Assay (Roche Applied Science, Indianapolis, IN). ASC telomere lengths were measured at early passages (4–9 PDs), mid-passages (22 PDs), mid to late passages (51–55 PDs), and at senescence (73–78 PDs). UCMSC telomere lengths were measured at early passages (3–7 PDs), mid-passages (14 PDs), mid to late passages (38–41 PDs), and at senescence (54–79 PDs). BMSC telomere lengths were measured at initial passage (4 PDs), early-mid passage (11 PDs), mid-passage (18 PDs), and at senescence (27–33 PDs). Genomic DNA (2.5 μg) from each sample was digested using 1 μL of restriction enzymes (Hinf1, Rsa1), then electrophoresed on a 0.8% agarose gel, and transferred to positively charged nylon membranes (Hoffmann-La Roche Ltd., Basel, Switzerland) by overnight capillary transfer using 20×standard high salt buffer (3M NaCl, 0.3M sodium citrate, pH 7.0). DNA was cross-linked to the membranes that were then hybridized with a telomere-specific, digoxigenin-labeled probe. Blots were then incubated with an anti-digoxigenin-labeled alkaline phophatase-coupled antibody that was detected by chemiluminscence. Blots were exposed to HyBlotCL autoradiography film (Denville Scientific, Metuchan, NJ) that was developed and then scanned for analysis. Scanned images were analyzed using AlphaEaseFC software (Alpha Innotech, San Leandro, CA). A grid divided into 32 equal rectangles was overlaid on each lane, and spot densitometry was performed. Mean telomere length for each lane was then calculated according to the manufacturer's instructions. Changes in telomere length for each cell line were calculated relative to that of initial passage measured.
SA-β-Gal staining
A senescence β-galactosidase staining kit (Cell Signaling Technology, Danvers, MA) was used according to the manufacturer's instructions for SA-β-Gal staining MSCs at various early, mid-, and late passages in culture (as described for telomere assay). The assay was scaled down to 24-well plates, and MSCs were seeded in replicates of 4 wells at 5,000 cells/cm2 grown until 70% confluent. Wells were rinsed twice with DPBS containing Ca2+ and Mg2+, then fixed, and stained. Cells were examined for development of blue color under an inverted microscope (Leica DMI6000B Microsystems Inc., Bannockburn, IL), and 20× brightfield images were captured with a digital camera (Leica DFC340FX Microsystems Inc., Bannockburn, IL) using Image-Pro MDA 6.3 software (Media Cybernetics, Bethesda, MD).
Immunocytochemistry
Cells were collected for immunocytochemistry at early passage (3–4 PDs), mid-passage (11–22 PDs), and at senescence (27–33 PDs for BMSCs, 54–80 PDs for ASCs and UCMSCs). 1×105 MSCs were pelleted onto microscope slides using a Cytospin 4 (ThermoShandon, Pittsburgh, PA). Slides were air-dried, acetone-permeabilized, and stored with dessicant at −20°C before staining. Immunocytochemistry was performed with a routine streptavidin-biotin detection system as described elsewhere [8,36]. Cells were stained for vimentin (Dako Corporation, Carpinteria, CA; mAb clone 384; 1:100), smooth muscle actin (Biogenex Corporation, San Ramon, CA; mAb clone 1A4; 1:200), and osteonectin (Biogenex Corporation; mAb clone OST1; 1:300). A frozen section of equine tissue was used as a positive control sample; the section contained the specific antigen and was run in parallel with the MSC samples. Omission of the primary antibody and substitution of normal goat serum were used as a negative control sample. Slides were microscopically examined (Olympus CH30, Olympus, Center Valley, PA), and the number of MSCs stained and the staining intensity for each sample was assessed blindly by one author (D.L.B.).
RNA extraction and quantitative polymerase chain reaction
MSCs (2×106) were lysed (RLT buffer; Qiagen), snap frozen in liquid N2, and stored at −80°C. Total RNA was collected according to the manufacturer's instructions (RNeasy Mini kit; Qiagen) including on-column digestion of genomic DNA. RNA quality was assessed by the ratio of absorbance at 260 and 280 nm. Total RNA (1 μg) was reverse-transcribed with a QuantiTect Reverse Transcription kit (Qiagen). Quantitative reverse transcription with the polymerase chain reaction (PCR) was performed by using the TaqMan Universal PCR Master Mix (Applied Biosystems, Foster City, CA) on a Mastercycler realplex2 (Eppendorf, Hauppauge, NY); proprietary primers/probes for the SRY-related HMG box protein, Sox-2 (assay ID: Ec03818470_s1) were purchased from Applied Biosystems (Carlsbad, CA). Amplification conditions were 50°C for 2 min, 95°C for 10 min, followed by 40 cycles at 95°C for 15 s, and 60°C for 30 s. Quantitative PCR results were first normalized to 18s ribosomal RNA transcript level to yield ΔCt and expressed as 2−ΔCt.
Statistical analysis
Data were assessed for normality with the Shapiro Wilk test. Analysis of variance was performed by using Proc GLM (SAS 9.1.2; SAS Institute, Cary, NC) with a Tukey's test for post-hoc comparisons. A Student's t-test was used for pair-wise comparisons of the least square means to examine interaction effects. The immunocytochemistry data were analyzed using ranked ANOVA analysis. Statistical analyses and the type I error was maintained at <0.05 for all comparisons. Data are reported as arithmetic means±SEM.
Results
MSC proliferation
MSC proliferation was assessed from passage 3 (BMSCs and ASCs) and passage 4 (UCMSCs) onward. MSCs were considered senescent, and cultures were discontinued when their cell doubling time increased ≥3 to 5-fold compared with that of early passages. Cell doubling times (days/cell doubling) of BMSCs, ASCs, and UCMSCs during early passages (4–26 PDs) averaged 1.4±0.1, 0.9±0.01, and 1.1±0.02, respectively (Fig. 1). Cell doubling times increased significantly between the early passages (4–9 PDs) and P10 (27 PDs) for BMSCs (P=0.022), P20 (73 PDs) for ASCs (P<0.0001), and P18 (49 PDs) for UCMSCs (P=0.0256) (Fig. 1). Cell doubling times thereafter were either erratic or cell proliferation ceased altogether. BMSCs stopped proliferating after 27–33 PDs (Table 1), whereas ASCs and UCMSCs stopped proliferating after 68–80 PDs (Table 3) and 54–79 PDs (Table 2), respectively. BMSCs achieved 33 total population doublings between P3–P12, which was significantly lower (P<0.0001) than those from ASCs (78 total population doublings between P3–P22) and UCMSCs (58±8 total population doublings between P4 and P25). ASCs underwent significantly more total cell doublings than did UCMSCs (P=0.0115).
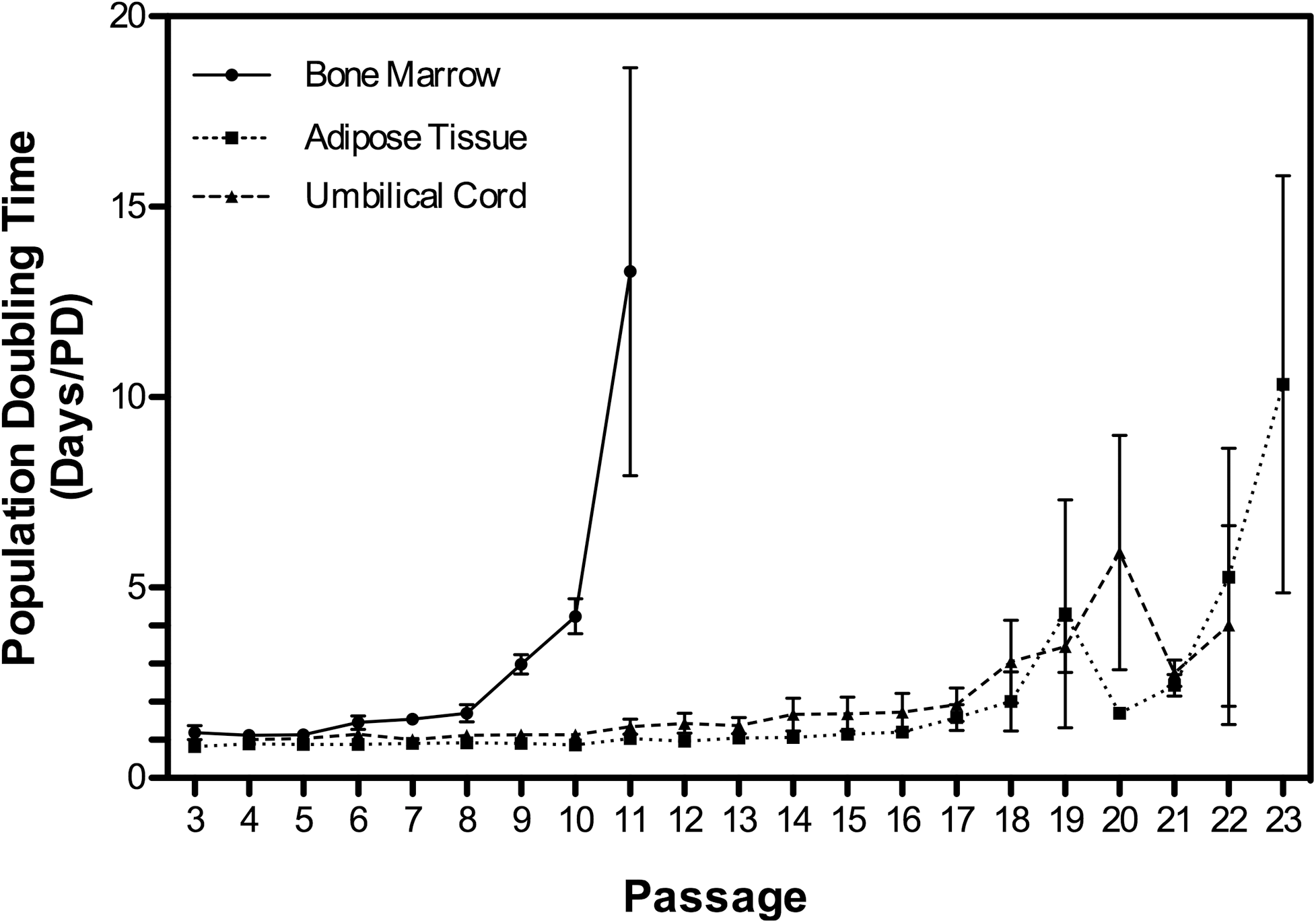
Cell doubling times of equine BMSCs, ASCs, and UCMSCs increased significantly (P<0.05) between early and senescent passages for all 3 cell types. BMSCs showed significantly increased PD times (P<0.01) after 27 PDs (P10), whereas population doubling increased (P<0.01) in UCMSCs and ASCs after 54 PDs (P20) and 73 PDs (P22), respectively. All data are presented as the arithmetic mean±SEM. BMSC, bone marrow-derived mesenchymal stem cell; UCMSC, umbilical cord tissue-derived mesenchymal stem cells; ASC, adipose tissue-derived mesenchymal stem cells.
SA-β-gal staining
Senescent cells were identified by a flat and hypertrophic phenotype (Fig. 4B, D). MSCs in senescent cultures were often sparser in cell numbers and were less adherent to plastic. BMSCs showed increased SA-β-gal staining as early as after 22 PDs but showed consistent marked accumulation after 27 PDs (Fig. 2). UCMSC SA-β-gal staining was detected in some cells after 41 PDs. However, SA-β-gal staining was noted in all ASC cultures at 73 to 80 PDs and all UCMSC cultures at 54 to 79 PDs (Fig. 2).

Senescence-associated β-galactosidase activity in aging BMSC, ASC, and UCMSC populations. SA-β-gal activity was consistently noted in ASCs by P20, in USMSCs by P21, and in BMSCs by P10. The figure is representative of cells grown from one donor for each cell type.
Telomere length
Telomere length consistently shortened in all MSCs with increasing passage number (Fig. 3A). The number of telomere base pairs from MSCs in early passages was significantly higher compared with late passages for ASCs, and a trend was also found for UCMSCs (Fig. 3B). The average base pair length among all cell types at the senescent stages was 5,393±326 bp, and there was no significant difference between cell types (Table 4). The percent decrease in base pairs between early and senescent passages was 32% for BMSCs, 54% for ASCs, and 45% for UCMSCs.
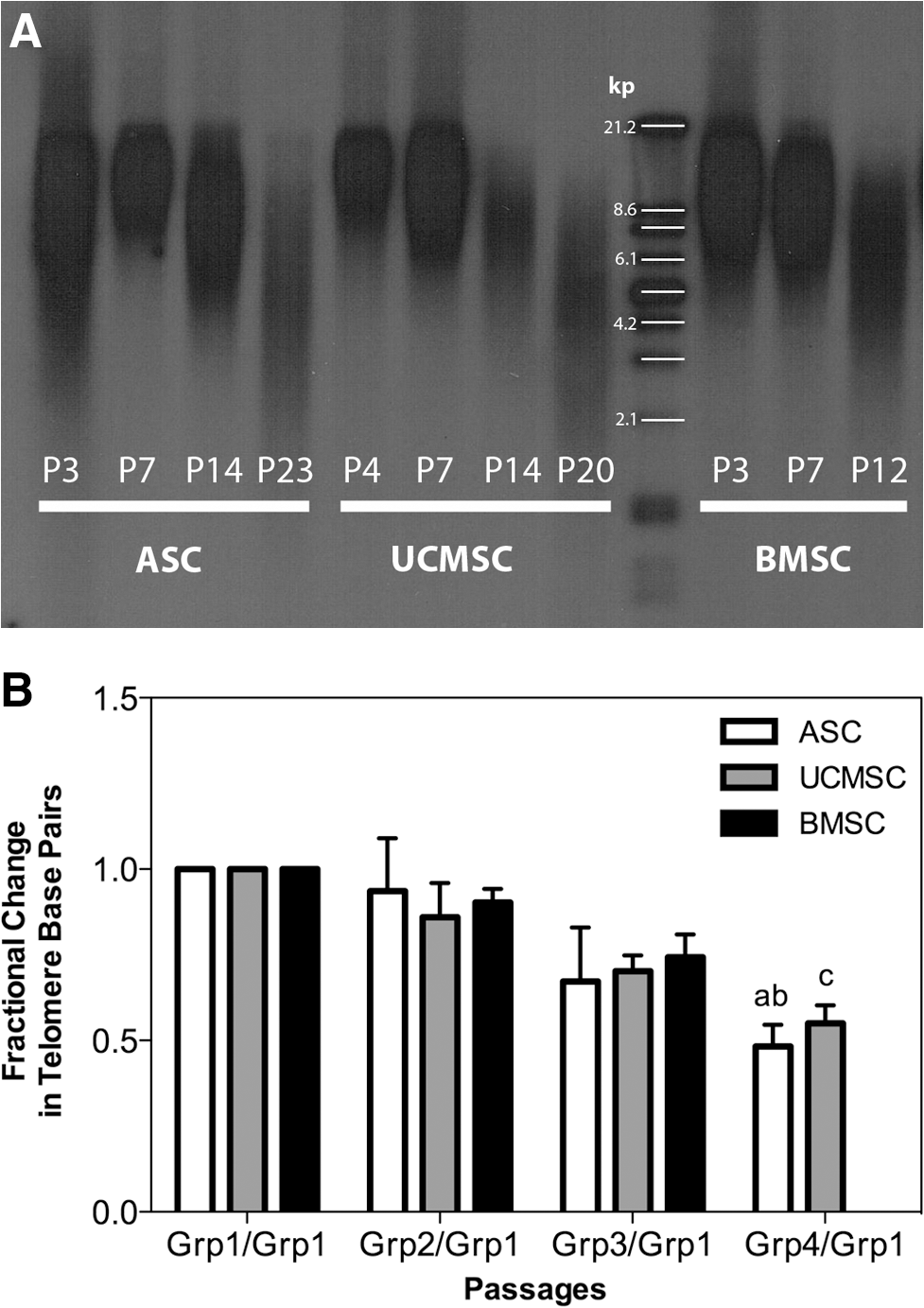
Telomere length analysis in aging BMSC, ASC, and UCMSC populations. Panel (A) shows a representative Southern blot analysis of ASCs, UCMSCs, and BMSCs from one donor. Panel (B) represents the quantitative analysis of telomere length from all 3 cell types derived from 3 to 4 donors analyzed by groups. Groups assigned for BMSCs were Grp1 (P3), Grp2 (P7), and Grp3 (P10); for ASCs were Grp1 (P3), Grp2 (P7), Grp3 (P14; P15), and Grp4 (P20; P22; P23); and for UCMSCs were Grp1 (P3; P4), Grp2 (P7), Grp3 (P14; P15), and Grp4 (P20; P22; P26). Data are represented as the fractional change of telomere base pairs, and the letters “a” (P=0. 0.015) and “b” (P=0.0.44) indicate the significant decrease in telomere base pairs of ASCs relative to Grp1/Grp1 and Grp2/Grp1, respectively. A decreasing trend (“c” P=0.0764) relative to Grp1/Grp1 was also found for UCMSCs. All data are presented as the arithmetic mean±SEM.
The data represent the arithmetic mean±SEM.
BMSC, bone marrow-derived mesenchymal stem cell; UCMSC, umbilical cord tissue-derived mesenchymal stem cells; ASC, adipose tissue-derived mesenchymal stem cells.
Replicative senescence in equine BMSCs, ASCs, and UCMSCs was evident at a telomere length threshold averaging ∼5.2 to 5.5 kb. The range of telomere length shortening for these 3 cell types was ∼247 to 374 bp per passage. During the first 10 passages, MSCs from BM, AT, and UCT underwent 3.2 to 4.3 PDs per passage, which translates to a loss of about 70 to 116 bp for every cell division.
Immunocytochemistry
Osteonectin staining intensity increased significantly at senescence in all cell types (Fig. 4A–D). Osteonectin was also expressed at significantly higher levels in BMSCs at the early passages (P3–P5) compared with ASCs and UCMSCs (Fig. 4E). No significant differences were seen in smooth muscle actin (Fig. 4F) and vimentin (Fig. 4G) expression across all passages in all cell types.
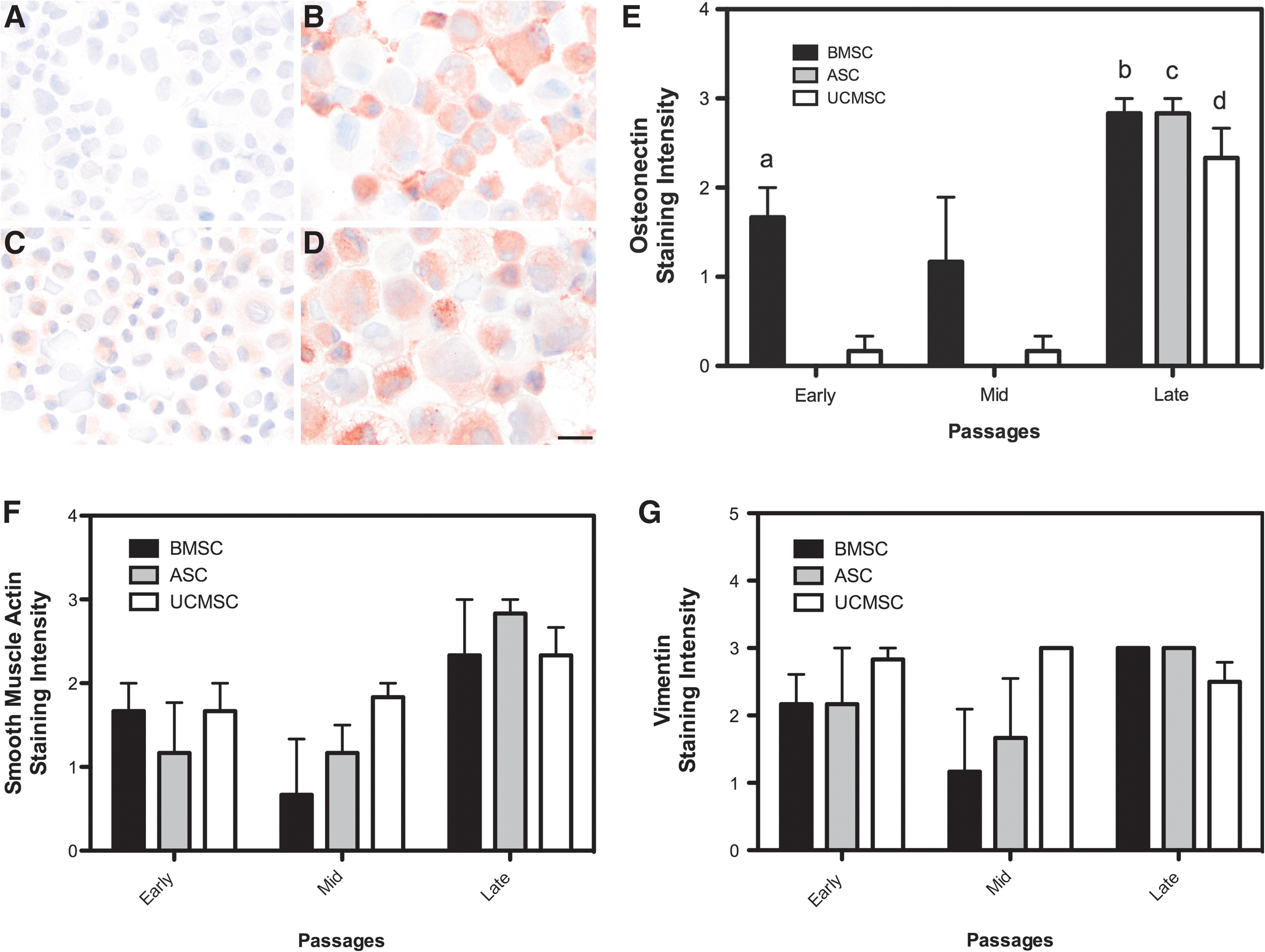
Immunocytochemistry of osteonectin, smooth muscle actin, and vimentin in aging BMSC, ASC, and UCMSC populations.
Sox2 expression
Sox2 was expressed in all cell types but although there was a trend for higher expression levels in ASCs compared with BMSCs and UCMSCs, these differences were not significant (Fig. 5).
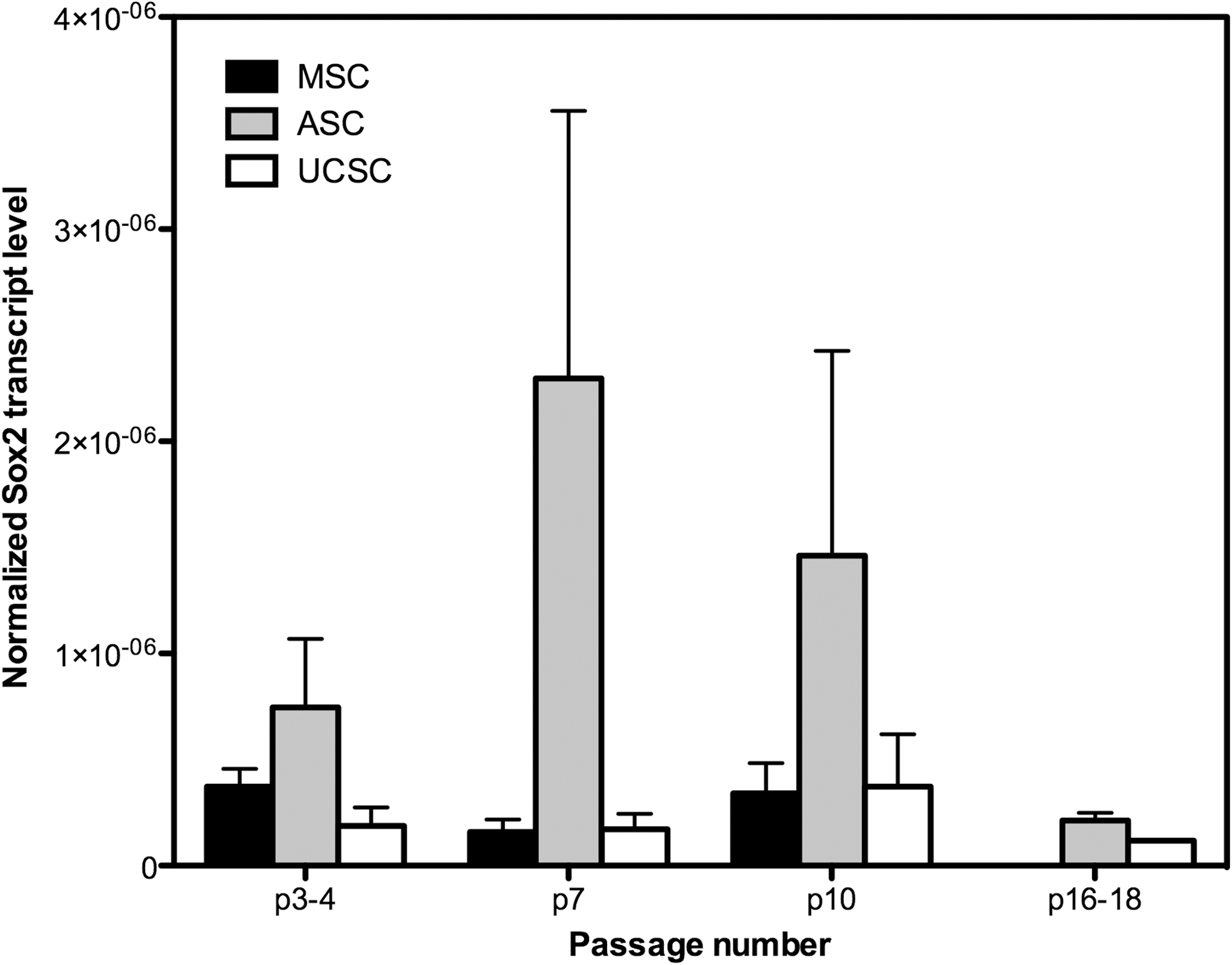
Sox-2 expression in aging BMSC, ASC, and UCMSC populations. Sox-2 was expressed in all cultures, but no significant differences between cell types were noted. All data are presented as the arithmetic mean±SEM.
Discussion
The present study demonstrates that all aging equine MSC cultures from BM, AT, and UCT exhibited characteristics of proliferation arrest, SA-β-gal+ cell accumulation, and telomere shortening as well as an increase in osteonectin expression. Interestingly, there was a significant difference in the onset of senescence between BMSCs, ASCs, and UCMSCs.
An important aspect of regenerative medicine is the need for replication of large cell numbers for the purpose of seeding scaffolding materials. In horses, MSCs are currently used mostly in the form of cell-based therapy for tendon and ligament injuries as well as for joint therapy. There are no published data, however, that suggest ideal cell numbers to optimize tissue healing, as endogenous stimulation of tissue repair, immune-modulatory effects, angiogenesis, antiscarring, and other potential roles of MSCs in tissue repair [37] have not yet been described and quantitated for horses. As such, MSC-based therapies have employed various MSC doses, usually tens of millions of cells, for large tendon and ligament lesions. However, BMSCs are present at very low numbers in BM and require extensive subculturing, which raises the concerns of cell senescence and its potential effect on tumorigenesis.
Senescent human fibroblasts have been shown to stimulate malignant epithelial cell proliferation due to putative factor secretion from the senescent cells [38]. Also, murine BMSCs (P6) injected into nude or SCID mice after long-term confluent cultures formed soft tissue sarcomas and had chromosomal abnormalities [39]. Reports on human AT derived MSCs have shown that senescent cells have a normal karyotype and may be free of genetic abnormalities [40], and human BMSCs do not appear to be prone to tumor formation [41]. Morphological signs of chromosomal instability have been shown in human fibroblasts such as chromosomal loss and fusion as well as anaphase bridges [42]. Therefore, the frequency of chromosomal instability may depend on the cell type and species.
The senescent cell phenotype is characterized by growth arrest in the G1 cell cycle phase and an increase of SA-β-Gal expression [43 –45]. Most MSCs in this study showed variable but very low expression of SA-β-Gal at the early passages. However, once senescence occurred, all cultures across different donors dramatically increased SA-β-Gal expression within 1–2 passages. Equine BMSCs showed early conversion to a senescent phenotype within ∼30 population doublings compared with those from ASCs (78 PDs) and UCMSCs (54 PDs). However, since the experiments were initiated at P3 and P4, the total population doubling numbers for MSCs (the initial 10 to 15 population doublings during the early passages) were not accounted for. The consistent early onset in replicative senescence around 30 PDs after 197 days in culture has also been described for human BMSCs [46], although human cells appear to exhibit much slower population doubling times compared with equine BMSCs, which only required 60 days for 30 PDs. Baxter and coworkers [46] reported that some BMSC lines from younger donors achieved higher population doubling numbers; but in the present study, the limited donor numbers will not allow conclusions on potential age-related differences in population doubling parameters.
Human BMSCs have also shown rapid 4-fold increase in SA-β-Gal- positive cells from ∼20% at passage 7 to ∼80% at passage 9 in senescent MSC cultures [47]. Also, molecular changes between passage 3 and 12 showed consistently decreased expression of CD13, CD29, CD44, CD73, CD90, CD105, CD146, and CD166 in senescent MSC cultures [47]. Accumulation of another marker associated with senescence in human BMSCs is p16INK4a , which under physiological conditions is down regulated by the polycomb group (PcG) protein BMI1, which affects self-renewal and cell senescence [28].
Senescent equine MSCs became flat and hypertrophic and dissociated more readily from plastic surfaces. Other senescent characteristics include changes in cellular morphology due to accumulation of cell debris, excess actin fiber expression, and increased autofluorescence due to lipofuscin accumulation [26]. The increase in cell size and granularity has been well documented for human MSCs [26,47]. Alterations in cellular morphology were also noted in this study.
BMSC yield and proliferation rate may depend on isolation technique [48]. BMSCs used in this study were isolated using a commercial centrifugation technique, and proliferation was similar to that described for BMSCs isolated over a Ficoll gradient [18]. A recent study by Braun and coworkers [25] reported senescence in ASCs as early as passage 5 and markedly slower cell doubling rates. The potential differences in methodology in this study due to culture media or other conditions is unknown but illustrates the importance of precise method descriptions and raises the question whether optimization and standardization of cell culture conditions specific to the various MSC types may be required.
Human BMSCs appear to tolerate freezing in liquid nitrogen level in media containing 10% DMSO without affecting their proliferative or differentiation capacity [49]. Proliferation and differentiation also appear to be preserved after long-term cryo-preservation in equine peripheral blood MSCs [50], canine ASCs [51], and humans ASCs [52]. Consistent with these reports, MSCs used in this work had been cryopreserved in media containing 10% DMSO and have shown good proliferative potential to high passages; and, therefore, neither isolation technique nor cryopreservation was responsible for the early BMSC senescence.
Telomere length thresholds averaged ∼5.2 to 5.5 kb for equine BMSCs, ASCs, and UCMSCs, consistent with reported kb values of 5.8 to 10.5 in human MSCs [24,46]. The loss of ∼70 to 116 bp for every cell division during the first 10 passages from BMSCs, ASCs, and UCMSCs is consistent with other nonstem cells (30–120 bp/cell doubling) [53] and those reported by others for human MSCs, chondrocytes [54 –56], and fibroblasts [57].
Cytoskeletal markers, such as vimentin for mesenchymal cells and smooth muscle actin and osteonectin, representative of differentiated cell lineages, were used in this study to show potential changes in differentiation during subculturing and the onset of senescence. The expression of vimentin and smooth muscle actin did not change in senescent cultures, unlike those recently reported for equine umbilical cord blood MSCs [8]. Umbilical cord blood derived MSCs did not express vimentin in early passages, but >80% of senescent umbilical blood-derived MSCs expressed vimentin and >50% expressed smooth muscle actin compared with MSCs of the early passages [8].
The increase in immunocytochemical expression of osteonectin at late passages, corresponding with upregulation of SA-β-Gal, was similar for MSCs from all 3 tissue types and consistent with Schuh's work [8], in which >50% of senescent MSC stained for osteonectin compared with early passage cells. These data suggest that osteonectin may be upregulated during in vitro senescence, which has also been reported for osteonectin mRNA expression in human periodontal ligament cells during both in vitro and in vivo senescence [58]. Osteonectin, also known as secreted protein, acidic and rich in cysteine (SPARC), modulates in vitro cell migration, proliferation, and synthesis of extracellular matrices [59]. Studies on SPARC-null mice have shown distinct phenotypical patterns during aging of these mice, which include osteopenia, increased skin laxity, and increased fat stores [60]. Therefore, it is believed that SPARC affects cell differentiation and homeostasis of normal tissues that are injured, stressed, or aging [61]. However, the precise cause for the increase in SPARC expression in this study around the time of senescence remains unknown. Further investigation is required to determine whether SPARC is expressed because a proportion of aging cells may undergo spontaneous osteogenic differentiation or whether it is associated with cell senescence through other pathways.
SRY-related HMG box protein (Sox-2) belongs to a number of endogenous genes such as Nanog and OCT4, which are essential for cellular pluripotency and self-renewal, and is, therefore, an important transcription factor during organogenesis. So far, only a few studies have reported the expression of Sox-2 in equine BMSCs and umbilical cord blood MSCs [3,35]. We determined Sox-2 gene expression to determine whether MSCs lose their expression of Sox-2 during the onset of senescence, thus reducing their pluripotency. Our data suggest that Sox-2 is consistently expressed well into senescence in all 3 MSC types.
Further work is required to assess changes in the differentiation potential of BMSCs, ASCs, and UCMSCs at the higher passages. It has been described that senescence cells will lose their differentiation potential. The present work, however, suggests that equine BMSCs appear to enter senescence as early as passage 8. So far, there are no reports that have examined the ability of equine MSCs to undergo osteogenesis, adipogenesis, or chondrogenesis after 8 passages. BMSCs have been reported to reliably undergo osteogenic [18,19], adipogenic [18,19], and chondrogenic differentiation [25] at passage 4. ASCs maintain their tripotential [62] by passage 8 or 9, and umbilical cord matrix cells are reportedly tripotential at passage 4 [2]. It has been reported that differentiation potential will cease as well but more recently, it has been shown that senescent cells are still capable of differentiation.
In summary, the present study shows that equine BMSCs become senescent at much earlier passages during subculturing compared with ASCs and UCMSCs. Similar to other species, senescent equine MSCs show decreased proliferation, alterations in cellular morphology, telomere shortening, and accumulation of SA-β-Gal. These data provide a benchmark, suggesting that BMSCs beyond passage 6 or 7 should be avoided for research and clinical purposes.
Footnotes
Acknowledgments
The authors would like to acknowledge Dick and Carolyn Randall who made this work possible through private funding to the UC Davis Center for Equine Health, which supports the Stem Cell Regenerative Medicine Group.
Author Disclosure Statement
No competing financial interests exist for any of the authors.