
Abstract
Mesenchymal stem cells (MSCs) are multipotent progenitor cells exerting immunomodulatory effects on cells of the innate and adaptive immune system. It has been shown that an inflammatory milieu is required for the activation of MSC-mediated immunomodulation, and interferon-γ (IFN-γ) plays an important role in this process. We determined the influence of IFN-γ on human adipose-derived stem cells (ASCs) and human amniotic mesenchymal stromal cells (hAMSCs). We further evaluated the effect of MSCs on stimulated T-cells and peripheral blood mononuclear cells (PBMCs) in a cell-contact independent setting. On IFN-γ treatment, ASCs and hAMSCs possessed significantly higher antiproliferative properties and showed surface characteristics of nonprofessional antigen presenting cells (HLA-DR+CD40med+CD54high) with a possible regulatory phenotype (PD-L1+PD-L2+). The effect of ASCs and hAMSCs on cytokine secretion and T-cell activation was dependent on stimulation method and cellular context. Although ASCs and hAMSCs highly inhibited cytokine secretion of stimulated PBMCs, this was not observed in the case of purified T-cells. The presence of ASCs even favored the secretion of pro-inflammatory cytokines including IFN-γ by T-cells, although T-cell proliferation was efficiently inhibited. Further, ASCs enhanced the number of CD69+ T-cells independent of the stimuli and cellular context. Interestingly, ASCs significantly suppressed CD25 expression on phytohemagglutinin stimulated PBMCs but had no effect on αCD3/αCD28 stimulated cells. Depending on the stimulation method and cellular context, immune cells create a specific cytokine milieu in vitro, thus differently influencing MSCs and, in turn, affecting their action on immune cells.
Introduction
M
Great effort has been put into elucidating the mechanisms associated with immunomodulatory properties of MSCs. Different molecules and signaling pathways have been proposed to play a role, depending on the species and tissue from which MSCs were isolated, the type of immune cells used in cocultures with MSCs, and the coculture setting employed. So far, several soluble factors have been implicated in the immunomodulatory mechanism exerted by human MSCs including but not limited to prostaglandin E2 (PGE2) [25,27 –29], indolamine-2,3-dioxygenase (IDO) [30 –33], leukemia inhibitory factor (LIF) [34,35], and transforming growth factor beta (TGF-β) [21,25,36]. Although human BM-MSCs constitutively secrete a number of soluble factors [32,37] potentially influencing an immune response, it has been found that supernatants from MSC mono-cultures do not suppress proliferation of stimulated peripheral blood mononuclear cells (PBMCs) [36]. However, conditioned supernatants from direct and indirect PBMC/MSC cocultures had an antiproliferative effect on mixed lymphocyte reactions (MLR), which was caused by monocyte-secreted IL-1β [36]. Thus, MSCs require activation before gaining their full immunosuppressive potential. In this context, interferon-γ (IFN-γ) has also been assigned an important role in MSC-mediated immunosuppression. Presence of IFN-γ induces expression of HLA-DR, PD-L1 (B7-H1), and IDO by MSCs and upregulates secretion of PGE2 [30,32,33,38]. Further, it does not enhance immunogenicity of MSCs [30,39] but promotes their antiproliferative properties [32,39] and is even required for the induction of MSC-mediated immunosuppression [40] and treatment of GVHD [41,42].
In this regard, we studied the effect of exogenous IFN-γ on antiproliferative properties, surface marker expression, and cytokine secretion of human adipose-derived stem cells (ASCs) and human amniotic mesenchymal stromal cells (hAMSCs). Further, we determined the influence of MSCs on T-cells and PBMCs in an inflammatory milieu achieved by exogenous activation of immune cells. We systematically analyzed proliferation, cytokine secretion, activation marker surface expression, and gene expression of T-cells and PBMCs cocultured with MSCs of different sources.
Materials and Methods
The collection of placentae, adipose tissue, and peripheral blood was approved by the local Ethical Board. All cells were cultured at 37°C, 5% CO2, and 95% air humidity.
Isolation and culture of MSCs
Subcutaneous adipose tissue (n=7) was obtained during outpatient tumescence liposuction under local anesthesia, and human placentae (n=6) were obtained on cesarean sections. ASCs were isolated from liposuction material and hAMSCs from amniotic membrane as previously described [43]. Briefly, liposuction material was washed with phosphate-buffered saline (PAA, Pasching, Austria) and digested with 1.5 mg/mL collagenase I (Biochrom, Vienna, Austria) for 60 min at 37°C under vigorous shaking. After centrifugation, the cell pellet was treated with erythrocyte lysis buffer [154 mM NH4Cl, 10 mM KHCO3, and 0.1 mM EDTA (Sigma, Vienna Austria) in aqua bidest.] for 10 min at 37°C. For isolation of hAMSCs, amniotic membrane was minced and digested with 1 mg/mL collagenase I for 2 h at 37°C. ASC and hAMSC cell suspensions were filtered through 100 μm strainers (BD, Vienna, Austria), centrifuged, and cultured in endothelial growth medium-2 (Lonza, Verviers, Belgium) to a subconfluent state. Cells were used for experiments at passages 2–6.
To observe the influence of an inflammatory milieu on ASCs and hAMSCs, cells were also kept in endothelial growth medium-2 supplemented with 200 U/mL recombinant human IFN-γ (R&D Systems, Minneapolis, MN). After 4 days of culture, untreated or IFN-γ treated cells were used for direct coculture experiments, subjected to flow cytometry analysis, and cytokine secretion profile was determined in conditioned supernatants.
Isolation of PBMCs and T-cells
Human PBMCs were isolated from buffy coats (n=17, Red Cross Blood Transfusion Service of Upper Austria) by density gradient centrifugation using Biocoll (specific gravity=1.077 g/mL; Biochrom) and cultured in RPMI-medium (RPMI 1640, 10% fetal calf serum, 2 mM L-glutamine, 100 U/mL penicillin, and 0.1 mg/mL streptomycin; all reagents were obtained from PAA) overnight. CD3+ T cells were isolated from PBMCs using the Pan T-cell Isolation Kit (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer's instructions. Purity of isolated CD3+ T-cells was determined by flow cytometry as described in the section “Flow cytometry analysis.”
PBMC and T-cell stimulation
Human CD3+ T-cells and PBMCs were cultured in RPMI-medium for 6 days. T-cell proliferation was induced on day 0 by stimulating cells with 8 μg/mL surface bound αCD3 and 3 μg/mL soluble αCD28 (both Biolegend, San Diego, CA). PBMCs were either stimulated by αCD3/αCD28 as described for T-cells or in MLR with an equivalent number of γ-irradiated 3rd-party PBMCs on day 0 or by 5 μg/mL phytohemagglutinin (PHA; Sigma) added on day 3. Cells were harvested on day 6 of culture and subjected to further analysis.
Cocultures
For direct cocultures, untreated and IFN-γ pretreated allogeneic ASCs or hAMSCs were seeded into a 96-well plate with an equivalent number of PBMCs. For indirect cocultures, allogeneic ASCs or hAMSCs were seeded in the upper chamber and an equivalent number of either CD3+ T-cells or PBMCs in the lower chamber of a 24- or 6-well Transwell® plate (Corning, Corning, NY). Thus, cell populations were physically separated by a semi-permeable membrane (0.4 μm pore size). T-cell and PBMC proliferation was induced according to the section “PBMC and T-cell stimulation.” Stimulated CD3+ T-cells and PBMCs alone served as positive controls. ASCs and hAMSCs cultured alone in RPMI-medium served as controls for proliferation assays and cytokine secretion. Immune cells were cocultured with MSCs for 6 days and then subjected to further analysis. In case of 96- and 24-well mono- and cocultures, proliferation of immune cells was measured. Further, PBMCs and T-cells from 6-well coculture experiments were subsequently subjected to flow cytometry- and gene expression-analysis.
Proliferation assay
On day 5, 10 μM bromodeoxyuridine (BrdU) were added to mono- and cocultures in 96- and 24-well plates, respectively; and on day 6, PBMC and T-cell proliferation was assessed by BrdU ELISA (Roche Applied Science, Vienna, Austria) according to the manufacturer's instructions. In case of direct coculture calculation, PBMC and T-cell proliferation was performed as previously described [43].
Flow cytometry analysis
Monoclonal antibodies (mAbs) conjugated to fluorescein isothiocyanate, phycoerythrin, peridinin chlorophyll protein, or alexa fluor 647 against human CD3, CD4, CD8, CD25, CD69, HLA-DR, HLA-ABC, CD73, CD90, PD-L1 (programmed death-ligand 1), PD-L2, CD80, CD86, CD40, CTLA-4, ICOS-L, CD106 (VCAM-1), CD166 (ALCAM), CD54 (ICAM-1), CD29, CD44 (all BD Biosciences, Schwechat, Austria), GITR-L (eBioscience, San Diego, CA), CD105, and HLA-G (both from Abcam, Cambridge, United Kingdom) were used to characterize ASCs, hAMSCs, T-cells. and PBMCs. Conjugated mouse IgG1 κ-chain (BD Biosciences) served as isotype control. Briefly, 1–2.5×105 cells in 50 μL phosphate-buffered saline containing 1% fetal calf serum (PAA) were incubated with mAbs according to the manufacturer's instructions. Cells were washed with Cell Wash™ and resuspended in Cell Fix™ (BD Biosciences). Samples were stored at 4°C in the dark for up to 24 hours until measured on a FACSCalibur using CellQuest Pro software (BD Biosciences). Percentage of positive cells was determined, and surface density of selected markers was expressed as mean ratio fluorescence intensity (MRFI), which represents the mean fluorescence intensity of specific mAb/mean fluorescence intensity of the isotype control.
Semi-quantitative cytokine array
Cell-free culture supernatants from ASCs and hAMSCs cultured in the absence or presence of IFN-γ were collected on day 4, filtrated (0.22 μm), and stored at −80°C until use. Semi-quantitative measurement of C5a, sCD40-L, G-CSF, GM-CSF, GRO-α, I-309, sCD54, IFN-γ, IL-1α, IL-1β, IL-1rα, IL-2, IL-4, IL-5, IL-6, IL-8, IL-10, IL-12p70, IL-13, IL-16, IL-17, IL-17E, IL-23, IL-27, IL-32α, IP-10, I-TAC, MCP-1, MIF, MIP-1α, MIP-1β, Serpin E1, RANTES, SDF-1, TNF-α, and sTREM-1 was performed using the Human Cytokine Array Panel A Array Kit according to the manufacturer's instructions (R&D Systems). Explanations for abbreviations used for soluble factors are provided in Table 1. Membranes were incubated with chemiluminescent detection reagent (Amersham™ ECL; GE Healthcare, Vienna, Austria) and exposed to x-ray film according to the manufacturer's instructions. The blots contain internal positive and negative control spots. Additionally, a negative control with medium only was performed. Semi-quantitative evaluation of results was performed using Quantity-One 1-D analysis software (BioRad, Vienna, Austria). Obtained values were expressed as Volume Intensity*mm2 and normalized to 106 cells.
PBMC, peripheral blood mononuclear cell; ASC, adipose-derived stem cell; hAMSC, human amniotic mesenchymal stromal cell.
Quantitative cytokine measurement
Cell-free culture supernatants from indirect cocultures and the respective controls were collected on day 6, filtrated (0.22 μm), and stored at −80°C until use. IL-1β, IL-2, IL-4, IL-5, IL-6, IL-8, IL-9, IL-10, IL-12p70, IL-12/23p40, IL-13, IL-17A, IL-21, IFN-γ, TNF-α, IP-10, MIG, MCP-1, MIP-1α, MIP-1β, RANTES, GM-CSF, LIF, sCD40-L, sCD54, sFAS-L (CBA-Flex Sets and CBA Master Buffer Kit, BD Biosciences), and TGF-β (CBA-Single Plex; BD Biosciences) were analyzed using a cytometric bead array system according to the manufacturer's instructions. Explanations for abbreviations used for soluble factors are provided in Table 1. Samples and standards were acquired on an FACSCalibur and analyzed with FCAP Array Software (BD Biosciences). Standards ranged from 10 to 2,500 pg/mL and from 40 to 10,000 pg/mL in case of TGF-β.
PGE2 was measured by competitive enzyme-linked immunosorbent assay (ELISA) using a commercially available ELISA kit (R&D Systems) according to the manufacturer's instructions. Concentrations were calculated from standards in the range of 39–2,500 pg/mL. Standards, samples, and controls were measured in duplicate on a microplate reader (TECAN, Grödig, Austria).
Quantitative real-time polymerase chain reaction
Gene expression levels of transcription factors specific for different T-cell subsets were evaluated by quantitative real-time polymerase chain reaction: FOXP3 (forkhead box P3, Hs01085835_m1), GATA3 (GATA binding protein 3, Hs00922328_m1), TBX21 (T-box protein 21, Hs00894392_m1), and RORC (RAR-related orphan receptor gamma, Hs01076119_m1). In the case of PBMC mono- and cocultures, CD3+ T-cells were isolated from PBMCs as described in the section “Isolation of PBMCs and T-cells” before RNA isolation. CD3+ T cells were transferred into TriReagent (Sigma), vortexed, frozen at −80°C, and fully thawed. RNA was isolated according to the manufacturer's instructions. Quality and quantity of RNA were assessed by Agilent 2100 Bioanalyzer using an RNA 6000 Nano Chip Kit (both from Agilent Technologies, Böblingen, Germany). Isolated RNA was processed as described by Stadler et al. [44] using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Brunn am Gebirge, Austria). Standard curves were prepared for quantification; expression values were normalized to the housekeeping gene RPLP0 (ribosomal protein P0, Hs99999902_m1), analyzed using the LightCycler 480 Relative Quantification Software (Roche Applied Science), and calculated as fold of induction relative to levels of day 6 positive controls. Only a 4-fold up- or downregulation was regarded as actual regulation. All markers were obtained from Applied Biosystems.
Statistical analysis
Data are expressed as mean and standard error of the mean calculated from 3 to 6 biological replicates. Statistical analysis was performed by 1-way analysis of variance, and comparison of different culture conditions was performed using Tukey's multiple comparison test. A P value <0.05 was considered to be significant. Cytokine concentrations were calculated from standard curves created by 4-parameter curve fit.
Results
Since various different culture conditions were used throughout this work, abbreviations were introduced and summarized in Table 1.
First, ASCs and hAMSCs were thoroughly characterized regarding their antiproliferative properties on stimulated PBMCs, surface marker expression profile, and cytokine as well as chemokine secretion under normal and inflammatory conditions. The latter was simulated by the addition of IFN-γ to ASC (“ASCs [IFN-γ]”) and hAMSC (“hAMSC [IFN-γ]”) cultures.
IFN-γ pretreatment enhances antiproliferative properties of ASCs and hAMSCs
The effect of IFN-γ pretreatment on antiproliferative properties of ASCs and hAMSCs was analyzed in direct cocultures. Proliferation analysis revealed that ASCs [IFN-γ] and hAMSCs [IFN-γ] possess significantly higher suppressive potential on stimulated PBMCs than untreated ASCs and hAMSCs (Fig. 1).
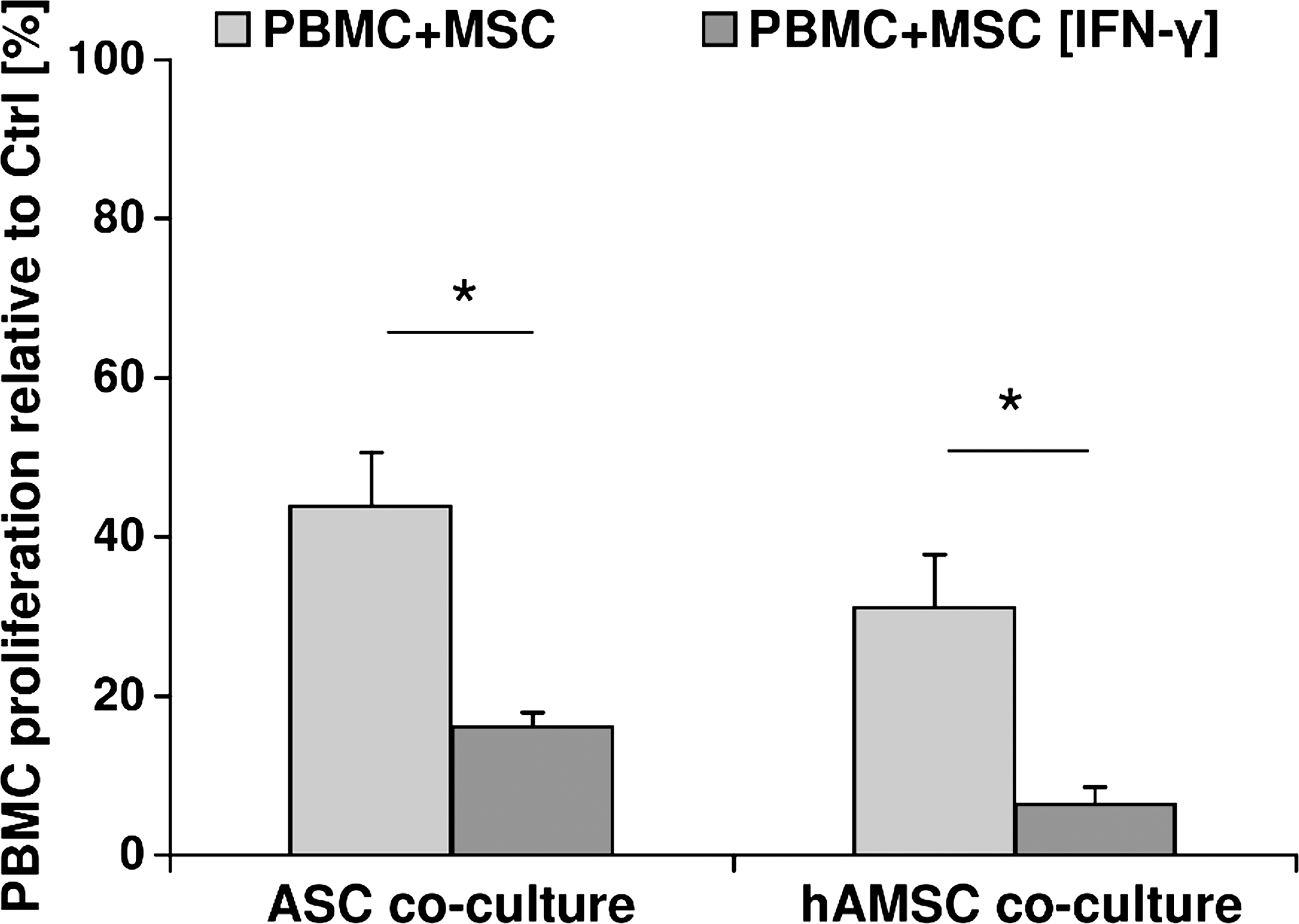
IFN-γ significantly enhances MSC-mediated antiproliferative properties on PBMCs. Phytohemagglutinin-stimulated PBMCs were directly cocultured with untreated or IFN-γ pretreated allogeneic ASCs and hAMSCs (light-gray bar: PBMC+MSC, dark-gray bar: PBMC+MSC [IFN-γ]). Stimulated PBMCs cultured alone served as positive control (100% proliferation). Proliferation of PBMCs was measured by bromodeoxyuridine ELISA on day 6 of culture and expressed as percentage relative to the positive control (Ctrl). Mean±SEM (n=3 biological replicates measured in triplicate), *indicates statistically significant difference with P<0.05. MSC, mesenchymal stem cell; hAMSC, human amniotic mesenchymal stromal cell; ASC, adipose-derived stem cell; IFN-γ, interferon-γ; PBMC, peripheral blood mononuclear cell; SEM, standard error of the mean.
IFN-γ induces HLA-DR expression and upregulation of PD-L1, PD-L2, and CD54 on ASCs and hAMSCs
ASCs and hAMSCs showed typical MSC surface marker profile characterized by positive staining for CD73, CD90, and CD105 and absence of CD34 and CD45 (Table 2). Further, ASCs and hAMSCs also stained positive for adhesion markers CD166, CD29, CD44, and CD54; and a low percentage of cells also expressed CD106 (Table 2). Under inflammatory conditions, significantly less hAMSCs [IFN-γ] (68.2%±4.2%) stained positive for CD105 compared with untreated hAMSCs (89.3%±4.4%) (Table 2).
Data are expressed as percent positive cells, mean (SEM) of n=3–8.
Significant difference to hAMSC with P<0.05.
Presence of IFN-γ led to downregulation of CD90 surface density on ASCs [IFN-γ] (MRFI: 18.0±5.6, P<0.05) and hAMSCs [IFN-γ] (MRFI: 80.9±60.7) (Fig. 2A). Interestingly, MRFI of intercellular adhesion molecule CD54 was highly upregulated on ASCs [IFN-γ] (115.5±48.7) and hAMSCs [IFN-γ] (173.9±102.0, P<0.05) as compared with untreated ASCs (5.7±2.5) and hAMSCs (12.4±8.1) (Fig. 2A). MSCs were also characterized regarding the expression of surface markers typically expressed by antigen presenting cells (APCs) and relevant for the interaction with immune cells (Fig. 2B). Under normal culture conditions, ASCs and hAMSCs did not or marginally express HLA-DR and CD40 on their surface. However, in the presence of IFN-γ, we observed an induction of HLA-DR and CD40 positivity on ASCs [IFN-γ] (HLA-DR: 98.8%±0.2%, CD40: 31.3%±7.6%) and hAMSCs [IFN-γ] (HLA-DR: 72.0%±6.0%, CD40: 20.6%±8.9%). Under normal culture conditions, ASCs did not express PD-L1, and 4%–13% expressed PD-L2. However, IFN-γ treatment significantly induced expression of PD-L1 (67.4%±6.9%) and highly enhanced positivity for PD-L2 (43.6%±10.1%) on ASCs [IFN-γ]. A high percentage of hAMSCs already constitutively expressed the inhibitory co-stimulatory molecules PD-L1 (73.1%±7%) and PD-L2 (56.6%±16.5%), which further increased to 92.8%±2.8% and 87.8%±6.8%, respectively, on IFN-γ stimulation. Regardless of the culture condition, a high percentage (mean: 48%–65%) of ASCs and hAMSCs also expressed surface HLA-G. Although the classical co-stimulatory molecule CD86 was present on 4%–8% ASCs and hAMSCs, other molecules including CD80, ICOS-L, GITR-L, and CTLA-4 were either present on <5% or completely absent regardless of the culture condition. Overall, IFN-γ induced surface properties of nonprofessional APCs (CD45-, HLA-DR+, CD40med+, and CD54high) with a possible regulatory phenotype (PD-L1+, PD-L2+, and CD86low) on both MSC populations.
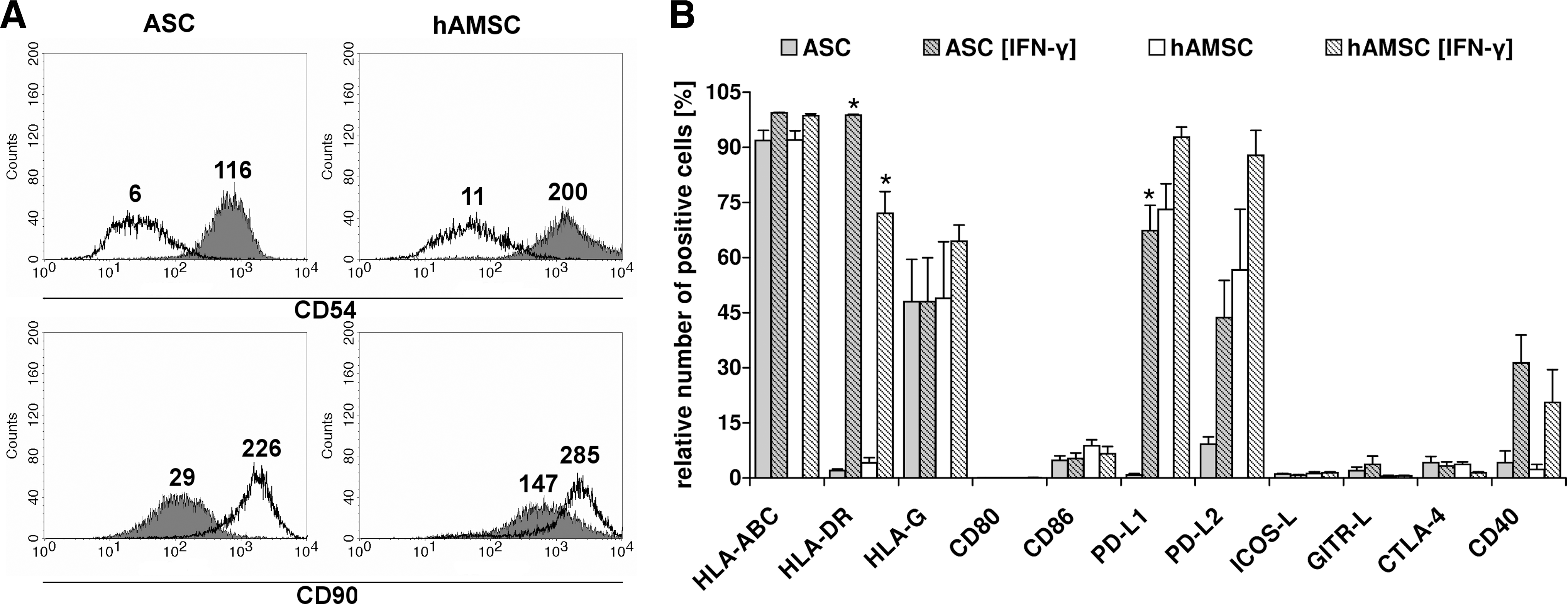
Presence of IFN-γ leads to upregulation of HLA-DR, PD-L1, PD-L2, and CD54 on ASCs and hAMSCs. ASCs and hAMSCs were cultured in the absence or presence of 200 U/mL rIFN-γ [ASC (IFN-γ), hAMSC (IFN-γ)]. Surface expression was analyzed by flow cytometry after 4 days.
ASCs and hAMSCs secrete chemokines
Culture supernatants of untreated and IFN-γ treated ASCs and hAMSCs were screened for the presence of cytokines and chemokines, which could possibly influence or bias an immune response. The sensitivity of the semi-quantitative assay allowed detection of 8 of 36 factors. ASCs and hAMSCs secreted Gro-α, IL-6, IL-8, MIF, and Serpin-E1, which was enhanced on IFN-γ stimulation (Table 3). MCP-1 was constitutively secreted by ASCs, and secretion levels decreased in the presence of IFN-γ. In contrast, MCP-1 was not detectable in hAMSC cultures but was induced in 2 of 3 hAMSC [IFN-γ] donors. IFN-γ also induced IP-10 secretion in 2 of 3 ASC and 2 of 3 hAMSC donors. Further, sCD54 was not detectable in normal ASC and hAMSC cultures, but IFN-γ highly induced its secretion by hAMSCs (Table 3). This observation relates to the increase in CD54 surface density on IFN-γ treatment.
Secretion profile was determined by a semi-quantitative proteome profiler array mean Volume Intensity*mm2 (n=3): +, 1–100; ++, 100–1,000; +++, 1,000–2,000; ++++, >2,000.
Induced in 2 of 3 IFN-γ treated donors.
Induced in 2 of 3 IFN-γ treated hAMSC donors.
ND, not detectable.
We have shown that the presence of an artificially created inflammatory milieu enhances immunomodulatory properties of MSCs. Therefore, we also analyzed the influence of MSCs on cocultured stimulated T-cells known to secrete pro-inflammatory mediators. T-cells were characterized regarding cytokine secretion, activation marker, and gene expression in cell-contact independent cocultures with MSCs by applying different stimulation methods (MLR, PHA, and αCD3/αCD28) and cellular contexts (CD3+ T-cells, PBMCs).
ASCs and hAMSCs suppress PBMC and T-cell proliferation in a cell-contact independent manner
In a 24-well indirect coculture setting, ASCs and hAMSCs efficiently suppressed proliferation of PBMCs stimulated in an MLR (MLR/ASC: 34%±4%, MLR/hAMSC: 33%±5%) and T-cells stimulated with αCD3/αCD28 (T/ASC: 57%±2%, T/hAMSC: 66%±2%) compared with their respective positive controls (Fig. 3A). Both ASCs and hAMSCs showed significantly higher capacity to suppress stimulated PBMCs than T-cells.
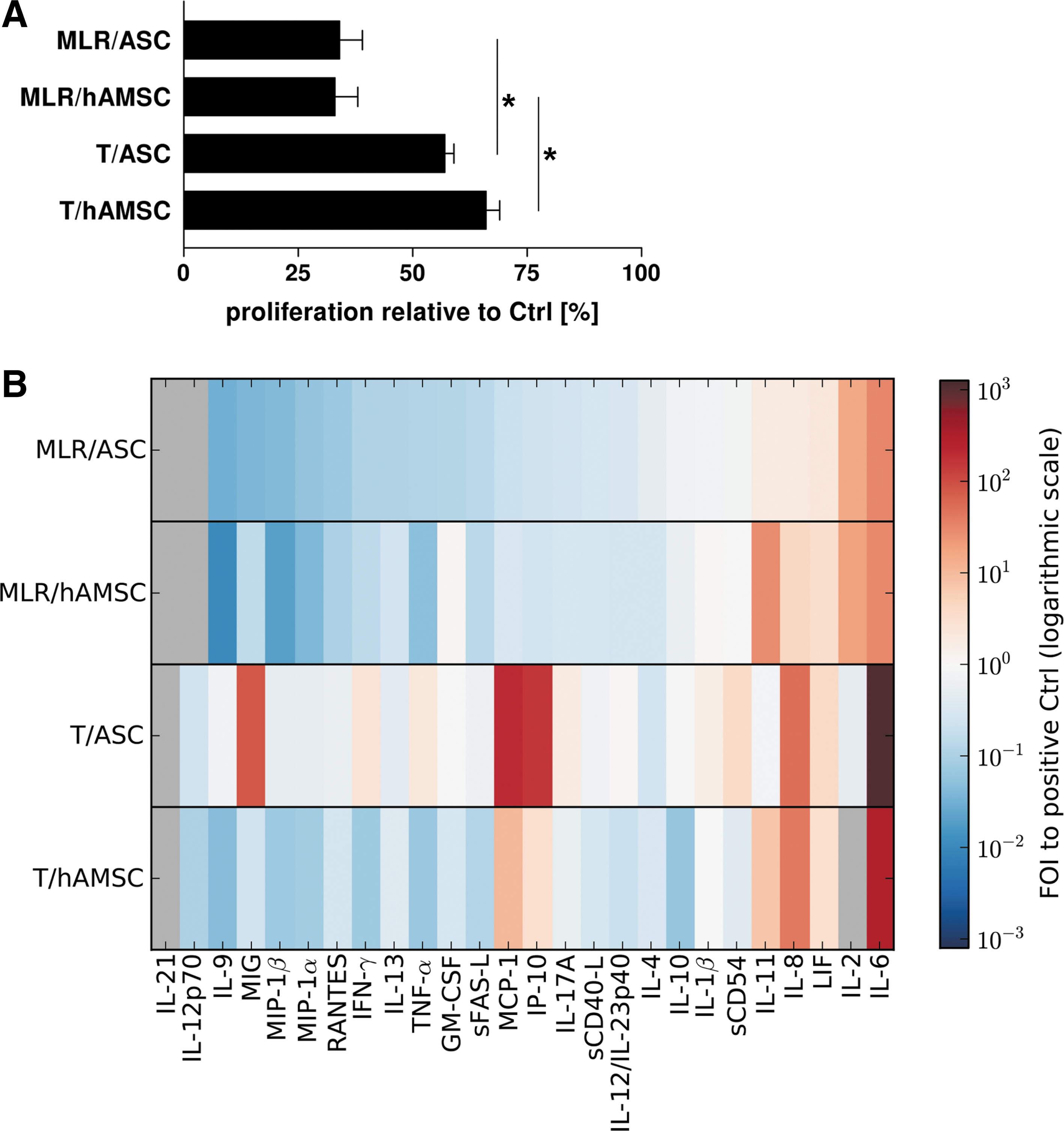
Suppression of T-cell proliferation and cytokine secretion by MSCs depends on stimulation method, cellular context, and MSC source. Allogeneic ASCs or hAMSCs were cocultured with an equivalent number of stimulated PBMCs or CD3+ T-cells in a 24-well indirect coculture system. PBMCs were stimulated in a mixed lymphocyte reaction (MLR) with 3rd-party PBMCs and T-cells (T) with αCD3/αCD28 on day 0. Stimulated immune cells cultured alone served as positive control (Ctrl).
ASCs and hAMSCs suppress cytokine secretion of cocultured stimulated PBMCs
To further elucidate the effect of ASCs and hAMSCs on immune cells, a cytokine screening of supernatants from 24-well indirect cocultures and the respective mono-cultures (ASCs, hAMSCs, PBMCs, and T-cells) was performed. Both MSC populations constitutively secreted moderate to high amounts of RANTES, MCP-1, MIP-1α, IL-6, IL-8, IL-11, LIF, sCD54, and GM-CSF (Supplementary Fig. S1; Supplementary Data are available online at
PBMCs activated in MLR cocultures (MLR/ASC, MLR/hAMSC) displayed a similar secretion pattern characterized by suppressed secretion of chemokines (RANTES, IP-10, MIG, MIP-1α, MIP-1β, and MCP-1), cytokines (IL-21, IL-12p70, IL-9, IFN-γ, IL-13, TNF-α, IL-17A, IL-12/IL-23p40, IL-4, and IL-10), sFAS-L, and sCD40-L and enhanced secretion of IL-11, IL-6, IL-8, and LIF compared with the positive control (Fig. 3B). The cytokine secretion pattern found in cocultures of stimulated T-cells with hAMSCs (T/hAMSC) showed similarity to MLR cocultures except for the increase instead of suppression of MCP-1 and IP-10 concentration (Fig. 3B). Interestingly, cocultures of stimulated T-cells and ASCs (T/ASC) showed a strikingly different cytokine secretion pattern compared with all other patterns. In coculture with ASCs, T-cells increased secretion of IFN-γ (2.6-fold), TNF-α (2.2-fold), IL-17A (2.0-fold), sCD54 (4.0-fold), MIG (86.0-fold), MCP-1 (207.0-fold), and IP-10 (163.0-fold) compared with the positive control (Fig. 3B). Similar to T/hAMSC and MLR cocultures, we also found higher levels of LIF, IL-6, and IL-8 in T/ASC cocultures compared with the positive control (Fig. 3B). This was, at least, partly caused by MSCs, which constitutively secreted all 3 factors. Taken together, although ASCs were able to suppress proliferation of stimulated T-cells (Fig. 3A), they did not inhibit but rather increased secretion of inflammatory mediators. Thus, we further analyzed the effect of ASCs on activation, cytokine secretion, and gene expression of stimulated T-cells and PBMCs in an upscaled (6-well) indirect coculture setting.
A higher number of T-cells express CD69 in presence of ASCs
Either enriched CD3+ T-cells or PBMCs were stimulated to undergo proliferation in the absence (positive control) or presence of allogeneic ASCs (T/ASC, P/ASC). ASCs caused a slight increase in CD4+ and a decrease in CD8+ T-cell populations (Fig. 4). On αCD3/αCD28 stimulation, surface expression of activation markers CD25, CD69, and HLA-DR was induced on T-cells. The presence of ASCs by trend suppressed HLA-DR expression on T-cells. In ASC coculture, the population of CD4+/CD25+ T-cells on the average slightly increased, whereas CD8+/CD25+ T-cells decreased compared with the positive control. Further, ASCs induced an up to 3.2-fold increase in CD4+/CD69+ and CD8+/CD69+ T-cells.
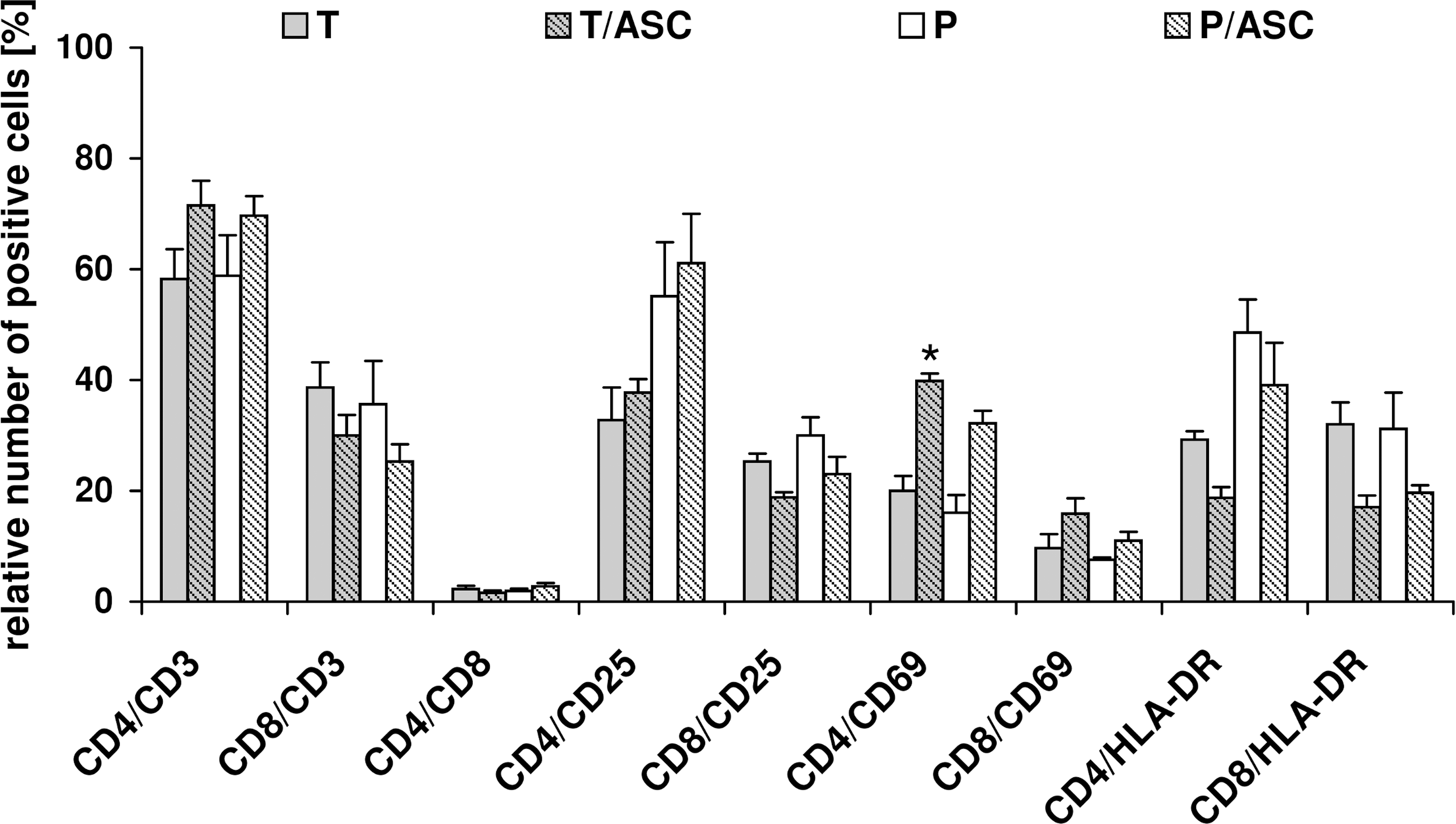
Presence of ASCs enhances acquisition of CD69 on CD4+ and CD8+ T-cells. Stimulated CD3+ T-cells or PBMCs were cultured in the absence (T, P) or presence of allogeneic ASCs (T/ASC, P/ASC) in a 6-well indirect coculture system. Immune cells were stimulated with αCD3/αCD28 on day 0. Surface expression analysis was performed by flow cytometry after 6 days of culture. Percentage of cells positive for the respective surface marker is displayed. Mean±SEM (n=3–4), *indicates statistically significant difference to the respective immune cells cultured alone with P<0.05.
When stimulating PBMCs with PHA in the presence of ASCs, the total number of CD25+ cells was significantly decreased (48.6%±7.9%) compared with the positive control (91.7%±1.9%) (Table 4). In contrast, coculture of αCD3/αCD28 stimulated PBMCs with ASCs did not or only marginally reduced the number of CD25+ cells (79.2%±9.8%) compared with PBMCs cultured alone (86.4%±6.6%). In contrast, CD69 surface expression on cocultured PBMCs was similar for both stimulation methods (Table 4).
Data are expressed as mean±SEM (n=4).
Significantly different to PBMC+PHA and to αCD3/αCD28 stimulated P/ASC (P<0.05).
ASCs differently influence cytokine expression of cocultured purified T-cells versus T-cells from cocultured PBMCs
Influence of ASCs on secretion of cytokines specific for T-cell subsets including Th2 (IL-4, IL-5, IL-13), Th1 and CTL (IFN-γ), Th17 (IL-17A), and Tregs (IL-10, TGF-β) was determined by quantitative cytometric bead array (Fig. 5). Concentrations of IL-4 were below the standard curve in all analyzed supernatants.
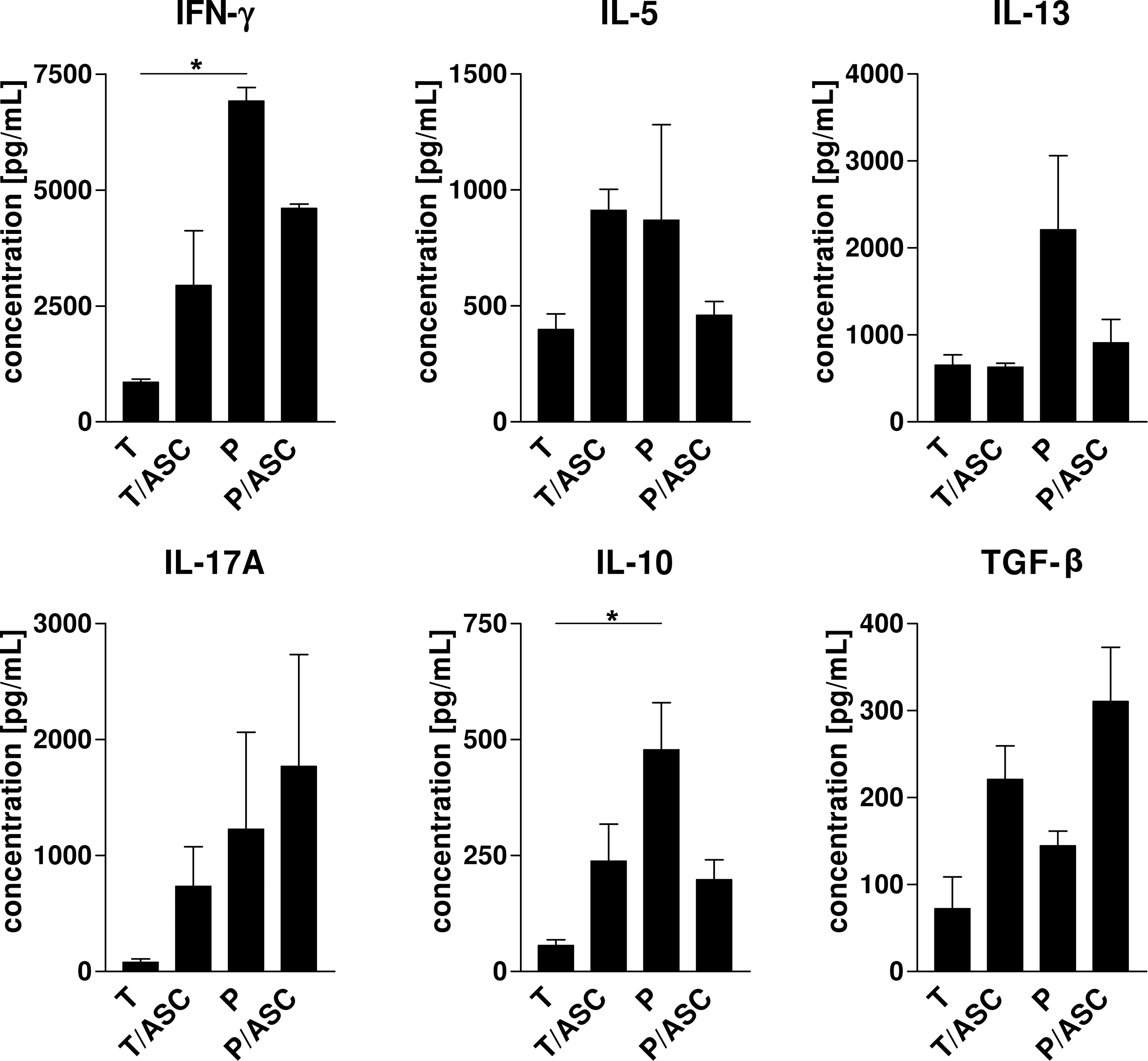
ASCs differentially influence cytokine expression by immune cells depending on the cellular context. CD3+ T-cells or PBMCs were cultured in the absence (T, P) or presence of allogeneic ASCs (T/ASC, P/ASC) in a 6-well indirect coculture system. Immune cells were stimulated with αCD3/αCD28 on day 0. Concentrations of IFN-γ, IL-5, IL-13, IL-17A, IL-10, and TGF-β were determined by cytometric bead array in cell-free supernatants after 6 days of culture. Mean±SEM (n=3), *indicates statistically significant difference with P<0.05. TGF-β, transforming growth factor beta.
Stimulated PBMCs and T-cells cultured alone showed apparent differences in cytokine secretion levels. PBMCs secreted significantly higher amounts of IFN-γ and IL-10 (6914.3±293.5, 477.7±101.7 pg/mL) than stimulated T-cells (850.6±73.0, 56.2±12.3 pg/mL). Further, the average concentrations of IL-5, IL-13, IL-17A, and TGF-β were also higher in PBMC mono-cultures.
In the presence of ASCs IL-13, secretion by cocultured PBMCs was suppressed; whereas its secretion by cocultured T-cells did not change. Whether MSCs induced or suppressed secretion of IL-5, IFN-γ, and IL-10 by immune cells was dependent on the cellular context (purified CD3+ T-cells compared with PBMCs). Interestingly, secretion of these 3 cytokines was suppressed in P/ASC and enhanced in T/ASC cultures compared with the control. Nevertheless, levels of IFN-γ and IL-10 were similar in both coculture settings. Concentrations of IL-17A and TGF-β were elevated in both ASC cocultures. In case of TGF-β, this might be an additive contribution of immune cells and ASCs, as ASCs constitutively expressed the factor (data not shown).
In addition to cytokine secretion, we analyzed whether ASCs also influence expression of subset-specific transcription factors by T-cells (data not shown). Interestingly, in contrast to the cytokine secretion profile, the presence of ASCs did not alter gene expression according to our analysis criteria.
PGE2 secretion is highly induced on coculture of ASCs with T-cells or PBMCs
Although PGE2 was only marginally secreted by one ASC donor and not detectable in the other ASC mono-cultures, we found high PGE2 concentrations in T/ASC (157.7±34.1 ng/mL) and P/ASC (145.4±39.6 ng/mL) cocultures (Fig. 6).
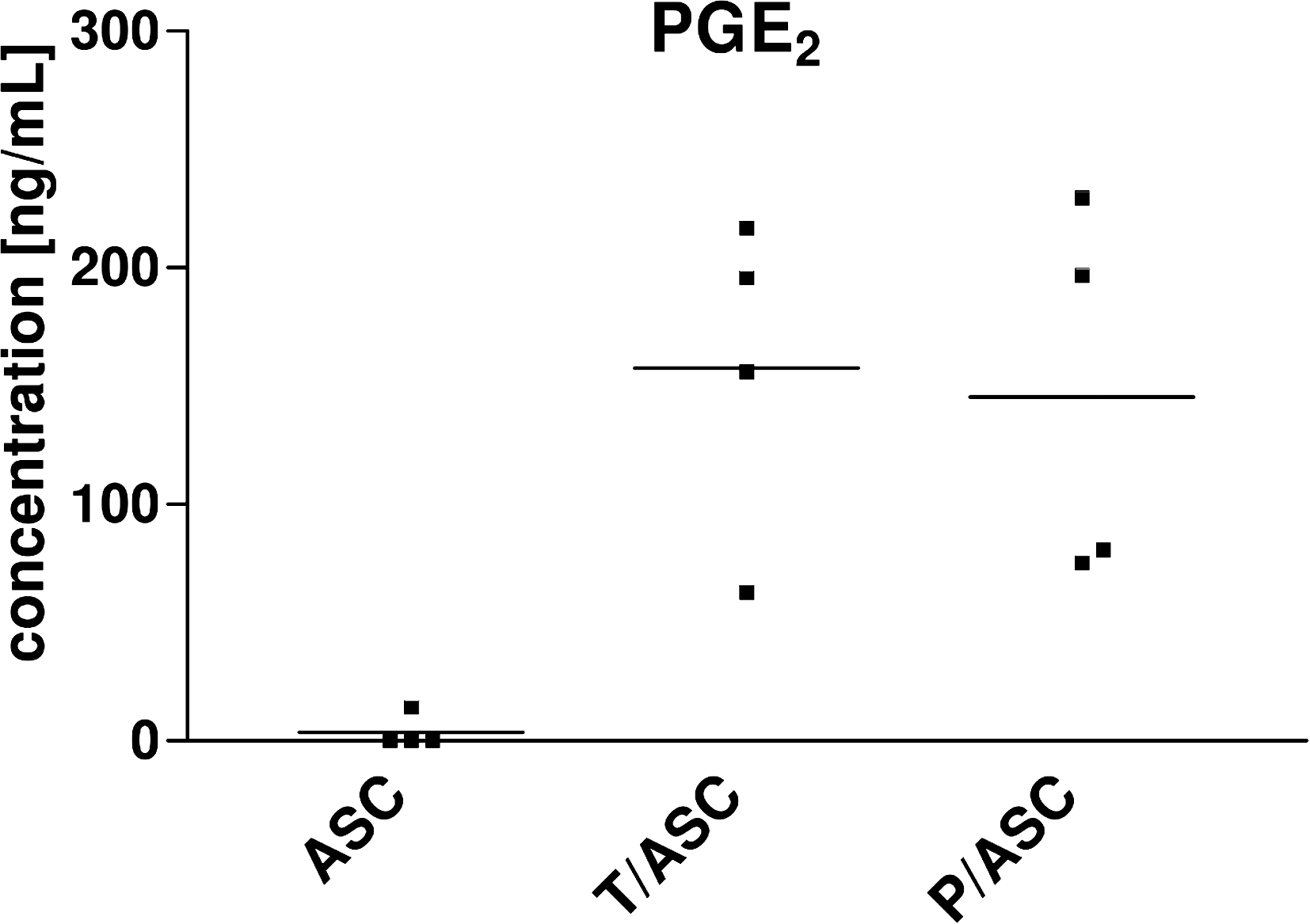
PGE2 is highly induced in cocultures. Allogeneic ASCs were cultured in the absence (ASC) or presence of CD3+ T-cells or PBMCs (T/ASC, P/ASC) in a 6-well indirect coculture system. Immune cells were stimulated with αCD3/αCD28 on day 0. Concentration of PGE2 was determined by ELISA in cell-free supernatants after 6 days of culture. Dots represent donors, bar represents mean (n=4). PGE2, prostaglandin E2.
Discussion
IFN-γ has been assigned a critical role in the activation and enhancement of MSC-mediated immunosuppressive properties. In a GVHD model, Polchert et al. [41] demonstrated that treatment of MSCs with high levels of IFN-γ increased survival of animals to 100%; and IFN-γ activated MSCs administered on day 0 could even prevent development of GVHD. It has been shown that BM-MSCs acquire HLA-DR [39], PD-L1, and IDO expression through exogenously added or lymphocyte-produced IFN-γ [30,32,38]. MSCs were not able to upregulate PD-L1 and to exert their antiproliferative properties in cocultures with lymphocytes lacking IFN-γ secretion [38]. Further, it has been found that inhibition of the IFN-γ receptor leads to abrogation of MSC-mediated suppressive activity [40]. In this study, we show that IFN-γ impacts ASCs and hAMSCs regarding phenotype and secretory profile and enhances their antiproliferative properties on stimulated PBMCs. Interestingly, ASCs and hAMSCs downregulated surface density of the typical MSC marker CD90 on IFN-γ treatment. It has been reported that MSCs showing reduced positivity for CD90 were not able to suppress PHA stimulated PBMC proliferation but initiated an alloproliferative response [45], which led to the conclusion that CD90 might be used as a predictive marker for MSC-mediated inhibitory properties. In contrast to this hypothesis, we observed that IFN-γ treated ASCs and hAMSCs more efficiently suppressed PBMC proliferation, although CD90 surface density was decreased.
Further, our results demonstrate that IFN-γ also induces ASCs and hAMSCs to acquire surface properties of nonprofessional APCs (HLA-DR+, CD40+) with a possible regulatory phenotype (PD-L1+, PD-L2+). Sheng et al. [38] demonstrated that siRNA knockdown of PD-L1 in murine BM-MSCs (mBM-MSCs) significantly reduced their ability to inhibit lymphocyte proliferation. We have demonstrated that an average amount of 73% and 57% hAMSCs expressed PD-L1 and PD-L2, respectively, under normal culture conditions. Similarly, Tipnis et al. [33] have also demonstrated constitutive expression of PD-L1 on 35%–42% human umbilical cord MSCs, which was further upregulated by IFN-γ. Indeed, PD-L1 expression has been found in cells of the human fetal-maternal interface (including trophoblast cells, decidua, chorion, and amniotic mesenchyme) [46] and has been proposed as a negative regulator of maternal alloimmune response against fetal antigens in mouse [47].
Adhesion molecules are also important mediators of inflammatory responses. ICAM-1 (CD54) and −2 (CD102) as well as VCAM-1 (CD106) are known key players in adhesion and transmigration of lymphocytes [48]. Ren et al. [49] recently demonstrated that CD54 and CD106 are critical for mBM-MSC-mediated immunosuppression. Both molecules were found to be inducible on mBM-MSCs by T-cell secreted IFN-γ concomitant with TNF-α or IL-1 [49]. We demonstrate that a high number of human ASCs and hAMSCs constitutively expressed CD54 and that surface density greatly increased on IFN-γ treatment. Both MSC populations also constitutively secreted low amounts of sCD54 (sICAM-1), and an induction of sCD54 secretion by IFN-γ pretreated hAMSCs was observed. However, we detected high levels of sCD54 in indirect cocultures with MSCs, which were similar to levels found in T-cell and PBMC controls and, thus, presumably contributed by immune cells rather than MSCs.
Since immune cells secrete IFN-γ on stimulation, immunomodulatory properties of ASCs and hAMSCs might be conferred by IFN-γ induced or enhanced expression of PD-L1, PD-L2, and CD54, at least in direct cocultures.
High levels of IFN-γ detected in ASC cocultures with αCD3/αCD28 stimulated immune cells might support ASC-mediated immunosuppression. PGE2 has been assigned an important role in MSC-mediated influence on effector functions of immune cells [25,29,50,51]. Human umbilical cord MSCs have been reported to enhance PGE2 secretion on culture with conditioned supernatants from stimulated T-cells and PBMCs, which was mediated by IL-1β and IFN-γ [52]. We have previously shown that ASCs only secreted low levels of PGE2 in coculture with monocytes stimulated to undergo dendritic cell differentiation [17]. In contrast, we demonstrated here that PGE2 was highly induced on coculture of ASCs with T-cells or PBMCs. Interestingly, we also detected high levels of IL-17A in ASC cocultures with αCD3/αCD28 stimulated immune cells, which might indicate the polarization of T-cells into Th17 cells in this coculture setting.
It has been demonstrated that human BM-MSCs inhibit upregulation of CD25, CD38, and CD69 on PHA-stimulated CD3+CD4+ lymphocytes in direct cocultures [36,53]. Ramasamy et al. [54] observed no effect of BM-MSCs on αCD3/αCD28 stimulated CD3+CD4+ T-cell activation status. In contrast to these reports, we show that positivity for CD69 was enhanced on cocultured stimulated CD3+ lymphocytes, which was even more pronounced in case of CD4+ compared with CD8+ T-cells. Further, we demonstrate that ASCs did not affect total number of CD25 positive cells in coculture with αCD3/αCD28 stimulated PBMCs, whereas ASCs strikingly inhibited acquisition of CD25 on PHA-stimulated PBMCs. Thus, ASCs differentially interfere with activation status of T-cells depending on the stimulatory agent. Although ASCs inhibited proliferation of T-cells, CD69 was enhanced in coculture. A possible explanation for the observed effect is that stimulated T-cells already downregulate the expression of early activation marker CD69 at time of analysis, whereas the presence of ASCs delays CD69 expression. Further, CD69 has been reported to play an immunoregulatory role depending on the specific cellular and pathophysiological context [55].
Contradictory results observed in our experiments and in literature can possibly be explained by differences in MSC sources, coculture settings, and stimulatory agents applied. Indeed, others have reported that MSCs differentially influence T-cell responses to allogeneic, mitogenic and virus-specific stimuli [56,57]. In this study, we tested MSCs from different sources under the same coculture conditions.
Our data confirm that MSCs differently act on PHA, MLR, and αCD3/αCD28 stimulated immune cells. Additionally, we also observed apparent differences in the action of ASCs on cytokine secretion of immune cells depending on the cellular context. When stimulating immune cells with αCD3/αCD28 in ASC coculture, IL-10 and IFN-γ secretion by PBMCs was inhibited, whereas their secretion by purified T-cells was enhanced. In the case of PBMCs, T-cells get stimulated in a multi-cell setting, where other immune cells including natural killer-cells, B-cells, and monocytes could be influenced by MSCs and, thus, contribute to the observed effect. It has been demonstrated that MSC-mediated induction of Tregs in coculture with CD4+ T-cells requires cell-cell contact in concert with soluble factors, whereas the induction of Tregs in PBMC/MSC cocultures is independent of the culture setting [25]. Especially, presence of CD14+ monocytes has been reported to enhance human umbilical cord MSC-mediated suppression of proliferation and IFN-γ secretion by T-cells [52]. On activation by exogenous stimulation, T-cells and PBMCs secrete a number of soluble factors in different concentrations depending on the cellular context (Supplementary Fig. S1 and Fig. 5) and activation stimuli [58]. Thus, the specific cytokine milieu created by T-cells and PBMCs might differentially influence cocultured MSCs and reciprocally affect their action on immune cells.
Overall, characterization of phenotype and secretory profile of human ASCs and hAMSCs under normal and inflammatory conditions demonstrates presence of surface markers (PD-L1, PD-L2, and CD54) possibly involved in immunomodulation and supports the role of IFN-γ in enhancing their immunosuppressive properties. Although ASCs and hAMSCs were able to suppress proliferation of both cocultured stimulated T-cells and PBMCs, their influence on cytokine secretion and activation status differed depending on stimulation methods employed and the type of cells cocultured. When employing MSCs in tissue engineering or cellular therapy, it is crucial to perform and thoroughly evaluate in vitro experiments regarding their immunomodulatory properties. Our data corroborate that specific experimental conditions including cell culture settings, MSC source, type of cocultured immune cells, and stimulation methods critically influence MSC-mediated action. Thus, such in vitro data have to be critically evaluated and might not correspond to the complex in vivo situation involving a variety of cell types and stimuli.
Footnotes
Acknowledgments
We thank K. Hofer, K. Schauer and S. Neussl for technical assistance; U. Dobramysl for artwork assistance; and J. Thalhamer for discussing the work. This work was carried out under the scope of the European NoE Expertissues (NMP3-CT-2004–500283). B.K. is a Ph.D. candidate at the Paris-Lodron University of Salzburg, Austria, and this work is submitted in partial fulfillment of the requirement for the Ph.D.
Author Disclosure Statement
The authors declare no competing financial interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.