
Abstract
Differentiation of human induced pluripotent stem cells (hiPSCs) and embryonic stem cells (hESCs) into the erythroid lineage of cells offers a novel opportunity to study erythroid development, regulation of globin switching, drug testing, and modeling of red blood cell (RBC) diseases in vitro. Here we describe an approach for the efficient generation of RBCs from hiPSC/hESCs using an OP9 coculture system to induce hematopoietic differentiation followed by selective expansion of erythroid cells in serum-free media with erythropoiesis-supporting cytokines. We showed that fibroblast-derived transgenic hiPSCs generated using lentivirus-based vectors and transgene-free hiPSCs generated using episomal vectors can be differentiated into RBCs with an efficiency similar to that of H1 hESCs. Erythroid cultures established with this approach consisted of an essentially pure population of CD235a+CD45− leukocyte-free RBCs with robust expansion potential and long life span (up to 90 days). Similar to hESCs, hiPSC-derived RBCs expressed predominately fetal γ and embryonic ɛ globins, indicating complete reprogramming of β-globin locus following transition of fibroblasts to the pluripotent state. Although β-globin expression was detected in hiPSC/hESC-derived erythroid cells, its expression was substantially lower than the embryonic and fetal globins. Overall, these results demonstrate the feasibility of large-scale production of erythroid cells from fibroblast-derived hiPSCs, as has been described for hESCs. Since RBCs generated from transgene-free hiPSCs lack genomic integration and background expression of reprogramming genes, they would be a preferable cell source for modeling of diseases and for gene function studies.
Introduction
R
Embryonic stem cells (ESCs) are a logical alternative source for blood cells. ESCs can grow in unlimited numbers and differentiate into hematopoietic cells of various lineages [5,6]. Recently, several groups described efficient differentiation of human ESCs (hESCs) into RBCs and demonstrated that erythropoiesis derived from hESCs recapitulates hemoglobin switching observed during early embryonic development in humans [7 –10]. Generation of human pluripotent stem cells, the so-called induced pluripotent stem cells (hiPSCs), from adult somatic cells by reprogramming with pluripotency factors [11 –13] provides another source of stem cells for RBC production. In addition, these cells can be used for the in vitro modeling of pathologic processes if hiPSCs are made from cells bearing genetic abnormalities affecting RBC development or function. Recent reports of successful production of functional RBCs from hiPSCs provided evidence for the feasibility of such approach [14]. However, it remains unclear whether hiPSCs differentiate into RBCs with efficiency similar to hESCs. Since prior studies demonstrate impaired differentiation of hiPSCs toward erythroid cells using embryoid body method [15], it is important to determine whether the observed differences could be attributed to the intrinsic properties of iPSCs or a differentiation method used for induction of RBCs from hiPSCs. Because the original reprogramming methods employ a virus-based delivery of reprogramming factors, permanent genomic integration of transgenes remains the major limiting factor surrounding this technology. In spite of transgene silencing after reprogramming [11 –13], we were able to detect a background of transgene expression in differentiated cells obtained from transgenic hiPSCs [16,17] and showed that it can affect their properties [18]. To overcome this problem, we developed a method for generation of hiPSCs free of viruses and transgene sequences by employing Epstein-Barr Virus (EBV)-based episomal vectors [19]. Here we characterize erythroid differentiation from transgene-free and transgenic hiPSC lines [11,19] as compared to hESCs.
Materials and Methods
Cell lines
The hESC H1 cell line (passages 31–47) was obtained from WiCell Research Institute, Madison, WI. Transgenic iPSCs (SK46)-M4-10 (TiPSC1) and Foreskin-1 (TiPSC2) (passages 9–35) were obtained by reprogramming human adult and fetal fibroblasts with POU5F1, SOX2, and NANOG lentiviral vectors [11,17]. The hiPSC lines 19-9-7T (TFiPSC1) and 4-3-7T (TFiPSC2) free of viral and transgene sequences have been obtained through reprogramming of neonatal fibroblasts with episomal vectors to express OCT4, SOX2, NANOG, LIN28, MYC, KLF4, and LT (SV40 large T gene) genes [19]. The hESCs were maintained in an undifferentiated state by coculture with mouse embryonic fibroblasts in the presence of 4 ng/mL of human basic fibroblast growth factor [20]. The iPSCs were kept as undifferentiated cells using the same culture conditions, but with 100 ng/mL of zebrafish basic fibroblast growth factor. The OP9 mouse bone marrow stromal cell line was obtained from Dr. Toru Nakano (Research Institute for Microbial Diseases, Osaka University, Japan). OP9 cells were maintained on gelatinized 10-cm dishes (BD) in α-MEM (Invitrogen) with 20% fetal bovine serum (Hyclone). Mouse MS5 stromal cell line was obtained from DSMZ and maintained on 10 cm dishes (BD) with α-MEM (Invitrogen) and 10% fetal bovine serum (Hyclone).
RBC production
To induce hematopoietic differentiation, hESCs and hiPSCs were harvested by treatment with 1 mg/mL of collagenase IV (Invitrogen) and cultured on a monolayer of OP9 stromal cells. Cells were plated at approximately 1.5×106 cells per 10 cm dish, in 20 mL of a-MEM (Invitrogen) supplemented with 10% fetal bovine serum (Hyclone) and 100 μM monothioglycerol (Sigma) and cultured without cytokines as previously described in details [21,22]. Differentiated hESCs and hiPSCs were harvested between day 7 and 8 of culture with treatment of 1 mg/mL collagenase (Invitrogen) for 25 min, followed by 0.05% trypsin–0.5 mM EDTA (Invitrogen) for 20 min at 37°C. Cells were washed with α-MEM with 10% FBS and filtered through a 70 μm cell strainer (BD). In one approach (Fig. 1, Method A), we used isolated CD34+ or CD31+ cells to generate and expand erythroid cells. CD34+ or CD31+ cells were positively selected using corresponding direct microbeads (Miltenyi Biotech) by magnetic-assisted cell sorting (Miltenyi Biotech) as described [22]. The isolated cells were then washed twice with serum-free expansion medium (SFEM; Stem Cell Technologies) and cultured at 1×106 cells/mL in SFEM with 0.3% Ex-Cyte (Millipore
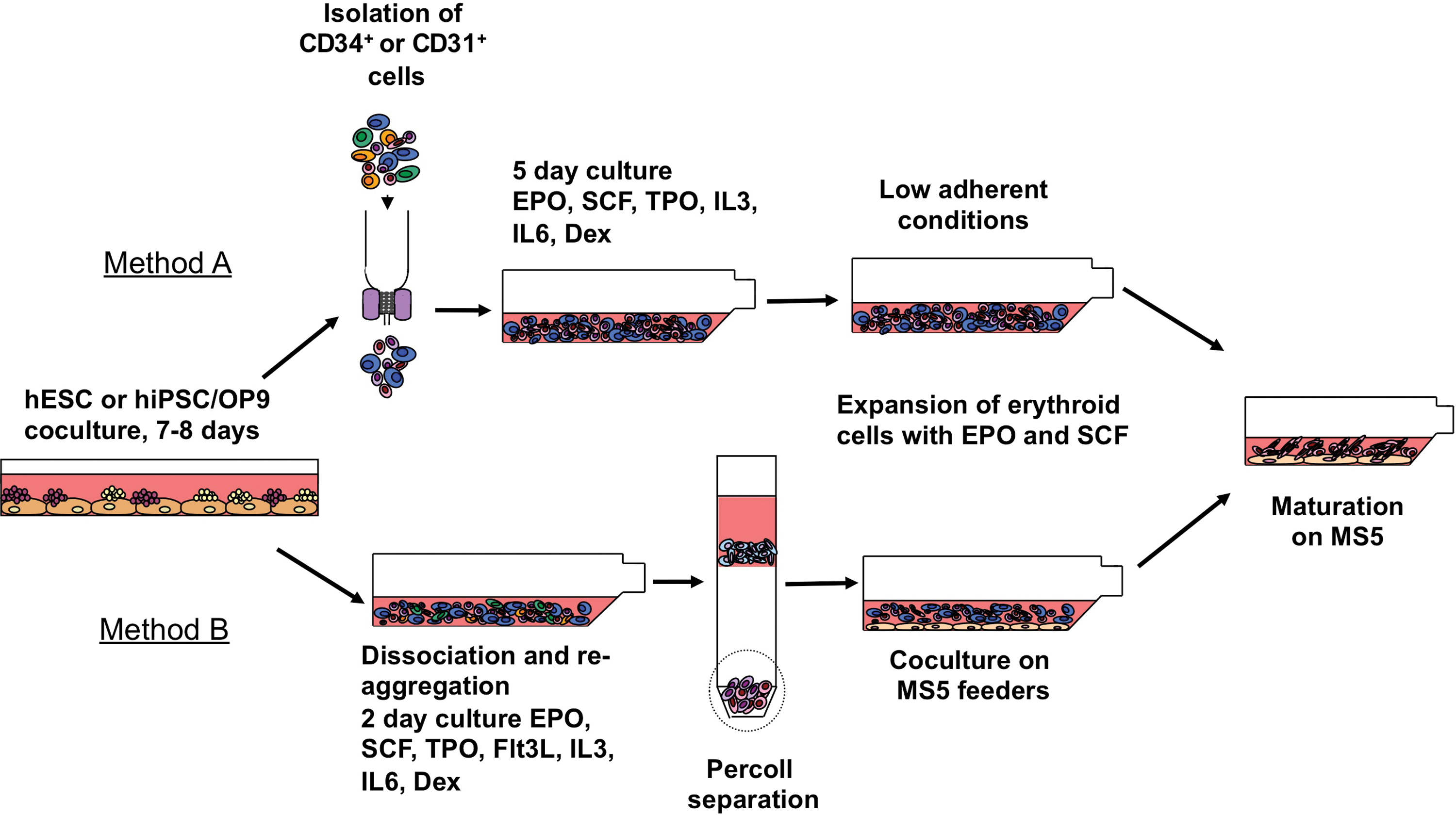
Schematic diagram of the protocol used for production of red blood cells from hESCs/hIPSCs. To induce hematopoietic differentiation, hESC/hiPSCs were cocultured with OP9 for 7–8 days. Two methods were used to produce red blood cells from differentiated PSCs. In Method A, CD34+ or CD31+ hematopoietic progenitors were isolated from differentiated PSCs and expanded/differentiated into red blood cells in low adherent conditions. In Method B, erythroid progenitors were selectively expanded from bulk cultures and maintained on MS5 feeders. Red blood cells were induced to mature in coculture with MS5 without cytokines. hESC, human embryonic stem cell; hiPSC, human induced pluripotent stem cell; SCF, stem cell factor; EPO, erythropoietin; TPO, thrombopoietin; IL, interleukin. Color images available online at
Assessment of erythroid differentiation by fluorescence-activated cell sorting analysis
Cells were prepared in PBS with 3% fetal bovine serum with 0.05% sodium azide and 1 mM EDTA. Samples were labeled with multicolor monoclonal antibody combinations and analyzed using FACSCalibur (BD) and FlowJo software (Tree Star Inc.). The monoclonal antibodies used for identification and characterization of cell subsets included CD41a, CD43 (Clone 1G10), CD45, CD71, and CD235a (BD Biosciences). Human cells in OP9 cultures were identified using TRA-1-85 APC monoclonal antibody (R&D Systems), which detects the OK blood group antigen expressed by virtually all human cells [25]. 7-Aminoactinomycin Via-Probe staining solution (BD) was added to samples just before analysis to allow for dead cell exclusion. Intracellular staining to determine hemoglobin expression was performed using FIX&PERM cell permeabilization reagents (CALTAG-Invitrogen). Embryonic hemoglobin was detected using anti-human hemoglobin epsilon FITC-conjugated monoclonal antibodies (Fitzgerald), fetal using anti-human fetal hemoglobin PE-conjugated monoclonal antibodies (CALTAG-Invitrogen), and adult using antihuman hemoglobin beta FITC-conjugated monoclonal antibodies (Santa Cruz).
Quantitative reverse transcriptase–polymerase chain reaction
Total RNA was isolated using TRI Reagent® solution (Applied Biosystems) and then treated with TURBO^TM (superscript) DNA-free DNase (Applied Biosystems). cDNA synthesis was carried out using SuperScript™ III First-Strand Synthesis System for RT-PCR (Invitrogen) according to the manufacturer's instruction protocol. Quantitative polymerase chain reactions (qPCR) were performed with Platinum SYBR-Green qPCR master mix (Invitrogen), DNA Engine Opticon real-time thermal cycler (Bio-Rad), and specific primers for each globin mRNAs: ɛ-globin forward (GCCTGTGGAGCAAGATGAAT) and reverse (GCGGGCTTGAGGTTGT); γ-globin forward (CTTCAAGCTCCTGGGAAATGT) and reverse (GCAGAATAAAGCCTACCTTGAAAG); β-globin forward (GGCACCTTTGCCACACTG) and reverse (CACTGGTGGGGTGAATTCTT). Expression levels were estimated by minimal cycle threshold values (Ct) normalized to the reference expression of ribosomal protein L13A (RPL13A) in each sample [26].
Results
To induce hematopoietic differentiation, we used coculture hESC and hiPSCs with OP9 bone marrow stromal cell line [17,21,22]. As we and others have shown previously, the first hematopoietic colony-forming cells arising from hESCs and iPSCs have CD34+CD43+ phenotype, coexpress CD41a and CD235a, and possess erythroid and megakaryocytic potentials [17,21,27,28]. The lin-CD34+CD43+CD45+/− multipotent progenitors with broad lympho-myeloid potential emerge 1–2 days later. These cells have granulocyte, erythroid, macrophage, megakaryocyte (GEMM)-colony-forming cells (CFC) potential, but are essentially lacking erythroid (E)-CFC potential [27]. In one approach, we used hESC-derived CD34+ cells to generate RBCs (Fig. 1, Method A). Because E-CFCs lose CD34 expression with advanced differentiation, we isolated CD34+ cells on days 7 of coculture with OP9 when almost all erythroid progenitors express CD34 and 15%–30% of CD34+ cells coexpress CD43, a panhematopoietic marker present on all hESC-derived CFCs (Supplementary Fig. S1A; Supplementary Data are available online at
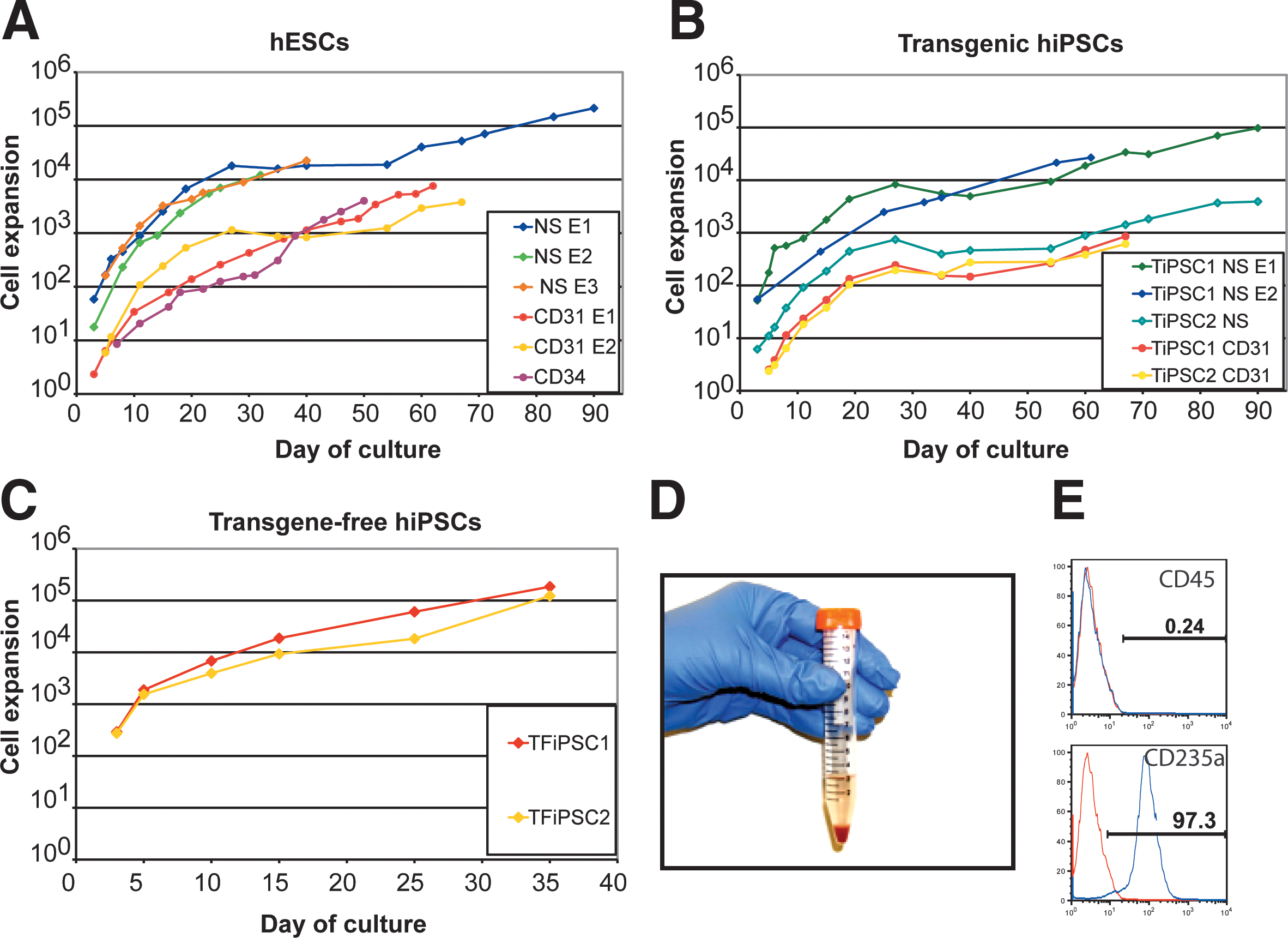
Expansion profiles of individual erythroid cultures from hESCs
Previously, we noticed that dissociation of hESC/OP9 cocultures and culture cells in nonadherent conditions in the presence of granulocyte-macrophage colony stimulating factor (GM-CSF) leads to cell re-aggregation and substantial expansion of myeloid progenitors [17]. To increase the production of RBCs from hESCs, we applied a similar approach, but used cytokines essential for erythroid cell expansion instead of GM-CSF (see Fig. 1, Method B). We collected differentiated hESCs from OP9 coculture on day 8 of differentiation and cultured them in the presence of cytokines in nonadherent conditions. After dissociation of cell aggregates and removal of dead cells by density gradient centrifugation, we were able to obtain a population of cells containing >70% of CD235a expressing erythroid progenitors. While these cells could be expanded in nonadherent feeder-free conditions as described in Method A, we found that more efficient expansion was achieved by culture of these cells with MS5 bone marrow stromal cells. The most efficient expansion occurred during the first 40 days of culture. When we continued to culture erythroid cells beyond 40 days, we found that most cell cultures continued to expand, though less robustly, and reached senescence around 70 days of expansion. However, some erythroid cultures continued to grow for up to 90 days. As shown in Fig. 2A, up to 2×105 of RBCs could be generated from 1 hESC using optimized conditions. When we tested the 2 described methods for differentiation of transgenic hiPSCs, we found a similar expansion pattern (Fig. 2B). Erythroid cells could be also produced and expanded from transgene-free iPSCs with a similar level of efficiency to that of hESCs (Fig. 2C). Congruent with our prior studies demonstrating variability in hematopoietic differentiation of transgenic iPSCs, we observed some differences in expansion potential between transgenic iPSC lines using Method B. In contrast, 2 tested transgene-free iPSCs produced very similar results consistent with our recent observation of minimal variability in hematopoietic differentiation between different transgene-free fibroblast-derived iPSCs [29].
Analysis of the erythroid cultures by flow cytometry and morphologic analysis of Wright-stained cytospins revealed that after 10–15 days of expansion more than 97% of the cells in cultures were CD235a+ erythroid cells (Figs. 2D and 3). Essentially, no myeloid cells were detected by morphologic analysis or by flow cytometry using CD45 antibodies (Figs. 2D and 4). As has been previously shown, most CD235a+ cells generated from hESCs coexpress CD41a and represent bipotential megakaryocyte-erythroid progenitors [27,28]. Culture in erythroid conditions shifted differentiation of CD235a+CD41a+ cells toward CD235a+CD41a− erythroid lineage cells (Fig. 3). Although CD235a−CD41a+ megakaryocytic cells could be detected on day 7 of expansion, these cells essentially disappeared after 10–15 days of culture. At this stage more than 98% of cells were CD235a+CD41a− erythroblasts as defined by the expression of a high level of transferrin receptor CD71 (Fig. 3). Morphologically, cells expanded for 5–20 days had the cytologic features of basophilic erythroblast with blue gray cytoplasm and nucleus with clumped chromatin (Fig. 4). Extended cocultures revealed RBCs with a developmentally more mature morphology. While occasional basophilic erythroblasts could be found even after 60 days of expansion, most cells had the appearance of polychromatic and orthochromatic erythroblasts at this point. No significant enucleation was observed when RBCs expanded in our conditions in the presence of SCF and EPO. To facilitate maturation and enucleation, erythroid cells expanded for 20–25 days were cultured on MS5 feeders without EPO and SCF. These cultures yielded smaller cells with bright pink cytoplasm, low nuclear cytoplasmic ratio, and very condensed nucleus. Although enucleation was obvious, the rate of enucleation was variable and relatively low 2%–10% (Fig. 4). Note that the differentiation pattern from hESCs and hiPSCs was essentially similar as demonstrated by flow cytometry and morphology of generated RBCs (see Figs. 3 and 4, and Supplementary Fig. S1B, C).
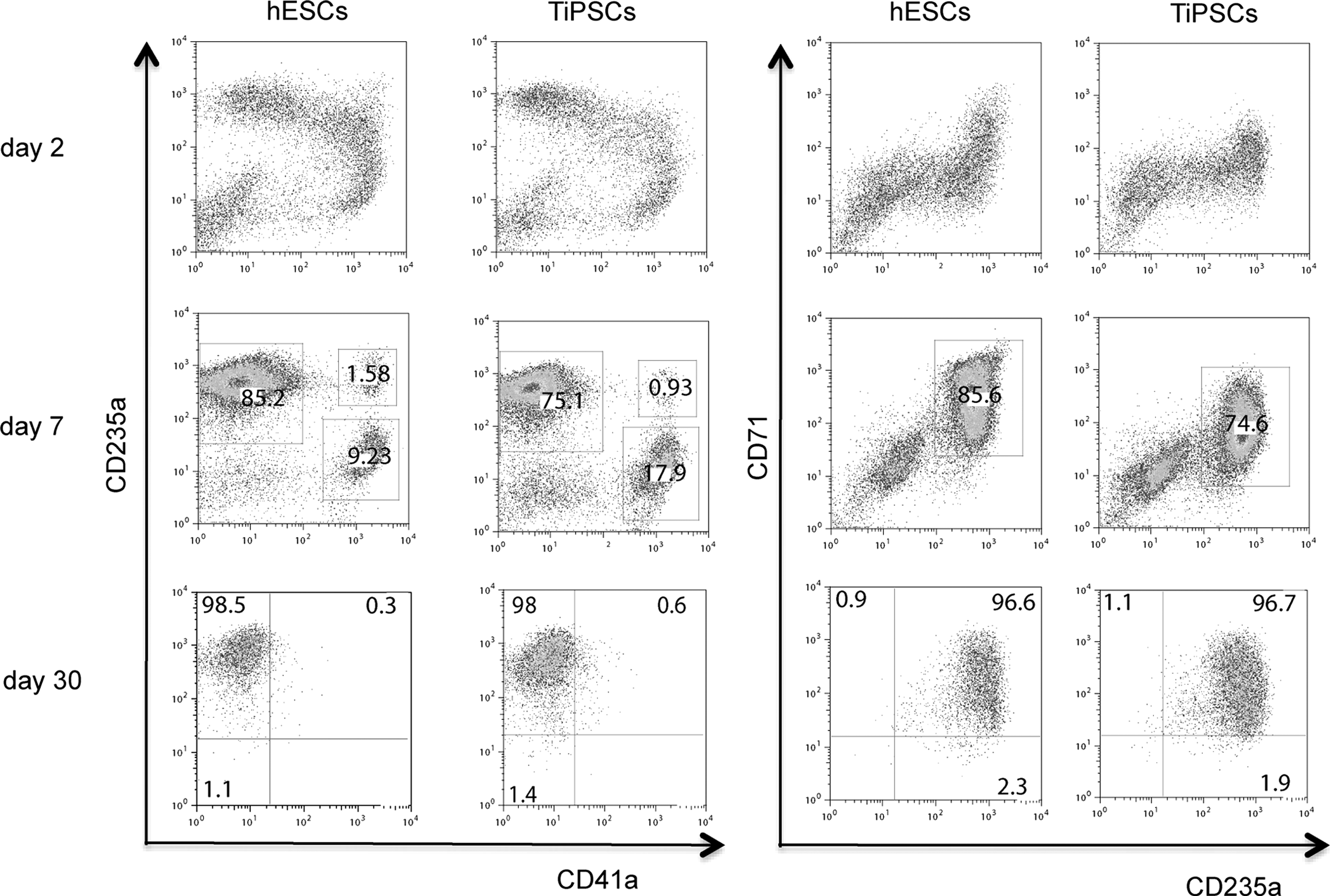
Flow cytometric analysis of erythroid cultures at different time of expansion. Representative experiments demonstrating H1 hESCs and TiPSC1 hiPSCs are shown. TiPSC, transgenic iPSC.
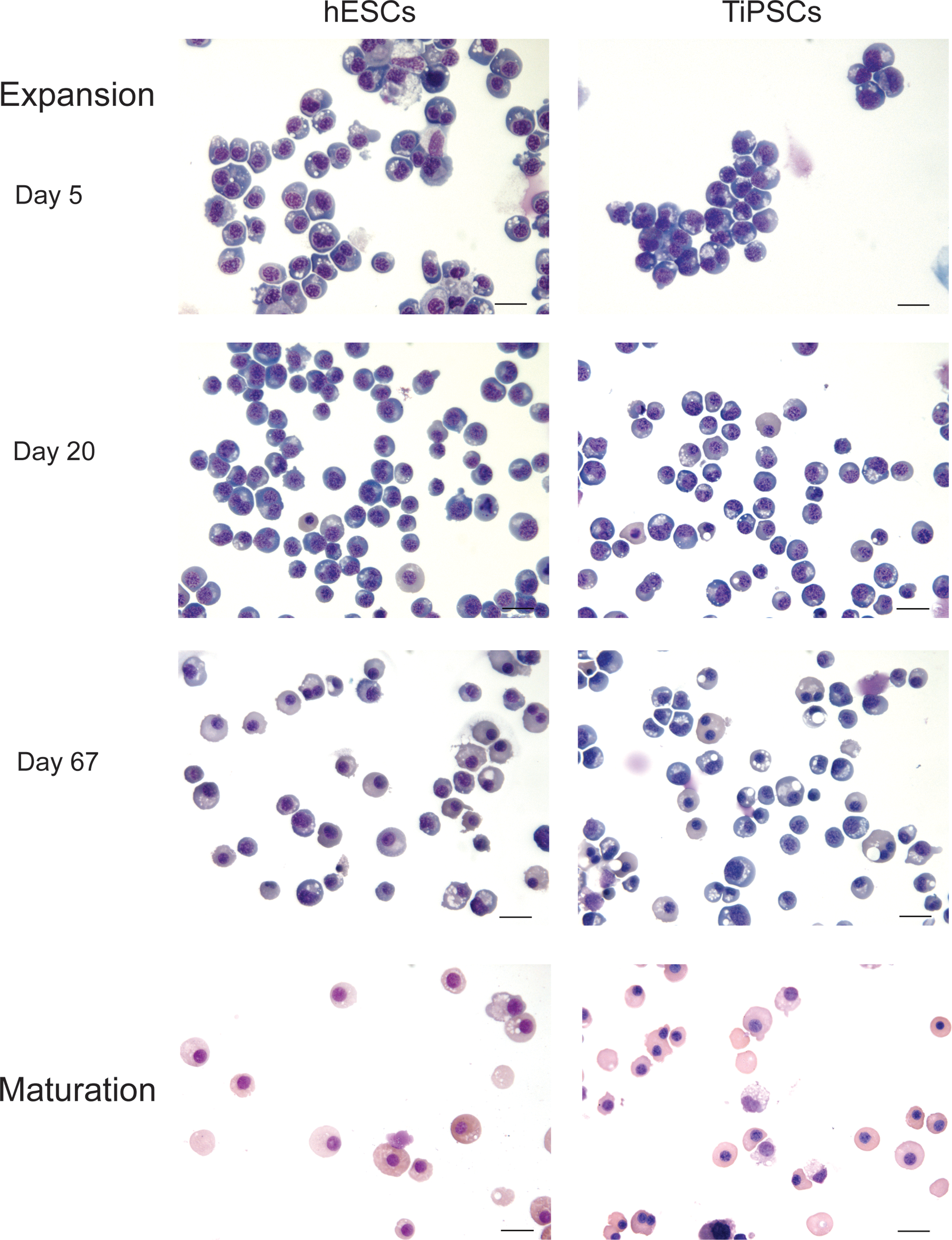
Morphology of PSC-derived erythroid cells at different stages of expansion and maturation. Wright-stained cytospin of H1 hESC and TiPSC1 are shown. Bars are 40 μm (expansion) and 20 μm (maturation). Color images available online at
To determine the type of hemoglobin expressed by hESC/hiPSC-derived erythroid cells, we analyzed cultures using qPCR and flow cytometry with primers and antibodies specific for hemoglobin ɛ (embryonic), γ (fetal), and β adult). This revealed that RBCs generated from iPSCs, similar to hESCs, produced predominately fetal and embryonic hemoglobin, consistent with complete reprogramming of globin locus. However, adult hemoglobin was detectable in our cells, as well (Fig. 5). As detected by real-time PCR, expression of all types of hemoglobin were upregulated from day 10 to day 20 of expansion probably reflecting activation of hemoglobin synthesis following advanced development of erythropoiesis in cultures. However, we noted that transgenic TiPSC1 produced less adult hemoglobin when compared with transgene-free cells.

Characterization of hemoglobin expressed in erythroid cells obtained from PSCs by quantitative polymerase chain reaction
Discussion
The in vitro system for RBC production from hESCs and hiPSCs provides a unique opportunity to study RBC development and function. This system would be ideal for loss- and gain-of-function studies to analyze the effect of specific genes on RBC development, differentiation, and function. hiPSCs generated from patients with inherited RBC diseases could be used to obtain hematopoietic and RBC progenitors carrying a particular genetic trait at the cellular level, thus offering a novel opportunity for in vitro studies of pathogenesis and drug therapies for RBC diseases. iPSCs and hESCs could be helpful to advance studies of a major malaria pathogen Plasmodium vivax. Although P. vivax is a major cause of malaria outside of Africa, and with concern for development of drug resistance, studies of this parasite are limited by lack of efficient culture technique because it infects only reticulocytes, which are difficult to obtain [30]. Since hESCs and iPSCs can be expanded indefinitely, they can be used as a constant supply of reticulocytes to allow for continuous cultures of P.vivax. In addition these cells can be engineered to express P. vivax receptor Duffy antigen/receptor for chemokines to improve parasite culture. As fetal hemoglobin may potentially inhibit malaria parasite growth [31], downregulation of hemoglobin γ through conditional BCL11A expression in erythroid cells [32] can be explored as another option to optimize hESC/hiPSC-based techniques for P. vivax cultures.
Transfusion of RBCs is the first and the most widely used form of cellular therapy in clinical practice. In addition to trauma victims, RBC transfusions are needed for surgery especially organ transplants, and for the treatment of burn victims, leukemia, cancer, sickle cell diseases, and thalassaemia major. With advances in medical treatment and an aging population, demand for blood products continues to increase [33]. Currently, RBCs utilized for transfusion therapy are obtained through voluntary donation, which creates periodic shortages and concerns about disease transfer. In addition, there is a shortage of universal donor type RBCs that can be safely transfused to anyone in an emergency situation, regardless of blood type. Manufacturing of RBCs from hESC/iPSCs provides several advantages. It can improve the continuity of blood supply, eliminate risk of infection transmission, reduce incidence of nonhemolytic transfusion reactions, and provide an opportunity to generate RBCs that fit specific clinical needs by using genetically engineered hESCs or hiPSCs with rare blood groups [34]. However, important steps should be accomplished to translate pluripotent stem cell-based technologies into the clinic. They include development of completely defined serum and animal protein free conditions for pluripotent stem cell derivation and differentiation, bioreactor-based technology for further scaling-up production, and evaluation of the therapeutic efficiency and safety of manufactured blood products in animal models [34].
Several studies described the successful production of RBCs from hESCs [7 –9,14,35]. In one approach, these cells have been cultured with fetal liver stromal cells to induced hematopoietic differentiation and subsequent production of RBCs [7,9,35]. Another approach is based on generation of blast colonies (hemangioblasts) and subsequent expansion of hemangioblast-derived RBCs using recombinant HOXB4 protein in addition to cytokines [8]. It is reported that up to 3.9×103 [14] and 1.2×104 [8] RBCs can be generated from 1 hESC [8,14]. Our differentiation approach allows for generation of RBCs with similar (Method A) or higher efficiency (Method B), and it does not require use of cytokines for induction of hematopoietic differentiation or recombinant HOXB4 for RBC expansion. Two groups recently obtained RBCs from transgenic hiPSCs [14,15]. Interestingly, Feng et al. [15] found significant (approximately 1,700-fold) differences in the expansion potential of erythroid cells from blast colonies derived from hESCs and hiPSCs, using the embryoid body method. In our studies we show that RBCs can be generated from transgenic and transgene-free hiPSCs with efficiency similar to hESCs. The reason for these discrepancies is not clear. Since Feng et al. [15] observed early senescence in nonhematopoietic lineages as well, it is possible that the limited expansion potential of hematopoietic derivates may be related to intrinsic properties of the employed hiPSC lines or specific conditions used for their maintenance. Because a recent study indicates that hESCs and different hiPSC lines require different concentrations of cytokines for efficient induction of mesodermal lineages [36], it is possible that embryoid body conditions are optimal for erythroid differentiation from hESCs but suboptimal for hiPSCs, whereas OP9 coculture supports an efficient induction of blood cells from both, hESCs and reprogrammed fibroblasts.
In summary, the findings reported here demonstrate that RBCs could be produced from transgenic and transgene-free iPSCs using the OP9 coculture method with efficiency comparable to hESCs. The hiPSC-derived erythroid cells show a hemoglobin expression profile similar to hESC-derived RBCs, indicating complete reprogramming of β-globin gene locus after fibroblast conversion to pluripotent state. The hiPSC-based technology described here provides an opportunity to generate an essentially homogenous population of erythroid cells for in vitro modeling of RBC diseases, drug screening, malaria studies, and evaluation of the role of specific genes in erythroid lineage development. In addition, hiPSC-based technologies could pave the way toward manufacturing of RBCs for transfusion therapies.
Footnotes
Acknowledgments
We thank Toru Nakano for providing OP9 cells and Joan Larson for editorial assistance. This work was supported by funds from the National Institutes of Health (R01 HL081962, P01 GM081629, and P51 RR000167), DARPA grant 7003-UWM, and Charlotte Geyer Foundation.
Author Disclosure Statement
J.A.T. owns stock, serves on the board of directors, and serves as chief scientific officer of Cellular Dynamics International. J.A.T. also serves as scientific director of the WiCell Research Institute. I.I.S. owns stock and is the scientific founder of Cellular Dynamics International.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.