
Abstract
While multipotent mesenchymal stromal cells have been recently isolated from adult lung (L-MSCs), there is very limited data on their biological properties and therapeutic potential in vivo. How L-MSCs compare with bone marrow-derived MSCs (BM-MSCs) is also unclear. In this study, we characterized L-MSC phenotype, clonogenicity, and differentiation potential, and compared L-MSCs to BM-MSCs in vivo survival, retention, paracrine gene expression, and repair or elastase injury after transplantation. L-MSCs were highly clonogenic, frequently expressed aldehyde dehydrogenase activity, and differentiated into osteocytes, chondrocytes, adipocytes, myofibroblasts, and smooth muscle cells. After intravenous injection (2 h), L-MSCs showed greater survival than BM-MSCs; similarly, L-MSCs were significantly more resistant than BM-MSCs to anchorage independent culture (4 h) in vitro. Long after transplantation (4 or 32 days), a significantly higher number of CD45neg L-MSCs were retained than BM-MSCs. By flow cytometry, L-MSCs expressed more intercellular adhesion molecule-1 (ICAM-1), platelet derived growth factor receptor alpha (PDGFRα), and integrin α2 than BM-MSCs; these proteins were found to modulate endothelial adherence, directional migration, and migration across Matrigel in L-MSCs. Further, L-MSCs with low ICAM-1 showed poorer lung retention and higher phagocytosis in vivo. Compared with BM-MSCs, L-MSCs expressed higher levels of several transcripts (e.g., Ccl2, Cxcl2, Cxcl10, IL-6, IL-11, Hgf, and Igf2) in vitro, although gene expression in vivo was increased by L-MSCs and BM-MSCs equivalently. Accordingly, both L-MSCs and BM-MSCs reduced elastase injury to the same extent. This study demonstrates that tissue-specific L-MSCs possess mechanisms that enhance their lung retention after intravenous transplantation, and produce substantial healing of elastase injury comparable to BM-MSCs.
Introduction
M
Methods
Animals and cell lines
All studies were approved by the Institutional Animal Care and Use Committee at Tufts University. Female C57BL6/J mice were used as recipients for transplantation assays in vivo. These mice were delivered elastase (1.5 IU porcine pancreatic elastase) intratracheally at 5 weeks of age to induce emphysema as previously described [36] and cells were delivered 6–7 weeks later.
L-MSCs were isolated from minced lung tissue of donor male C57BL6/J mice (8 weeks age). Lungs were flushed to remove blood and tissues minced into fragments (∼0.5 mm3) for culture in polystyrene plates coated (300 μl) with basal media (alpha minimum essential media [MEM], 15% FBS, L-glutamine 2 mM/L, penicillin 100 IU/mL, streptomycin 100 μg/mL, and amphotericin B 0.25 μg/mL). After 12–16 days of outgrowth, cells were passaged onto large dishes (150 cm2) using trypsin 0.25%/ethylenediaminetetraacetic acid (EDTA) 0.01% or trypsin-free reagents (TrypLE Express, Invitrogen; Enzyme Free, Millipore) as indicated in the text. Passage 7(P7) L-MSCs were used for in vitro studies of phenotype and function, and in vivo transplantation assays. Cryo-preserved BM-MSCs (P5) were derived from male C57Bl/6-TgN(ACTbEGFP)1Osb mice obtained from the Texas A&M Health Science Center and used for in vitro assays and in vivo retention studies at passage 7. Non-GFP (wild-type) male BM-MSCs from C57BL6 mice (kind gift from Dr. Marc Hershenon, University of Michigan) were employed at passage 7 for in vitro and in vivo studies of paracrine signals and treatment of elastase-injured mice.
Clonogenic assays, PDT, and PDs
Clonogenicity of L-MSCs was analyzed by plating cells onto 100 mm dishes (2,000 cells/plate, n=3) for enumeration of colony-forming units (CFU) defined as colonies with ≥50 cells at 10–12 days of subculture in basal media. Limiting dilution was used to further characterize clonal efficiency and to subculture clones at P4. Population doubling time (PDT) was measured in log phase cultures (P7, n=4), where PD=log2 (Ch/Cs) and PDT=time elapsed/PD, whereas PD is population doublings, Ch is number of cells harvested, and Cs the number of cells seeded.
Differentiation assays of L-MSCs
Subconfluent cells grown in adipogenic (StemX-Vivo; R&D systems), chondrogenic, or osteogenic (StemPro; Invitrogen) media for 21–28 days were fixed in formalin (4%) and stained appropriately. Cells were also grown on Matrigel (reduced growth factor, BD) diluted (1:1) with either basal media or endothelial growth media (EGM) containing recombinant human vascular endothelial growth factor (rhVEGF) (5 ng/mL), epidermal growth factor (EGF) (5 ng/mL), fibroblast growth factor (FGF) basic (5 ng/mL), and insulin like growth factor (IGF-1) (15 ng/mL) [American Type Culture Collection (ATCC)] to assess their capacity for tube formation and Dil-Ac-LDL (InVitrogen) uptake. They were also incubated in EGM for evaluation of endothelial differentiation based on quantitative real-time polymerase chain reaction (qPCR) including CD31/PECAM, Flk-1/Kdr, vWF, and Cad5/Vcad applying commercial primers (Supplementary Table S1; Supplementary Data are available online at
Surface protein expression in L-MSCs versus BM-MSCs
Immunophenotype was measured using flow cytometry (Accuri C6, Ann Arbor or FACS Calibur, BD). Cells were harvested using trypsin/EDTA and blocked with anti-FcR (CD16/32, eBioscience; 0.25 μg/mL, 15 min, 4°C). Cells were incubated with primary antibodies (2.5–10 μg/mL, 30 min, 4°C) conjugated to allophycocyanin (APC), phycoerythrin, or biotin. Antibody clones and manufacturers are detailed in the Supplementary Methods. Dead cells (7-aminoactinomysin D [7-AAD]pos) were excluded from analysis. Measurements were acquired using Cell Quest (v.4.0, BD) or CFlow (v.1.0.208.2, Accuri), and post hoc analyses with Summit (v.4.3, Beckman Coulter) or CFlow, respectively. Differentially expressed proteins (L-MSCs vs. BM-MSCs) were further corroborated by immunoblots, with protein concentrations normalized to beta actin (details of methodology are provided in the Supplementary Methods).
The effect of PKH26 staining on cell death (7-AADpos) and apoptosis (Annexin V, 5 μL/106 cells, eBioscience) was measured by flow cytometry. Anoikis resistance was evaluated by incubating L-MSCs or BM-MSCs at P7 (5×104) in polystyrene tubes subject to gentle agitation and measurement of cell death and apoptosis as above using flow cytometery (n=5/time point, 0–4 h).
Transplantation assays
All in vivo studies were approved by the Institutional Animal Care and Use Committee at Tufts University. Mice were transplanted with cells intravenously by tail vein injections 6 weeks after elastase delivery. Cells were labeled for studies of cell retention or in vitro assays with PKH26 (Sigma) according to the manufacturer's instructions. Lungs in the retention studies were processed by freezing after inflation (∼1–2 mL) with polyvinyl alcohol plus polyethylene glycol solution. For studies examining effects of cell transplantations on lung architecture, lungs were fixed intratracheally with 1% low melt agar in 10% formalin at 25 cm pressure before embedding and sectioning. Sections (5 μM) at each of 2 random cutting depths, 200 μM apart, were stained with hematoxylin and eosin and 30 randomly selected fields (15 per level) of alveolar parenchyma were photographed (200×) for measurement of mean linear intercept (MLI) [36].
For flow cytometric analysis in mice receiving cell transplants, single cell suspensions were generated from one lung lobe by collagenase/dispase (2 mg/mL) digestion. Cells were stained from each animal with APC-conjugated antibodies, including CD31, CD45.2, CD73, CD90.2, CD105, and CD11b (see Supplementary Methods for reagent details). Representative cryosections (n=3/animal) from L-MSC-injected mice were also immunostained for phenotypic proteins, including proSP-C, Ttf-1, aqp5, CD31, α-sma, vimentin, S100A4, or CD45 (detailed methods in the Supplementary Methods). Staining of tissue sections was analyzed using deconvolution microscopy and co-localization software (AxioVision v. 4.5, Carl Zeiss) that resolved digital image depth to 1uM, and measured pixel by pixel the Pearson correlation coefficients relating PKH to immunostain fluorescence of retained cells.
Endothelial adhesion assays
Adherence to endothelial monolayers (human umbilical vein endothelial cells [HUVECs]; ATCC) was evaluated in cells labeled with PKH for quantification with fluorescent microscopy or left unlabeled for counting under phase contrast (200×). Cells were adjusted to 106/mL and plated (5×104/well, 24 wells) onto HUVECs for 1 h. ICAM-1 function was antagonized using functional blocking antibody (clone Y1N/1.7.4, eBioscience, 30 μg/mL, 60 min, 4°C) or isotype. Wells were fixed in formalin and cell adherence measured as total fluorescence or total adherent cells counted manually per field (4 fields/well, 4 wells/group).
Migration assays
Migration assays were performed using 8 μM pore size transwells (Corning). The top well was not coated, or coated with Matrigel (Matrigel Invasion Chambers, BD, 2 mg/mL) using 6 replicates per group. Bottom wells were loaded with chemo-attractant [15% fetal bovine serum (FBS)], or control media (1% FBS, 56°C heat inactivated), or with specific chemo-attractants (human recombinant platelet derived growth factor AA [rPDGFAA] or FGF2, US Biologics, or rat recombinant platelet derived growth factor BB [rPDGFBB,] Sigma; 25 ng/mL). Cell suspensions (5×105 cells/100 μL) were loaded onto the top wells and migration to the underside was evaluated (24 h) by staining with crystal violet, dissolving with acetic acid (5%), and quantification of absorbance (560 nM). Migration across Matrigel-coated porous (8 μM) membranes was evaluated as follows: cells (2.5×104/well) were plated in the top well, and after 24 h the underside was fixed and stained (DipQuik) for counting of cells in 5 equidistant fields (100×) per well. In another experiment, Itg α2 blocking Ab (HMα2, eBioscience) or isotype (10 μg/mL) was introduced to the media at the time of seeding onto Matrigel-coated top wells, and cell migration across the Matrigel and porous layer was assessed (24 h) by cell counting as above.
ECM substrate-binding assays
The capacity for cells to adhere to extracellular matrix (ECM) substrates was tested in 96-well plates precoated (60 min, 23°C) with fibronectin, gelatin, collagen 1, poly-L-lysine, or heparin sulfate (20 μg/mL), or bovine serum albumin (20 μg/mL, negative control). Wells (n=6/substrate) were blocked with BSA (1%) for 30 min, then seeded with 2×104 cells, and evaluated after 1 h. Cells were stained with crystal violet, dissolved in acetic acid, and adherence measured by absorbance (560 nm).
Statistical analysis
Multiple comparisons between independent groups were analyzed using analysis of variance (ANOVA), and pairwise comparisons using Student's t-test. Time dependent data were analyzed using repeated measures ANOVA and post hoc Student's t-test. qPCR was analyzed using webtools (SABioscience) by published methods [38]. Otherwise, data were analyzed using commercial software (SPSS, v.13.0.1 or Excel SP3, Microsoft). Data are presented as mean±standard error of the mean unless otherwise indicated. A P value <0.05 was considered statistically significant.
Results
Outgrowth of minced lung yields multipotent MSCs
Explants grown on plastic in basal media consistently yielded large numbers of crescent-shaped fibroblastic cells with pronounced lamellopodia and filipodia (Fig. 1A, B). When plated at low density, intense colony formation was evident (Fig. 1C, D). Morphology of cells remained consistent over 29 passages (Supplementary Fig. S1A–F). The colony-forming efficiency [CFU, mean±standard deviation (SD)] at passage 3 (P3) was 6.8%±0.3%, which increased to 18.0%±0.9% (P5) and plateaued at 24.8%±1.8% (range 22.7%–26.4%) between P7 and P32 (Supplementary Fig. S1G). Clonogenic efficiency determined by limiting dilution at P7 was similar (36.2%±2.5%). Consistent with the CFU data, the frequency of aldehyde dehydrogenase-positive cells was relatively consistent between P8 (17.6%) and P30 (21.4%) (Supplementary Fig. S1H, I). PDT was 44.4±1.2 h at P7; there were 110 PDs between P2 and P32 (∼3.8 per passage). Cells in S phase comprised 35.1% and 36.5% of P7 and P29 cells, respectively, and ∼100% of P7 or P29 cell populations were diploid (Supplementary Fig. S1J, K). Individual clones sub-cultured from the parent population by limiting dilution (n=11) exhibited fibroblastic morphologies (crescent, spindle shaped) but a wide range of CFU, ranging 2%–35% at passage 7 (Supplementary Fig. S2A, B). The mean CFU efficiency of all 11 clones at P7 was 16.8%±8.6% versus 22.7%±2.7% for the parent population (N/S). Only 1 of 11 clones and none of the parent cultures exhibited a lipofibroblastic appearance (Supplementary Fig. S2A).
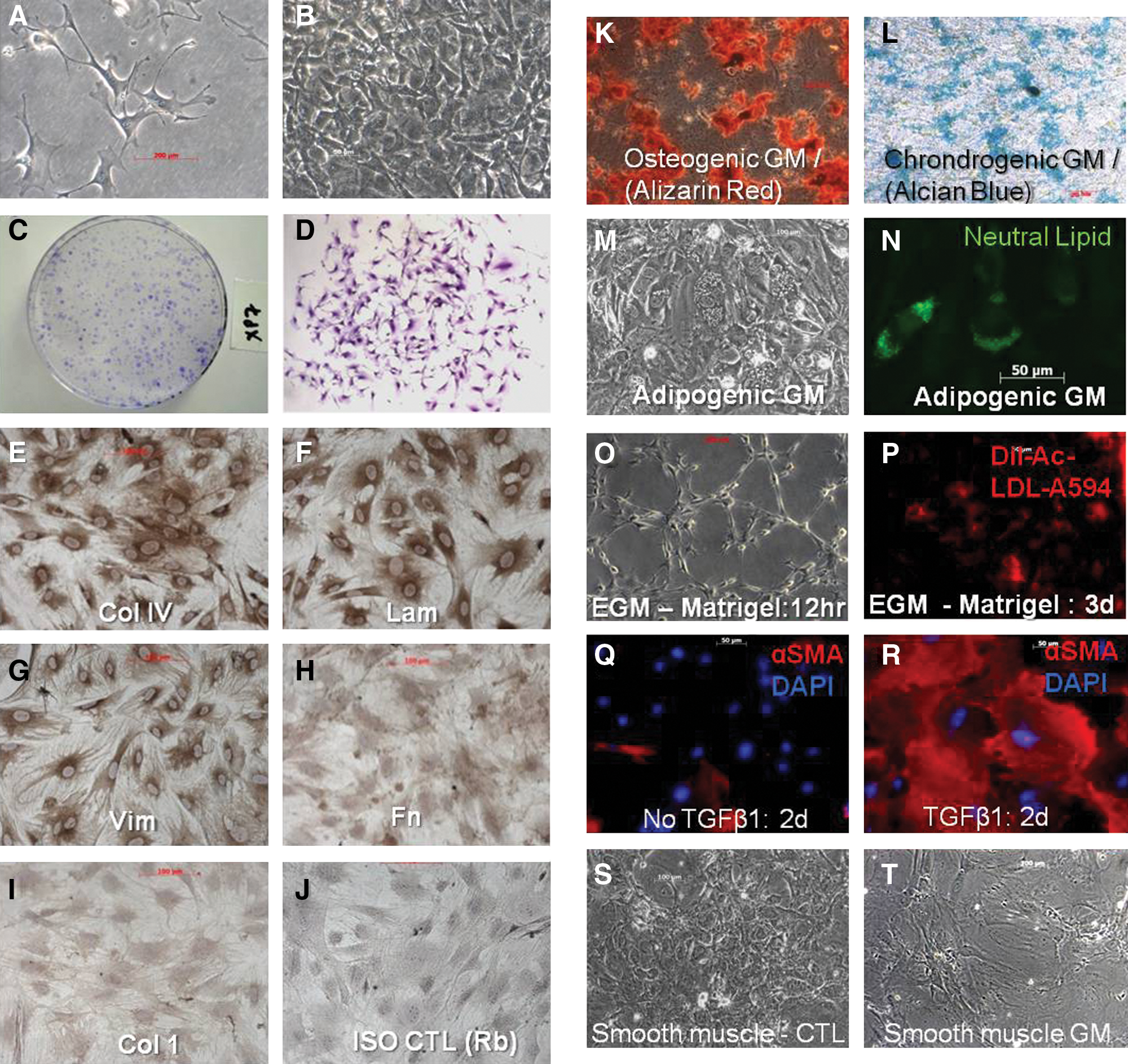
The culture phenotype and differentiation potential of L-MSCs. Lung-derived MSCs (L-MSCs) were cultured in basal media (alpha MEM, 15% FBS) by the outgrowth method on plastic.
L-MSCs grown on plastic in basal media uniformly expressed vimentin, collagen IV, and laminin, to a lesser extent fibronectin, but no collagen 1 (Fig. 1E–J). Cells could be differentiated into adipocytes, chondrocytes, or osteocytes based on histochemical staining (Fig. 1K–N). Adipocyte differentiation was further supported by significantly increased mRNA expression of Lpl and Pparγ (Supplementary Table S1). In EGM L-MSCs consistently formed tube-like structures (Fig. 1O) and initiated uptake of Dil-Ac-LDL (Fig. 1P); however, EGM did not induce expression of endothelial genes (Supplementary Table S1). In response to TGFβ1, L-MSCs developed a myofibroblastic morphology with increased expression of α-sma (Fig. 1Q, R), and increased expression of myofibroblastic genes (e.g., Acta2, Col 1A1, Lox, TnC, and Fn) and senescence genes (p53 and Cdkn1b), and decreased expression of survival genes (midkine and Birc5) consistent with a pronounced shift toward terminal differentiation (Supplementary Table S2). Cells did not increase expression of fibroblast activation proteins [39] such as FAPα, Dpp4, and S100A4 in response to TGFβ1 (Supplementary Table S2). Small airway growth media caused a significant increase in CCSP (Scgb1A1) and Aqp5 expression, but not Ttf-1 or proSP-C (Sftpc). L-MSCs exposed to the demethylating agent 5 Aza-cytidine displayed a smooth muscle cell phenotype (Fig. 1S, T) and significant upregulation of Des, Acta2, and MyoD1 (Supplementary Table S1). In sum, L-MSCs showed evidence of in vitro differentiation to mesenchymal lineage cells, including chondrocytes, osteocytes, adipocytes, myofibroblasts, and smooth muscle cells.
Retention after transplantation
At 32 days after transplantation into elastase-injured mice, L-MSCs comprised 0.53%±0.09% of all viable cells in the lungs versus 0.032%±0.022% for BM-MSCs, a 16.5-fold difference (P<0.001, Fig. 2A, B). The percentage of PKH-labeled cells that were identified as CD45neg in tissue sections was significantly higher for L-MSC-injected (mean±SD, 86.7%±2.5%) than BM-MSC-injected (57.4%±1.6%) animals (Fig. 2C, D lower left panel). Overall, L-MSCs that were CD45neg were retained at ∼25 times the rate of BM-MSCs. Moreover, L-MSCs formed contiguous clusters of ∼5–20 cells interspersed throughout damaged and healthy parenchyma; in contrast, BM-MSCs were found exclusively as singlets (Fig. 2D). Based on flow cytometry, retained L-MSCs were enriched for CD73 (76%±5.5%) and CD105 (73%±2.9%), markers that were present at high frequencies (>90%) on L-MSCs in vitro (Fig. 2E). Very few L-MSCs displayed CD31 or Sca-1.
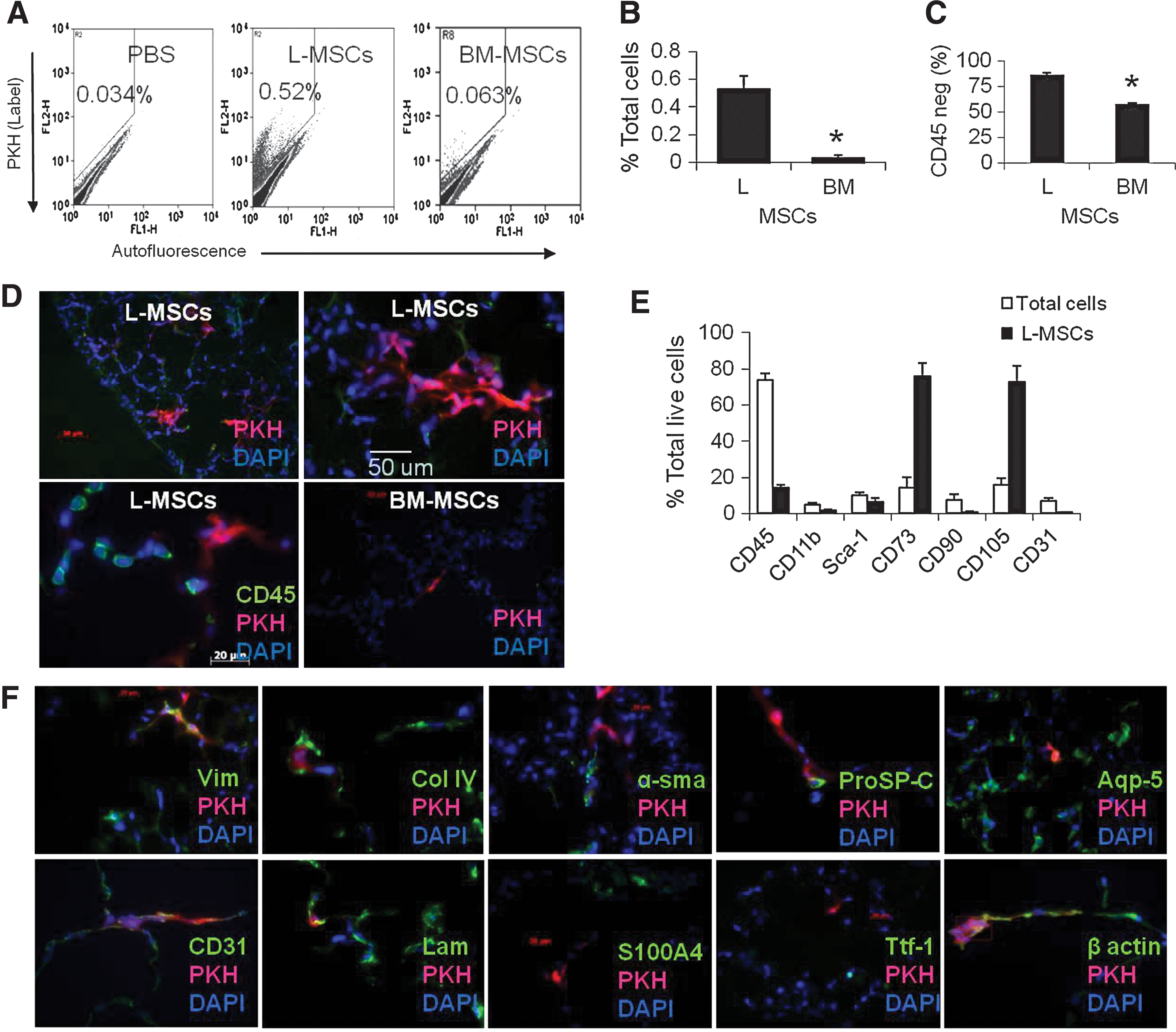
Retention of L-MSCs versus BM-MSCs was evaluated 32 days after syngeneic transplantation (1×106 cells IV) in a murine model of emphysema.
The PKH dye in L-MSCs was co-localized in tissue sections to vimentin (Pearson r 2=0.82) and beta actin (r 2=0.71), weakly co-localized with proSP-C (r 2=0.24) and CD31 (r 2=0.18), and showed no evidence of co-localization with Ttf-1 (r 2=0.0), aqp5 (r 2=0.0), α-sma (r=0.0), or S100A4 (r 2=0.06) (Fig. 2F). Therefore, transplanted L-MSCs did not show evidence of transdifferentiation to epithelial or endothelial cells, or differentiation to myofibroblasts or smooth muscle cells in vivo.
Retention of L-MSCs at 4 days postinjection was significantly (P<0.001) greater than BM-MSCs (Fig. 3A). Growth phase (log vs. stationary) had no impact on cells retained. The total L-MSCs was also the same for elastase-injured and noninjured mice. The total percentage of L-MSCs that were CD45 or CD11b (macrophage marker) positive was significantly lower than BM-MSCs 4 days after injection (Fig. 3A).

Transplantation assays comparing L-MSCS and BM-MSCs retention and evidence for phagocytosis (day 4), early survival (2 h), and in vitro assay to explore potential mechanism for survival (anoikis resistance).
The higher retention of L-MSCs at 4 and 32 days led us to inspect more immediate events after transplantation. There was no difference in entrapment at 2 h after injection (L-MSCs: 0.85%±0.10% vs. BM-MSCs: 0.87%±0.27%); distribution of each appeared stochastic within the alveolar parenchyma and mirrored distribution of inert spheres (Supplementary Fig. S3). At 2 h the viability of L-MSCs already exceeded BM-MSCs (P<0.05) and there was a trend toward fewer apoptotic L-MSCs (P=0.07) (Fig. 3B). The difference in viability or apoptosis was not caused by the labeling of cells with PKH (Supplementary Fig. S4). In vitro, L-MSCs better resisted apoptosis and cell death than BM-MSCs when incubated in anchorage independent conditions (Fig. 3C, D). These results suggest that active rather than passive mechanisms are likely to contribute to the superior survival and retention of L-MSCs in vivo.
Differential expression of surface proteins and functional significance
A variety of cell–cell and cell–matrix proteins were compared between L-MSCs and BM-MSCs using flow cytometry (Fig. 4A). L-MSCs were found to lack endothelial, epithelial, and hematopoietic cell markers, but displayed MSC markers consistent with their designation as MSCs (CD44, CD73, CD90, CD105, and CD106). Out of the 40 surface proteins assessed, the frequency of expression was substantially (>70%) higher in L-MSCs for ICAM-1 (CD54), PDGFRα (CD140a), and integrin α2 (CD49b), and these differences in expression were corroborated using western blots (Fig. 4B–E). The difference in expression of ICAM-1 persisted after stimulation with tumor necrosis factor alpha (rhTNFα, Sigma; 10–50 ng/mL, 24 h) (Fig. 4F) without altering cell viability (data not shown).
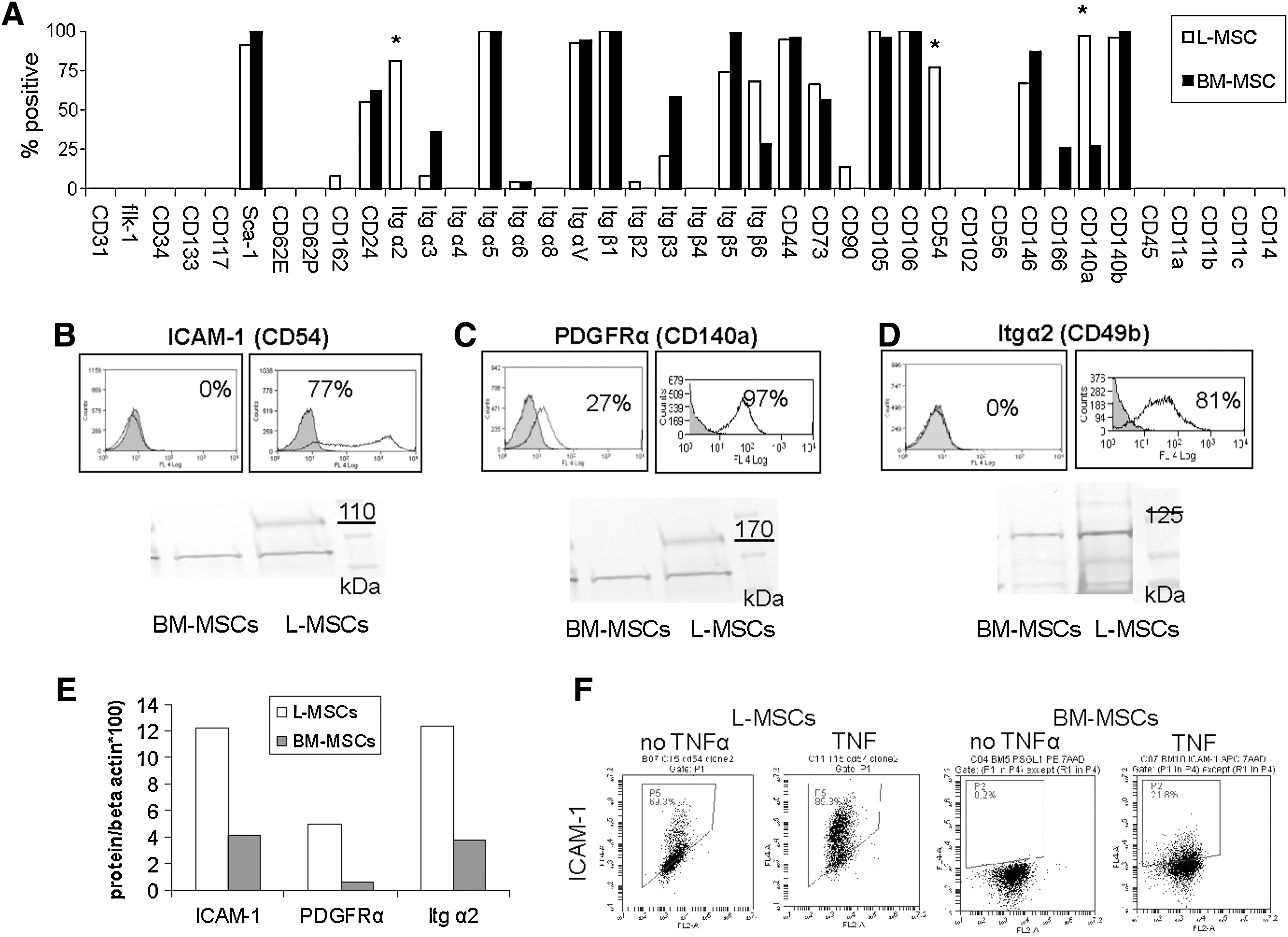
Comparison of surface proteins expressed on L-MSCs versus BM-MSCs used for transplantation assays by flow cytometry.
Since ICAM-1 is an important cell–cell adherence molecule in the lung, its role in endothelial adherence of L-MSCs versus BM-MSCs was examined specifically. Attachment to endothelium was significantly greater for L-MSCs. There was ∼5-fold greater adherence based on phase microscopy (Fig. 5A, D), and ∼2.5-fold greater adherence measured using fluorescence (Fig. 5B, E). Adherence to endothelial monolayers was significantly reduced (∼50%) by saturating concentrations of functional blocking antibody in L-MSCs, not BM-MSCs (Fig. 5A, B). Blockade of endothelial adherence by L-MSCs was also antibody concentration dependent (Fig. 5C).
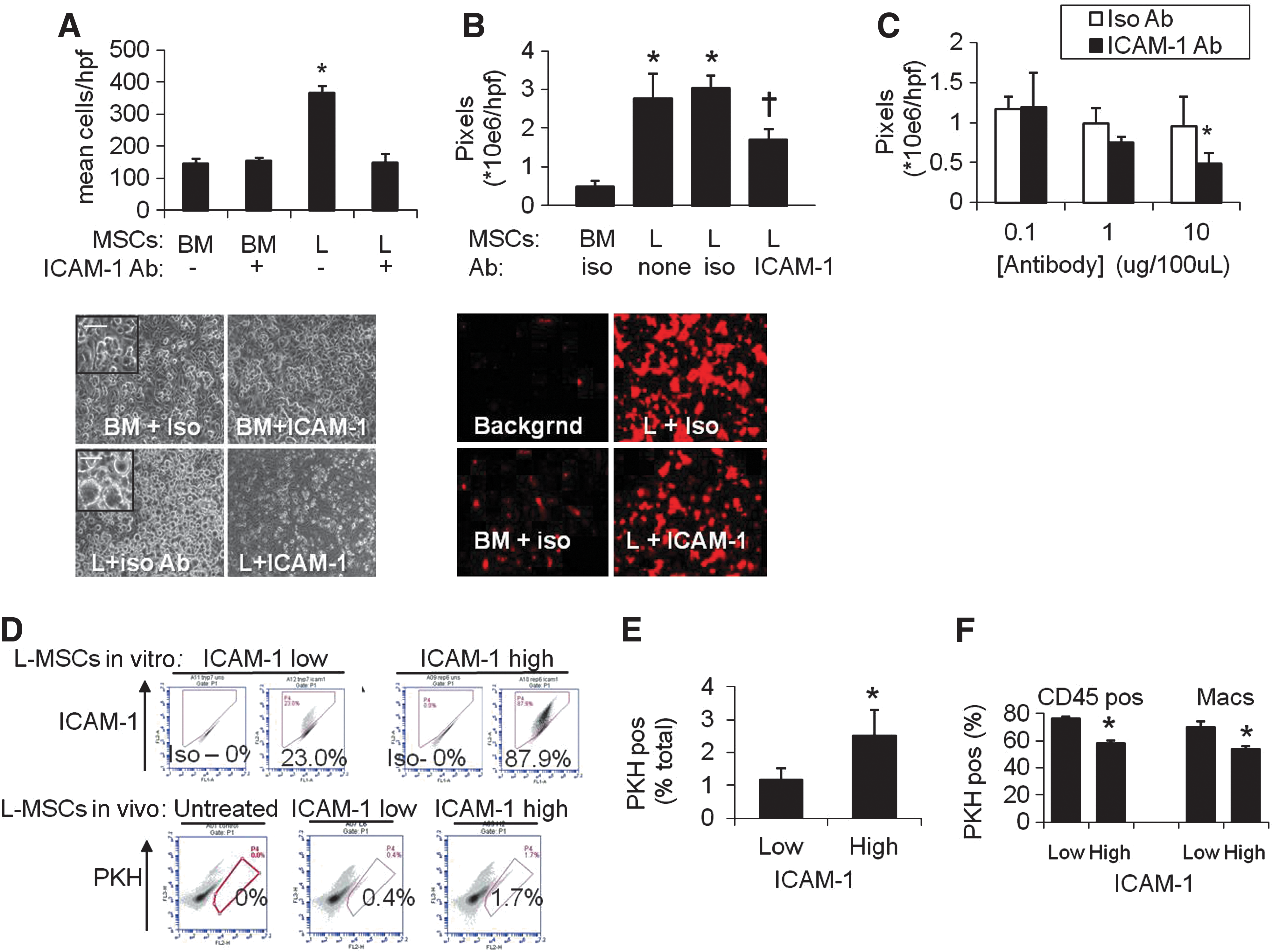
Endothelial attachment was compared at 1 h between L-MSCs and BM-MSCs.
We noted that repeated passage of L-MSCs using trypsin decreased ICAM-1 expression (Supplementary Fig. S5A, D), not PDGFRα or Itg α2 expression (data not shown). This was not due to an effect on antigen detection (Supplementary Fig. S5B). In contrast, ICAM-1 expression did not diminish if cells were passaged with trypsin substitute (TypLE Express). High versus low ICAM-1 cell lines (trypsin-free vs. trypsin passaged, respectively) were injected into elastase-injured mice. High ICAM-1 cells were retained at a significantly higher rate than low-ICAM-1 cells (Fig. 5F, G) and fewer high-ICAM-1 cells expressed CD45 or macrophage markers (Fig. 5H). These observations support the role of ICAM-1 in endothelial adherence and in vivo retention and survival of L-MSCs after transplantation.
Both random migration and directional migration induced by serum were significantly more efficient in L-MSCs than BM-MSCs (Fig. 6A). Matrigel reduced migration in both L-MSCs and BM-MSCs, but L-MSCs migrated randomly and directionally across Matrigel much better than BM-MSCs. Likewise, PDGFAA, PDGFBB, and FGF2-induced migration of L-MSCs more efficiently than BM-MSCs, with BM-MSCs responding only to PDGFBB (Fig. 6B). It was also observed that Itg α2 blockade significantly reduced migration across Matrigel by L-MSCs, not BM-MSCs (Fig. 6C).
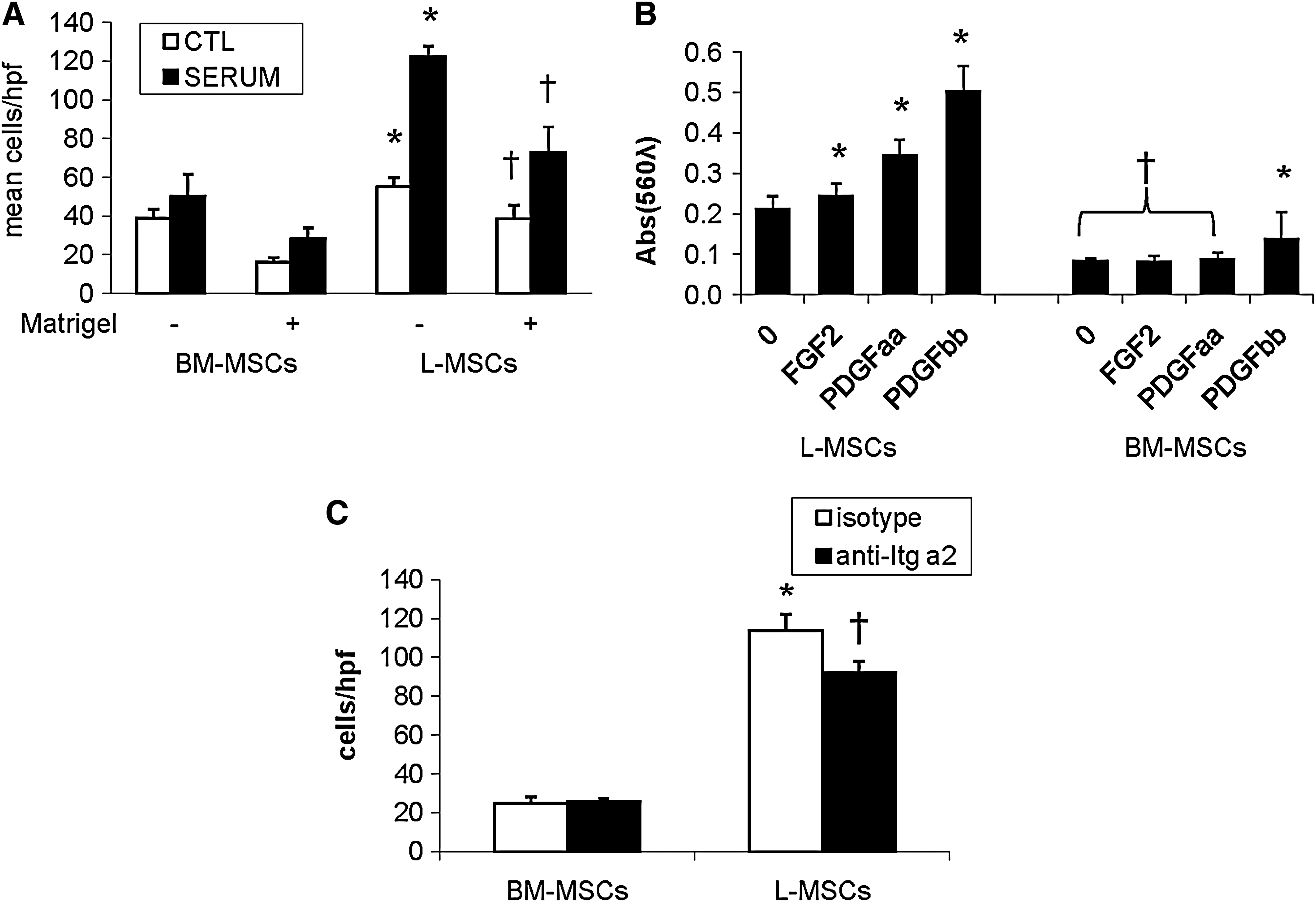
Migration and invasion assays in vitro.
Lung MSCs and BM-MSCs bound ECM substrates similarly, although BM-MSCs showed more avid binding to collagen (Supplementary Fig. S6).
Gene expression in vitro
A wide screen of ECM, growth factor, and chemokine and cytokine gene expression vitro revealed that L-MSCs significantly overexpressed many genes, including ICAM-1 (69-fold) and Itg α2 (3.6-fold) mRNA, consistent with flow cytometry and immunoblots. L-MSCs overexpressed ECM-related genes, including MMP-3, MMP-13, and MMP-14 (Supplementary Table S3), the latter of which is recognized as an important modulator of directional migration in fibroblasts [40]. Several growth factors and cytokines were markedly (>15-fold) overexpressed in L-MSCs, including Igf2, Figf, Hgf, Fgf18, IL-6, IL-11, and Lefty. Also upregulated were Vegfa-c. Several chemokines were markedly overexpressed in L-MSCs, including Ccl2, Ccl5, Ccl7, Cxcl1, Cxcl2, and Cxcl10 (>15-fold), as was chemokine receptor Cxcr7 (>60-fold), but not Cxcr4. In contrast, BM-MSCs overexpressed Inhibin A and B, and Spp1/osteopontin. After transplantation of L-MSCs, BM-MSCs, or phosphate-buffered saline (PBS), gene expression was measured in whole lung tissue at 6 and 24 h (n=4/group). While L-MSCs overexpressed most of the genes selected for analysis in vitro, L-MSCs and BM-MSCs (vs. PBS treatment) induced gene expression at similar magnitudes in the lung tissue in vivo (Fig. 7A).
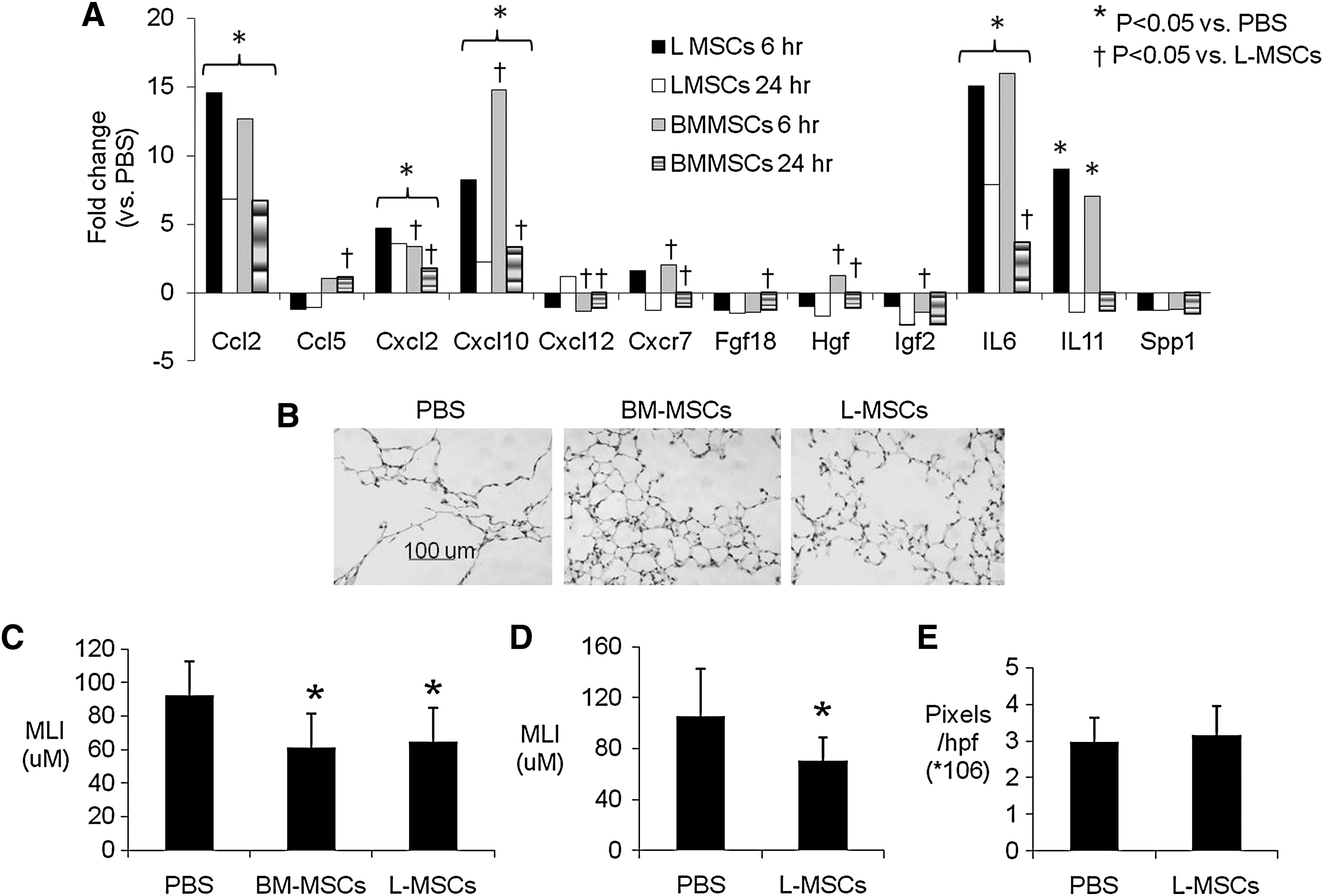
Effect of L-MSCs versus BM-MSCs on gene expression in vivo and repair of elastase injury.
Effects of cell transplantations on elastase injury
Both L-MSCs and BM-MSCs (0.5×106) caused a profound decrease in MLI 22 days after treatments (Fig. 7B, C). In a separate experiment, it was found that repeated intravenous injections of L-MSCs at lower concentrations (3 injections, 0.33×106 cells/injection) elicited a similar reduction in MLI in elastase-injured mice 28 days after the last injection (Fig. 7D). With concern that repeated injections of L-MSCs might incite fibrosis, picrosirius staining was quantified as a measure of collagen content. There was no difference in picrosirius staining between L-MSCs and PBS-treated mice (Fig. 7E).
Discussion
This study shows that lung outgrowths yield highly clonogenic, plastic-adherent, multipotent stromal cells that exhibit a broad range of matrix and growth promoting signals, persistent in the lung longer than BM-MSCs in the lung, and exert substantial reparative effects. These L-MSCs expressed unique surface proteins when compared with BM-MSCs, which accounted for differences in endothelial adherence and migration in vitro, important facets of lung retention and engraftment. Lung MSCs were also more resistant to anchorage independent culture conditions than BM-MSCs, which offers another potential mechanism by which L-MSCs avoid cell death and phagocytosis soon after transplantation. Differences in lung retention, however, did not predict differences in gene expression and repair of elastase injury, which were not different for L-MSCs versus BM-MSCs. The implication is that L-MSCs possess mechanisms that augment their persistence in the lung, but mechanisms of tissue healing are independent of this process in our model.
This study is unique in that methods traditionally used to isolate lung fibroblasts were used to isolate L-MSCs, and furthers the notion that MSCs and fibroblasts are inextricably related [41 –43]. In past studies, L-MSCs were isolated by direct sorting (ie, Sca-1high, C31neg, C45neg, EpCamneg) [44] or Hoechst dye efflux [24]. The end result of all methods has converged on isolation of plastic-adherent, fibroblastic cells with markers indicative of MSCs (e.g., C44pos, C73pos, C90pos, C105pos, C106pos) [28 –30,43,45]. Previous reports also describe MSCs as highly replicative and clonogenic, as observed here in passaged L-MSCs, and in past studies of outgrowth-derived L-MSCs [31,32]. After 3–4 passages, CFU efficiency reached ∼20%–25%. In passaged cells used for transplantation, the CFU best represents the self-renewal capacity of that population, which was substantial for L-MSCs. Consistent with this observation, replication capacity as gauged by PDs was extensive (n=110 doublings). This was even more numerous than is classically ascribed to lung fibroblasts (∼50 doublings, ie, Hayflick's limit) [46]. Moreover, a large fraction (17%–21%) of L-MSCs over multiple passages (P8 and P30) expressed aldehyde dehydrgenase activity, an energetic mechanism that has been repeatedly associated with progenitor or stem cells [47]. In addition, L-MSCs could be readily differentiated into adipocytes, chondrocytes, osteocytes, myofibroblasts, and smooth muscle cells in vitro, further refining our understanding of L-MSCs as highly undifferentiated fibroblasts, but committed mesenchymal lineage cells. Therefore, the lung may be similar to BM, possessing a population of MSCs that are heterogeneous with respect to their clonogenicity and level of commitment toward myofibroblasts [48].
Subtle differences between outgrowth L-MSCs and L-MSCs described in past studies are noteworthy. L-MSCs cultured from side population (Hoechst effluxing) cells [34,24] or directly sorted [23] showed more frequent expression (35% vs. 12% in this study) of CD90 (Thy-1), an antigen that is strongly expressed in lipofibroblasts [23], which were rare in outgrowth (parent or clonal) populations in this study. L-MSCs in this study were also CD34neg, which differed from L-MSCs isolated by FACS (CD31neg, CD45neg, Sca-1pos, CD34pos) [23]. Since the outgrowth method relies on repeated passage, it is plausible that expression or detection of these epitopes may be affected by this process. Outgrowth L-MSCs were CD146pos and side population MSCs in one study were CD146neg [34], suggesting that outgrowth L-MSCs, although lacking specific endothelial or pericyte markers, may be more closely affiliated with the endothelial compartment. Based on the observation that L-MSCs express CD146, form tubes upon exposure with VEGF, express VEGF in vitro, absorb LDL, and form complex structures resembling capillary networks in vivo after transplantation, it is possible that L-MSCs are responsible for supporting capillary structure in the alveoli, or that they promote angiogenesis through generation of adjacent stroma. In future experiments, the plausibility of stromagenesis induced by L-MSCs could be tested in 3D culture systems, repopulation assays, and more detailed analyses of transplantation assays in vivo.
Recent studies suggest that lung cells repopulate intact lung matrices according to their native configuration [49,50]. This line of evidence suggests that lung specificity may confer an advantage in engraftment assays in vivo, although no prior studies specifically addressed this question in the lung. Consistent with this notion, we found that lung retention efficiency (retained viable PKHpos cells/total viable lung cells) and evasion of phagocytosis was higher for L-MSCs than BM-MSCs on days 4 and 32 after transplantation, while baseline entrapment 2 h after injection was equivalent. Tissues other than lung were not examined, so it is unknown whether this pattern of retention pertains only to the lung.
The poor engraftment of BM-MSCs in this study is consistent with past studies using animal or human cells [14,20]. Several mechanisms have been proposed to explain why BM-MSCs do not engraft well in the lung, mainly focusing on the paucity of receptors on BM-MSCs for endothelial ligands such as P selectin [51,52]. In this study, the difference in retention of L-MSCs versus BM-MSCs presented a unique opportunity to probe mechanisms by analysis of differential expression. L-MSCs consistently expressed higher levels of 3 surface proteins (ICAM-1, PDGFRα, and Itg α2). Interestingly, these proteins were shown to modulate important engraftment-related functions (adherence, migration, and invasion) in L-MSCs (not BM-MSCs), which presumably contribute to tissue persistence. That cell–cell rather than cell–matrix interactions may better distinguish L-MSCs from BM-MSCs is evidenced by our data showing no difference in ECM substrate adherence except lower avidity of L-MSCs to collagen. These data suggest that Itg a2, which was expressed at high levels on L-MSCs, may modulate migration through Matrigel using mechanisms other than its role as collagen receptor such as protease activation [53]. Another mechanism that supports cell–cell interactions by L-MSCs was inferred by data showing greater endothelial adherence modulated by ICAM-1 in this study. More efficient adherence with pulmonary endothelium would provide L-MSCs with a greater survival mechanism of these anchorage-dependent cells. Alternative mechanisms by which L-MSCs increased lung retention are speculative but might include interactions between antigens found on L-MSCs (CD24, PSGL1, and Itgβ2) and ligands found on endothelial cells (e.g., P-selection, ICAM-1, and VCAM-1), either directly or through intermediates such as fibrinogen. Clearly, these mechanisms warrant further investigation in assays that incorporate appropriate shear stress and gain and loss of function in L-MSCs. The use of mouse pulmonary vascular endothelium would also improve specificity of these assays.
Another mechanism that may promote lung retention is more efficient transendothelial migration. Random migration, which was pronounced in L-MSCs compared with BM-MSCs, is known to promote trans-endothelial migration and homing [54]. Also, L-MSCs expressed more PDGFRα, an important receptor for PDGFAA that regulates cell motility [55 –57]. Similarly, directional migration as evidenced by chemo-attractant-driven responses to PDGFAA, PDGFBB, and FGF2 in transwell experiments was also more pronounced in L-MSCs than BM-MSCs, consistent with differences in receptors (e.g., PDGFRα).
Transplantation of L-MSCs into elastase-injured mice did not result in transdifferentiation of these cells based on co-localization studies. Rather, markers that were present on cultured L-MSCs (CD73, CD105, and vimentin) were strongly expressed on L-MSCs 32 days after transplantation. While L-MSCs show potential to differentiate into myofibroblasts or smooth muscle cells (ie, in response to TGFβ1), it is apparent from this study that the microenvironment of the elastase-injured lung does not promote these phenotypes. An unexpected finding was the loss of Sca-1 on L-MSCs 32 days after transplantation. Previous studies in skeletal myoblasts show that loss of Sca-1 is associated with reduced differentiation capacity [58]. Hence, future studies might address whether Sca-1 is a biomarker, or regulator of differentiation potential in L-MSCs.
Since L-MSCs exhibited higher retention and greater expression of genes encoding paracrine signaling proteins in vitro, we presumed that paracrine signaling induced in vivo would also be upregulated to a greater extent by L-MSCs. However, genes that were overexpressed in L-MSCs (vs. BM-MSCs) in vitro were not induced to a greater extent by L-MSCs in lung tissues after transplantation. In fact, both L-MSCs and BM-MSCs induced substantial (>15-fold) yet indistinguishable surges (P<0.05) in Ccl2, Cxcl10, IL-6, and IL-11 mRNA expression in lung tissues. This strongly suggests that homing and engraftment mechanisms that differed substantially between L-MSCs and BM-MSCs are distinct from mechanisms that promote gene expression and healing in elastase-injured tissue.
Consistent with the observation that L-MSCs and BM-MSCs induced similar mRNA expression in vivo, a single injection of either cell line reduced elastase injury to the same degree (∼25% reduction in MLI). A second study whereby L-MSCs were transplanted 3 times demonstrated a similar reduction in MLI (∼−33%). There was no effect of multiple L-MSC injections on collagen content based on analysis of picrosirius staining. This suggests that the relative undifferentiated L-MSCs that did not express collagen 1 in vitro or differentiate into myofibroblasts in vivo were not pro-fibrotic in this model.
As to the mechanism of tissue healing, IL-6 and IL-11 gene expression was upregulated in lung tissue after L-MSC or BM-MSC transplantation; IL-6 is a critical cytokine in liver regeneration [59,60], and there is a cooperative effect between IL-6 and HGF to stimulate hepatocyte DNA synthesis after partial hepatectomy [61], which is thought to involve IL-6 activation of nonparenchymal cells [62]. Recent studies also report that Hgf is an important mediator of the effects of MSCs to ameliorate elastase injury [7,63]. This suggests that induction of IL-6 and Hgf in this model was a relevant mechanism of tissue healing.
As to the mechanism by which MSCs in this study elicited these gene expression responses from lung tissue, our data are inconclusive whether MSCs acted directly through paracrine signals, indirectly through macrophage activation [8,64], or by activating other resident cells to produce these gene products. Many cells were associated with CD45; this suggests that many were phagocytosed, thus providing ample opportunity for MSC–macrophage interactions, but this would require macrophage knockdown to resolve. Whether MSCs in this study worked by direct paracrine mechanisms must be studied using conditioned media. Future studies are warranted to define the mechanisms by which these cell lines induce tissue healing and the role of IL-6 and other cytokines/chemokines that were overexpressed in cell-treated lungs in this study.
In conclusion, L-MSCs can be isolated from outgrowth of adult mouse lung tissue that exhibits phenotypic and functional characteristics that are distinct from BM-MSC. These distinctive features appear to predict retention, rather than reparative functions after transplantation, which was similar for both MSCs cell lines investigated. An intriguing phenomenon that deserves more attention is the phenotypic and functional diversity within passaged L-MSCs as evidenced by the wide range of clonogenicity of clones derived from the parent population of L-MSCs. This diversity in and of itself might have important implications for alveolar homeostasis and therapeutic efficacy.
Footnotes
Acknowledgments
The authors thank Dr. Dean Sheppard from the University of California at San Francisco for a generous supply of anti-Itg β5 and β6 monoclonal antibodies. We also thank Dr. Dan Weiss (University of Vermont) for his insight into the data. Some of the materials (BM-MSCs) employed in this work were provided by the Texas A&M Health Science Center College of Medicine Institute for Regenerative Medicine at Scott & White through a grant from NCRR of the NIH, Grant no. P40RR017447. We also thank Dr. Julie Abrams for technical assistance in data acquisition and analysis. Supported by NHLBI grant HL090145-01 (EI/AH), FAMRI 072161-CIA, and Alpha-1 Foundation/American Thoracic Society 07-035 (ST).
Author Disclosure Statement
The authors have no financial disclosure to declare.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.