
Abstract
In the field of tissue engineering, adult stem cells are increasingly recognized as an important tool for in vitro reconstructed tissue-engineered grafts. In the world of cell therapies, undoubtedly, mesenchymal stem cells from bone marrow or adipose tissue are the most promising progenitors for tissue engineering applications. In this setting, adipose-derived stem cells (ASCs) are generally similar to those derived from bone marrow and are most conveniently extracted from tissue removed by elective cosmetic liposuction procedures; they also show a great potential for endothelization. The aim of the present work was to investigate how the cocommitment into a vascular and bone phenotype of ASCs could be a useful tool for improving the in vitro and in vivo reconstruction of a vascularized bone graft. Human ASCs obtained from abdominoplasty procedures were loaded in a hydroxyapatite clinical-grade scaffold, codifferentiated, and tested for proliferation, cell distribution, and osteogenic and vasculogenic gene expression. The chromosomal stability of the cultures was investigated using the comparative genomic hybridization array for 3D cultures. ASC adhesion, distribution, proliferation, and gene expression not only demonstrated a full osteogenic and vasculogenic commitment in vitro and in vivo, but also showed that endothelization strongly improves their osteogenic commitment. In the end, genetic analyses confirmed that no genomical alteration in long-term in vitro culture of ASCs in 3D scaffolds occurs.
Introduction
I
In mammals, the predominant adipose tissue is white, consisting mainly of mature adipocytes filled with a lipid droplet of variable size. Between these mature adipocytes are stroma, which consists of blood vessels, fibroblasts, leukocytes, macrophages, and lipidless preadipocytes. The vasculature associated with the stroma maintains the growth of the adipose tissue parenchyma, including both the hyperplastic and the hypertrophic growth of adipocytes, as well as the differentiation of the maturing adipocytes. The adipose tissue continues to expand and shrink throughout adulthood. Likewise, the vasculature of most adult tissues is quiescent, whereas adipose tissue angiogenesis is active during adipose tissue expansion. This being so, the adipose tissue must contain cell populations with a marked proliferative capacity and a high potential for differentiation, that is, ASCs [7,8].
Adipose tissue could be an ideal source for harvesting large quantities of MSCs. Isolated from the stroma of adipose tissue, ADSCs bear a strong resemblance to bone marrow stem cells (BM-MSCs), as demonstrated by their expression of the same cell surface markers, their similar gene expression profiles, and their similar differentiation potentials. Unlike BM-MSCs, however, ASCs can be obtained in large quantities at low risk. In addition to being more abundant and readily accessible, adipose tissue yields far more stem cells per gram than bone marrow (5,000 vs. 100–1,000). It is therefore reasonable to expect ASCs to become the adult stem cells of choice for future clinical applications and primarily for bone reconstruction [9].
The past decade has seen considerable advances in our understanding of bone development and maintenance, with a growing awareness that its remodeling demands a source of energy and is intimately linked to other homeostatic pathways, such as the adipogenic and vasculogenic [10].
There is also evidence to suggest that adipocytes are some of the earliest cells to appear during osteogenesis. It is therefore imperative to consider the array of cell–cell communications regulating the osteoblast's differentiation function and the fate of MSCs [11].
In the light of these considerations, in the work described here, we developed an in vitro model of a hard tissue constructed starting from ASCs. The properties of these adult stem cells enabled us to obtain a bone-like tissue containing the principal cell types required for its maintenance, that is, osteoblasts and endothelial cells. In an in vivo model, we then confirmed our results with both morphological and molecular analyses. In addition, to ascertain the safety of this tissue-engineered biological product, we adopted a novel cytogenetic approach, that is, a comparative genomic hybridization (CGH) array to test whether the in vitro amplification of ASCs could give rise to structural alterations that might prove the health of the cells once implanted.
Materials and Methods
Biomaterial
Cell loading in scaffolds
Hydroxyapatite (HA)-based scaffolds were supplied in 1-cm3 cubes or in granules (Orthoss Block 1×1×2 and Geistlich Bio-Oss® Spongiosa large granules; Geistlich Pharma AG). The cubes/granules were then coated with fibronectin (Sigma Aldrich Italia) by soaking in a solution containing 50 mg/mL fibronectin for 4 h at room temperature. The scaffolds were air-dried overnight in a sterile biosafety cabinet. They were placed in a cell suspension (1×106 cells per scaffold) under vacuum conditions for 60 s to facilitate the flow of the cells inside the pores. After 3 h of incubation at 37°C with 5% CO2, the scaffolds were cultured with the medium for 2–3 weeks at 37°C with 5% CO2. In vivo, the control scaffolds were treated identically, but without cells.
Cell culture
ASCs were extracted from human adipose tissues of 5 healthy women and 5 healthy men (age: 21–36; BMI: 30–38) undergoing cosmetic surgery procedures, following the guidelines of the University of Padova's Plastic Surgery Clinic. The adipose tissues were digested with 0.075% collagenase (type 1A; Sigma Aldrich Italia) in a modified Krebs-Ringer buffer [125 mM NaCl, 5 mM KCl, 1 mM Na3PO4, 1 mM MgSO4, 5.5 mM glucose, and 20 mM HEPES (pH 7.4)] for 60 min at 37°C, followed by 10 min with 0.25% trypsin. Floating adipocytes were discarded, cells from the stromal-vascular fraction were pelleted, rinsed with media, and centrifuged, and then a red cell lysis step in NH4Cl was run for 10 min at room temperature. The resulting viable cells were counted using the trypan blue exclusion assay and seeded at a density of 106 cells/cm2 for in vitro expansion in Dulbecco's modified Eagle's medium (DMEM; Sigma Aldrich Italia) supplemented with 10% FCS and 1% penicillin/streptomycin. On day 10, the cells were seeded onto HA scaffolds (Geistlich Pharma AG) at a density of 106 cells/cm2. For the osteogenic commitment, the osteoinductive medium used was DMEM supplemented with 10% fetal calf serum, 1%
For vasculogenic commitment, M199 (Seromed) supplemented with 10% fetal calf serum, 1%
For the co-commitment, ASCs were cultured with M199 (Seromed) supplemented with 10% fetal calf serum, 1%
In vivo model
In vivo rat calvarial defect models of bone regeneration using HA scaffolds were prepared for calvarial implantation in 24, 8-week-old, female nude rats (Winstar, -NIH-FOXN1; Charles River). Two defects were prepared for each rat: HA scaffolds alone (control) and HA scaffolds+ASCs [previously, briefly (5 days) in vitro codifferentiated at a density of 1×107 cells/mL]. All operations were performed under general anesthesia by intraperitoneal injection of ketamine hydrochloride (40 mg/kg; Ketaras, Yuhan Corp) mixed with xylazine (10 mg/kg; Rumpens Bayer Korea Ltd.). After disinfecting the calvarial skin with 10% Betadine (Potadines; Sam-Il Pharm.) and subcutaneous injection of 2% lidocaine containing 1:100,000 epinephrine (Lidocaine HCL Injs; Yuhan Corp.) into the calvarial bone, an incision was made along the sagittal suture. The periosteum was elevated and a calvarial bone defect with 5 mm diameter was created with a trephine burr without perforating the dura. The area of the defect was either left with HA scaffolds alone (control group) or filled with HA+ASC (experimental group). The animals were sacrificed at 3 weeks after surgery under formalin perfusion. The calvarial bone was removed from the skull and decalcified. The decalcified bone tissues were fixed in 10% formalin overnight and embedded in paraffin after dehydration in 70% ethanol. For histochemical analysis, the paraffin sections were fixed for 10 min with xylene and stained with H&E (Sigma) and Masson's trichrome for the detection of cells and bone structure. All the animals were treated and handled in accordance with the “Recommendations for Handling Laboratory Animals for Biomedical Research” compiled by the Committee on the Safe and Ethical Handling Regulation for Laboratory Experiments at the University of Padova. The animals were housed separately in thermostat-controlled cages (22°C) with a 12-h day/night cycle, unrestrained and with food available ad libitum.
Histological staining
The cellularized scaffolds were fixed in a 4% paraformaldehyde phosphate-buffered saline (Seroderm) for 72 h and then dehydrated in a graded series of ethanol and acetone steps. Serial 7-mm sections were cut perpendicular to the osseous defects and surrounding bone (Reichert-Jung 2050; Nussloch).
The bone sections were stained with Alzan Mallory staining and observed under a light microscope.
Oil-red staining (Sigma-Aldrich) of the cytoplasmic droplets of neutral lipids was done according to a modification from Ramírez-Zacarías et al. [12]. Cells were rinsed, fixed in 10% buffered formalin, stained with 0.3% oil red (oro) in isopropanol and water (3:2), photographed, and extracted with 4% Nonidet (Sigma-Aldrich) and isopropanol. The optical density of the solution was measured at 520 nm for quantification using a Victor 3 spectrometer (Perkin-Elmer).
MTT test
Cell proliferation rates were determined by the MTT (3-4,5-dimethylthiazol-2yl-2,5-diphenyltetrazolium bromide)-based cytotoxicity test using the Denizot and Lang method with minor modifications [13]. Briefly, supernatant was gently harvested from the multiwell tissue culture plate, and 1 mL of MTT solution (0.8 mg/mL in phosphate-buffered saline) was added. Cultures were returned to the incubator, and after 3 h, the supernatant was harvested again. Each scaffold was then transferred to an Eppendorf microtube and 1 mL of extraction solution (0.01 N HCl in isopropanol) was added. The Eppendorf microtubes were vigorously vortexed for 5 min to enable total color release from the scaffolds and then centrifuged at 14,000 rpm for 5 min, and the supernatants were read at 534 nm.
DNA content
DNA content was determined using a DNeasy kit (Qiagen) to isolate total DNA from cell cultures following the manufacturer's protocol for tissue isolation, using overnight incubation in proteinase K (Qiagen). The concentration of DNA was detected by measuring the absorbance at 260 nm in a spectrophotometer. Cell number was then determined from a standard curve (microgram DNA vs. cell number) generated by DNA extraction from counted cells. The standard curve was linear over the tested range of 5–80 μg DNA (r=0.99).
Real-time RT-PCR
Primers and probes were selected for each target gene using Primer3 software. Gene expression was measured using real-time quantitative PCR on a Rotor-GeneTM 3500 (Corbett Research). PCRs were carried out using the primers at 300 nm and the SYBR Green I (Invitrogen) (using 2 mm MgCl2) with 40 cycles of 15 s at 958°C and 1 min at 608°C. All cDNA samples were analyzed in duplicate. Fluorescence thresholds (Ct) were automatically determined by the software, with amplification efficiencies for the genes being studied in the range of 92%–110%. For each cDNA sample, the Ct of the reference gene L30 was subtracted from the Ct of the target sequence to obtain the DCt. The expression level was then calculated as 2ΔCt and expressed as the mean±SD of quadruplicate samples of 2 separate runs. Experiments were performed with 3 different cell preparations and repeated at least 3 times. As control, ASCs cultured in presence of no differentiative medium has been used.
Scanning Electron Microscope
Samples of reconstructed adipose tissues were fixed with 2.5% glutaraldehyde in 0.1 m cacodylate buffer for 1 h before being processed either with hexamethyldisilazane or at the critical point followed by gold–palladium coating. All micrographs were obtained at 30 kV on a JEOL 6360LV scanning electron microscope (SEM) (Jeol).
Array CGH analysis
Array CGH was conducted using the Agilent Human Genome CGH Microarray Kit 44B (Agilent Technologies) with a resolution of w75 kb. Labeling and hybridization were performed following the supplier's protocols. Briefly, 4 mg of purified DNA from the patient and DNA from a female/male DNA commercial control (Promega) were double-digested with RsaI and AluI for 2 h at 37°C. After column purification, 1 mg of each digested sample was labeled by random priming (Invitrogen) for 2 h using Cy5-dUTP for the patient's DNA and Cy3-dUTP for the control DNA. Labeled products were column purified and prepared according to the Agilent protocol. After probe denaturation and preannealing with 50 mg of Cot-1 DNA, hybridization was performed at 65°C under rotation for 40 h. After 2 washing steps, the array was analyzed with the Agilent scanner and the Feature Extraction software (v8.0). Graphical overview was obtained using the CGH analytics software (v3.1) (Agilent Technologies).
Results
Cell isolation, seeding, and proliferation
After enzymatic digestion (see Material and Methods section), cells derived from adipose tissue were amplified in monolayers. As shown in Fig. 1a, there were no adult adipose cells positive for oil-red staining in the culture. Only a few cells with red droplets inside the cytosol were detectable (black arrows). ASCs are capable of organizing themselves into typical colonies (black circle). Like all other stem cell populations, ASCs also show small shapes and are capable of 3D growth. When ASCs were cultured in presence of both osteogenic and vasculogenic medium, they acquired both phenotypes. As well reported in Fig. 1b, ASCs acquired an elongated feature when endothelial commitment was attained and a star-like feature when osteogenic commitment was attained.

After 15 days of in vitro amplification with no differentiative medium, cells were seeded onto HA scaffolds (Orthoss®) (Fig. 2a) and cultured for up to 21 days in the presence of osteogenic and vasculogenic factors. As shown by the SEM images of Fig. 2b, c, and d, for up to day 21, the cells were able to adhere to the inner side of the scaffolds (black circle), forming a continuous cell layer by day 21 (black arrows).
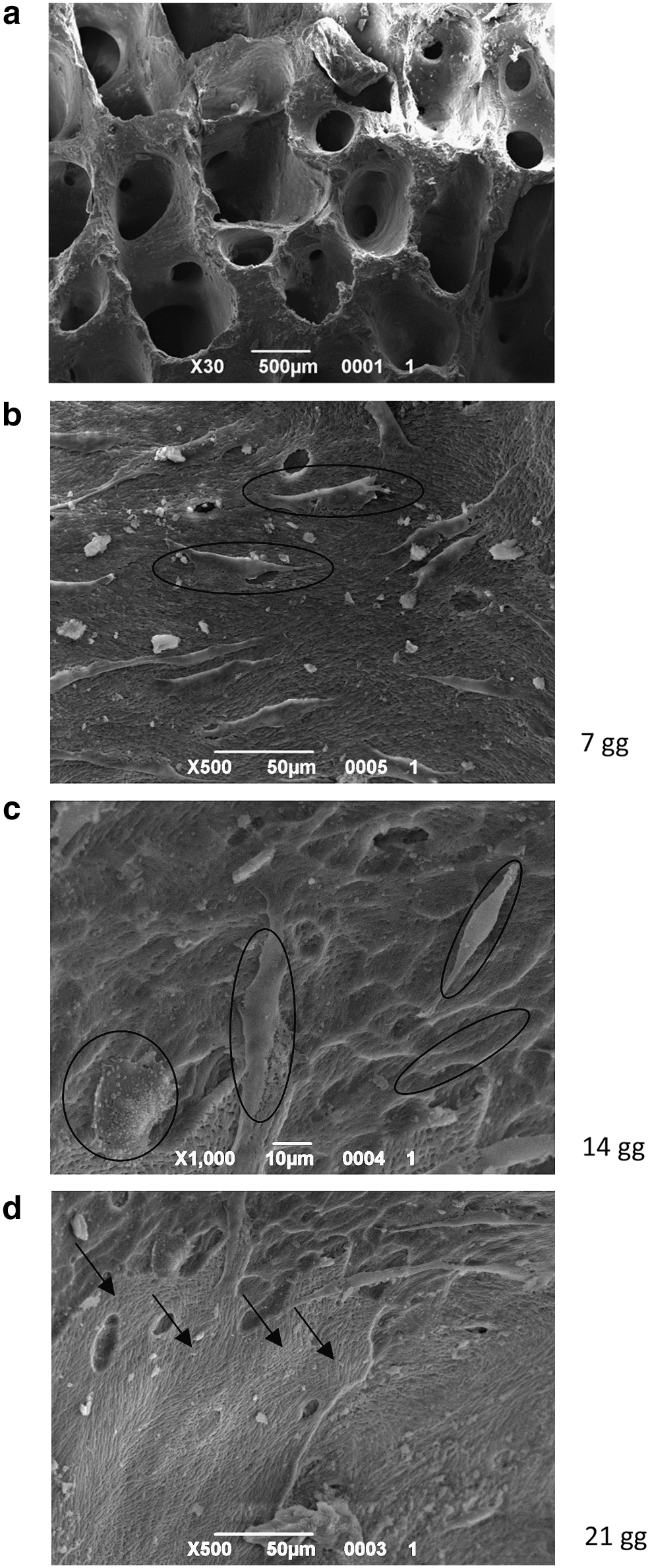
SEM analyses of hydroxyapatite (HA) scaffolds without cells
The cells were able to proliferate inside the scaffold, increasing in number as demonstrated by the MTT test (Fig. 3a) and the DNA content (Fig. 3b). The rising MTT values confirmed that the cells were alive, and the increase in DNA content confirmed that they were also able to proliferate.
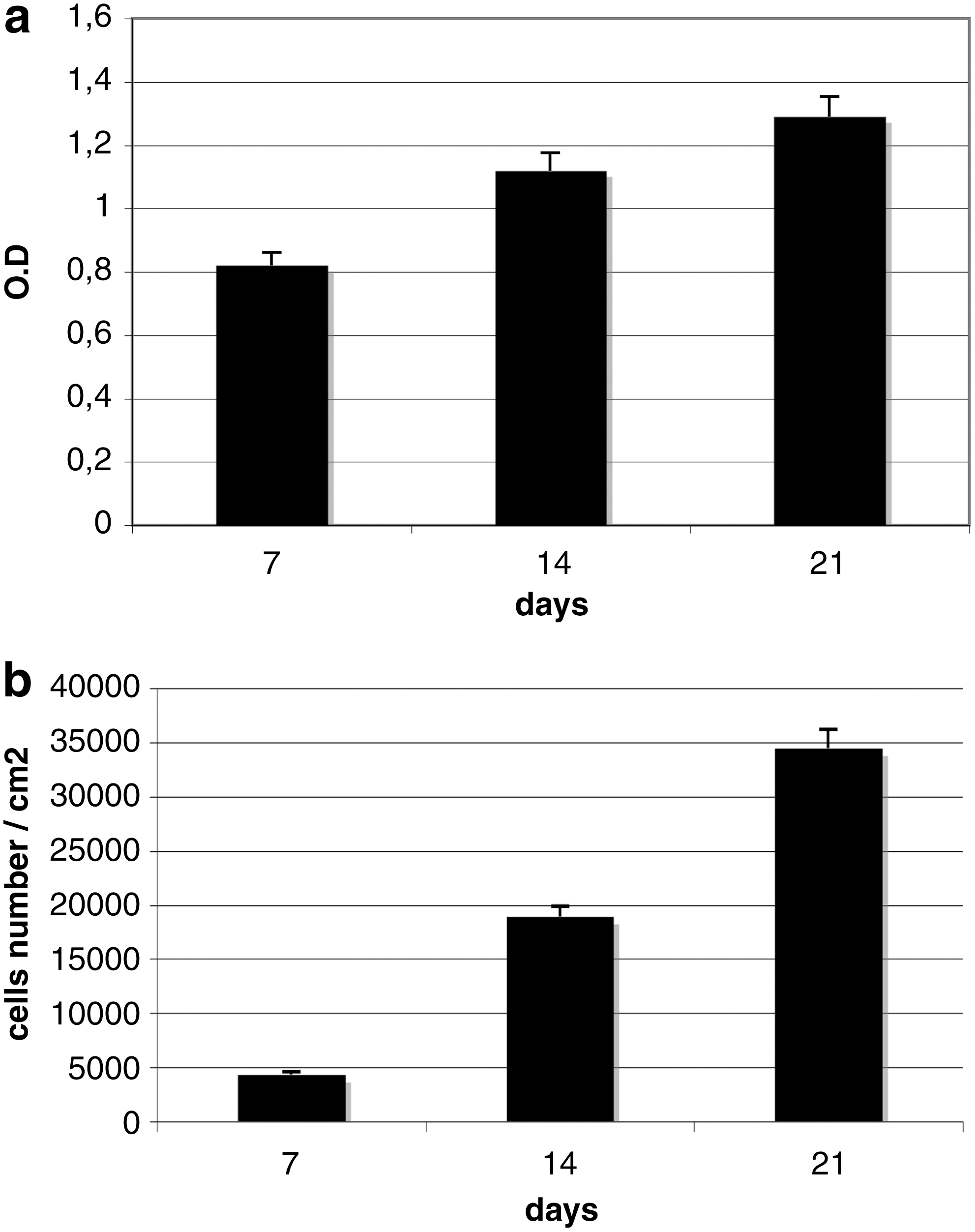
Proliferation test of ASCs seeded on HA scaffolds by means of MTT (3-4,5-dimethylthiazol-2yl-2,5-diphenyltetrazolium bromide) test
Gene expression
Real-time PCR was performed on 3D ASC cultures in the presence of osteoinductive factors (Fig. 4a), vasculogenic factor (Fig. 4b), or osteoinductive and vasculogenic factors (Fig. 4b). The gene expression detected by means of the molecular markers selected can provide information on the mature osteogenic phenotype (osteopontin, osteonectin, osteocalcin, collagen type I), the mature vascular cell phenotype [CD31, von Willebrand factor (vW), vascular endothelial growth factor (VEGF)], and factors regulating the osteogenic differentiation process (RUNX2) or adipogenic process (PPARγ) [14].
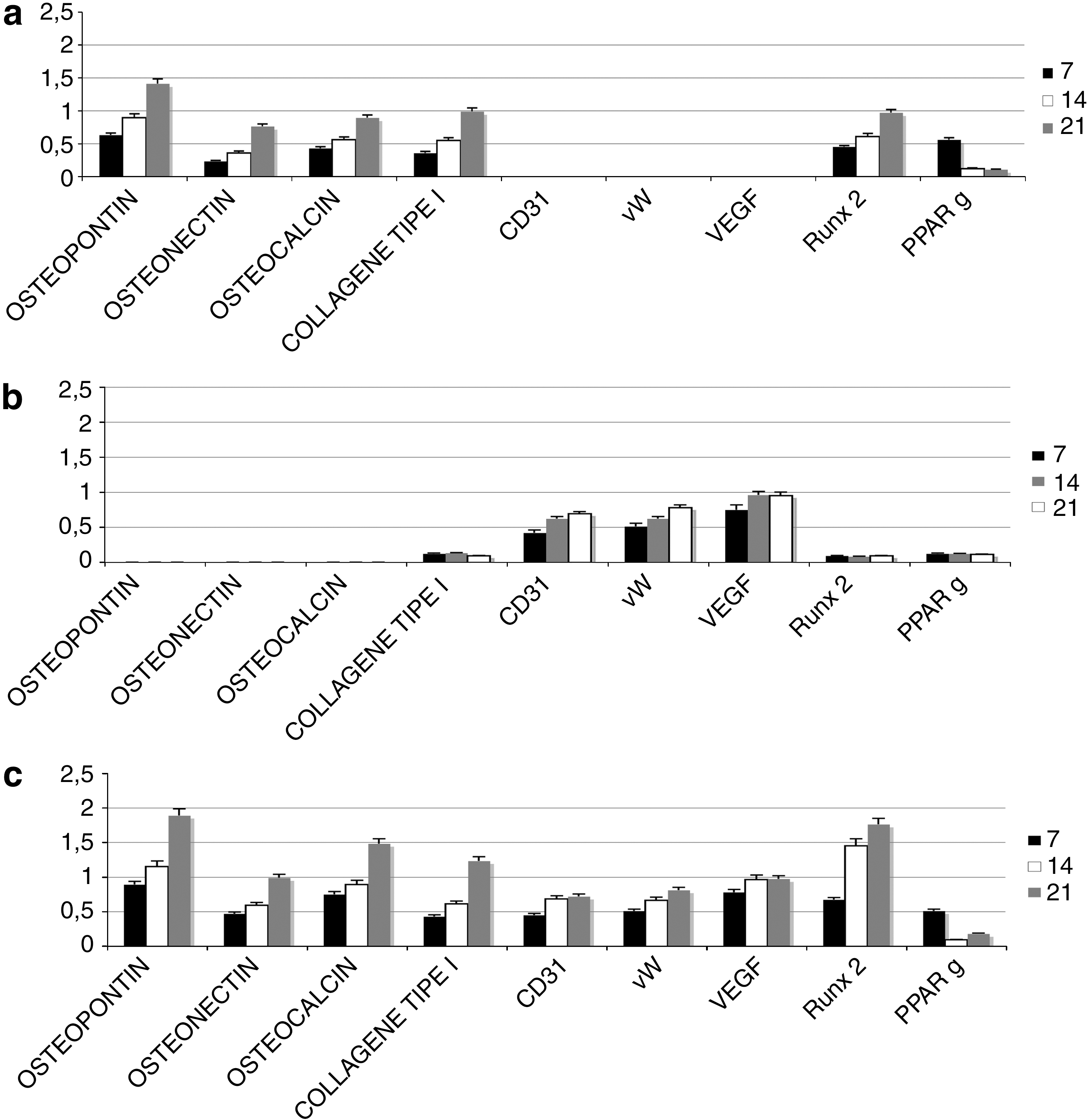
Time course of osteogenic (osteopontin, osteonectin, osteocalcin, collagen type I, RUNX2), vasculogenic [CD31, von Willebrand factor (vW), vascular endothelial growth factor (VEGF)], and adipogenic (PPARγ) mRNA expression analyzed by semiquantitative real-time PCR in ADSCs cultured on 3D HA scaffolds in osteogenic alone
As shown in Fig. 4a, the presence of osteoinductive medium was able to induce a good osteogenic commitment of the ASCs. Indeed, the expression profile for osteopontin, osteonectin, osteocalcin, and collagen type I increased with time. The expression of the specific osteogenic transcriptional factor RUNX2 also progressively increased, whereas PPARγ expression (specific for the adipogenic lineage) decreased. There was no detectable expression of endothelial cells in the presence of the cell medium used.
Figure 4b shows the gene expression of ASCs in the presence of both differentiative medium (osteogenic and vasculogenic). Here again, the osteogenic markers increased with time. Notably, the values are higher in this case than those obtained with osteoinductive medium alone. The same trend was seen for RUNX2 (an important transcription factor for osteocommitment), with a correlated decrease in PPARγ (a transcription factor correlating with the adipogenic lineage). The presence of mature endothelial cells on the tissue was confirmed by the expression of surface markers such as CD31 and vW and specific endothelial growth factors such as VEGF.
In vivo findings
The bone regeneration activity of the ASCs was assessed in vivo using a rat calvarial defect model. We considered 2 different groups to ascertain the influence of ASCs predifferentiated on new bone formation, that is, controls (Fig. 5a) and HA granules containing ASCs (Fig. 5b). Concerning the cell populations filling the implants, it is well evident that a sizable inflammatory cell population developed in the presence of HA granules alone (Fig. 5a, black arrow), whereas no inflammatory reaction around or inside the implant containing stem cells (Fig. 5b) occurred. In the implants enriched with stem cells, the HA granules were fully embedded with fibroblast-like cells capable of producing a good extracellular matrix consisting mainly of collagen type I, as revealed by Alzan Mallory staining (Fig. 5c, black arrows), and significantly, vessels (white arrows) could be found inside the scaffolds. A more detailed analysis of the cell population was performed using real-time PCR for osteogenic and endothelial markers. As shown in Fig. 5d, the presence of ASCs considerably improved the osteogenic population: the markers for osteopontin, osteonectin, osteocalcin, collagen type I, and RUNX2 were more strongly expressed (black bar) than in the implants containing no stem cells (gray bar). Clearly, there was also a marked improvement in the endothelial cell population in the presence of ASCs, and the concomitant presence of endothelial cells and osteogenic cells strongly improved the commitment of the latter.
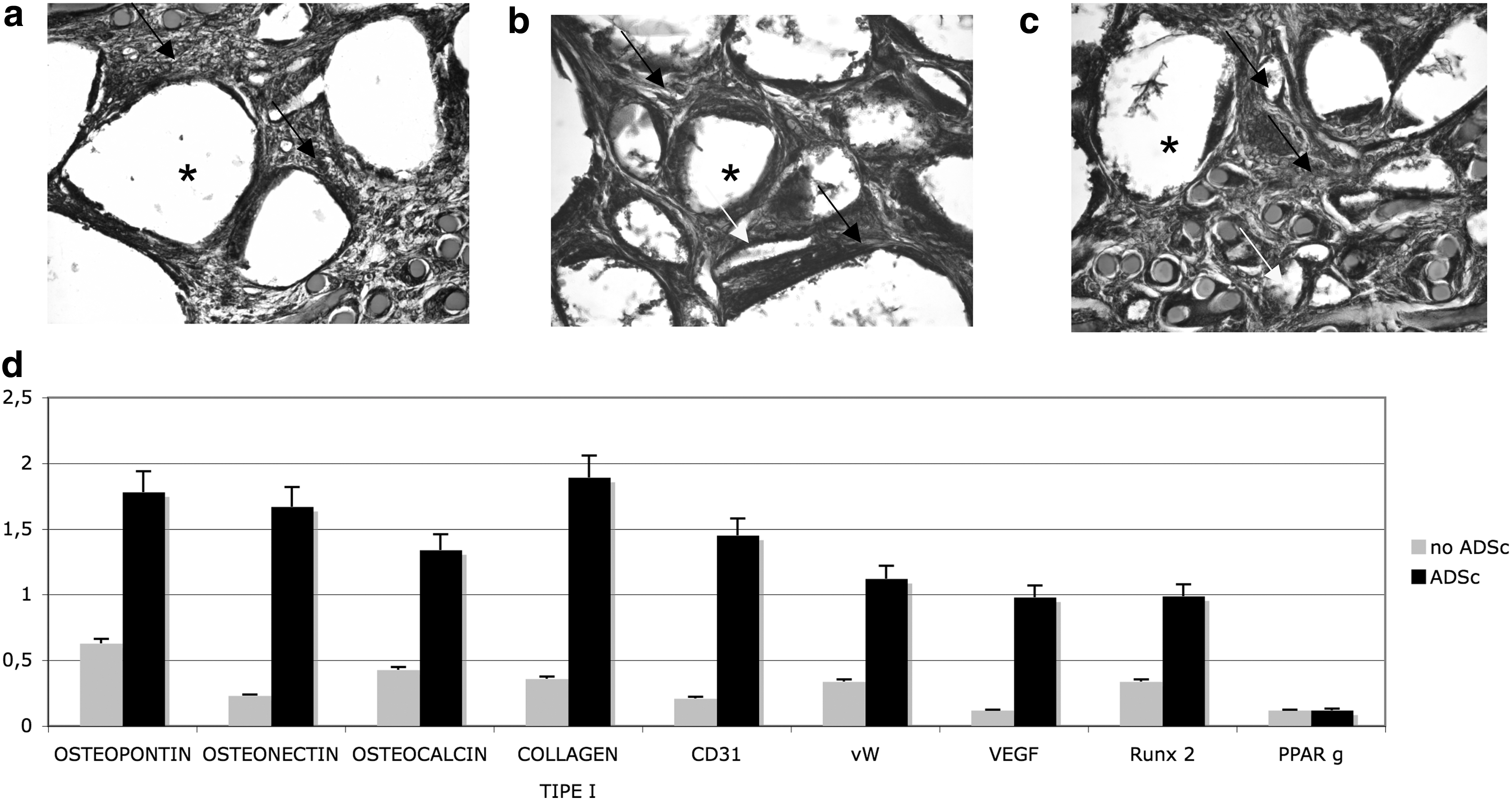
In vivo engraftment of HA scaffolds alone
Array CGH analysis
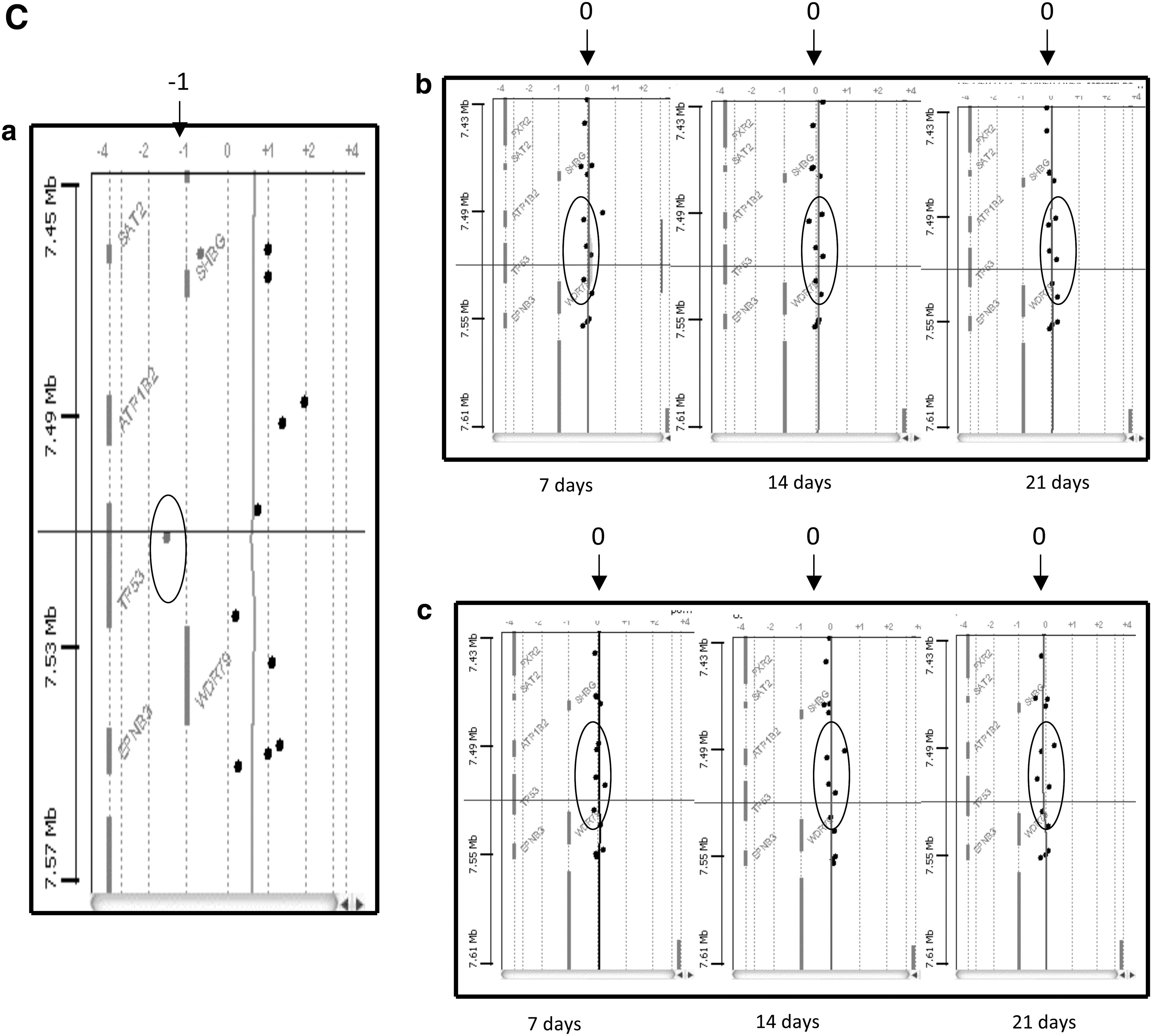
In the present work, we performed array CGH analysis on several prolonged 3D cultures of ASCs in the presence of both vascular and osteogenic factors. The DNA derived from 2 different donors and cultures on HA scaffolds at several time points (7, 14, and 21 days) were extracted and analyzed to identify any genomic alterations. As shown in Fig. 6, no chromosomal imbalance (duplications or deletions of DNA sequences) was detectable for either of the donors (female 46 xx; male 46 xy), confirming that neither long-term cultures nor the use of growth factors was able to induce structural DNA alterations.

Comparative genomic hybridization (CGH) array. Genomic alteration presents in the genome of a tumor cell line (HCTC). Alterations of genomic regions related to specific oncogenes are reported as follows:

Discussion
MSCs are adult stem cells well known to have proliferation, self-renewal, and multipotent differentiation capacities. They can be collected from many adult tissues such as bone marrow and also—as researchers have found in recent years—from adipose tissue [15,16]. In fact, adipose tissue is proving to be an interesting source of adult MSCs. The MSCs resident in bone marrow have great potential for differentiation, but they are rare: there is only 1 per 100,000 nucleated cells and their quantity declines with age [17]. The MSCs resident in adult adipose tissue (called ASCs) can be collected more easily from larger amounts of adult tissue, increasing stem cell availability. The chance to obtain larger quantities of cells using an easy clinical procedure, associated with the biological characteristics of ASCs (that enable them to become committed to several cell types), make ASCs an interesting option in the field of regenerative medicine. One of the most promising clinical applications of ASCs is in cell therapies because of their ability to ameliorate bone regeneration. Their osteogenic properties are well known and their potential for osteogenic differentiation (like the MSCs from bone marrow) has been also more recently demonstrated [18].
Another interesting property of ASCs is their plasticity toward endothelial cells, a characteristic strongly related to adipose tissue physiology. In fact, adipose tissue enlargement is the result of adipocyte hypertrophy and the recruitment and differentiation of regenerative precursors located in the stromal vascular fraction. The development of a capillary network is also required to ensure adipose tissue remodeling, however. First, the vascular pattern existing during embryonic development indicates that the formation of capillary convolutions is a specific, decisive phase in the development of fat lobules. Second, extensive vascularization is needed for adipose tissue to optionally function as a metabolic and endocrine tissue. Third, cells of the adipose lineage have been shown to release potent angiogenic factors [18].
In the light of such considerations, in the present work, we exploited the properties of ASCs for the in vitro reconstruction of a hard tissue with vasculogenic elements, with a view to improving the survival of the cell after their implantation in vivo.
First, we prepared an in vitro tissue-engineered product that has been characterized not only by its biological properties (tissue vitality, growth factors released, extracellular matrix component secreted, cell population) but also by its DNA stability (and then testing the safety) for the purposes of subsequent in vivo applications.
Proliferation tests such as MTT and DNA quantification confirmed that ASCs seeded onto HA scaffolds increase in number and give rise to a vital tissue. Morphological analyses using SEM showed that the cells were able to adhere to the niches of the biomaterial, forming a thin monolayer, as well as occur in vivo by osteoblasts, osteocyte, and endothelial cells.
Subsequent gene expression tests for osteogenic markers strongly supported a correct commitment to mature osteocytes. Detailed real-time PCR confirmed the presence of extracellular matrix components such as osteopontin, osteocalcin, and osteonectin, which have a fundamental role in cell interaction with bone matrix and in matrix mineralization, already after 1 week of 3D culture. The expression of collagen type I, which is essential to the formation and maturation of HA crystals, was also clearly detectable, confirming a correct extracellular matrix composition. As for the control over the commitment at genome level, RUNX2 and PPARγ were used as transcription factors for the commitment pathways considered; indeed, lineage commitment of MSCs is determined by expression and/or activation of specific transcription factors, such as RUNX2 in the case of osteoblasts and PPARγ in the case of adipocytes. Many evidences indicate an important role of PPARγ in bone metabolism. [19].
PPARγ favors adipogenesis and suppresses osteoblast genesis, partly by inhibiting RUNX2 function, resulting in fewer osteoblasts in the bone marrow. Vice versa, increasing RUNX2 levels inhibit adipogenesis, favoring osteogenesis [14].
In our 3D cultures, RUNX expression clearly increased with time in the presence of osteogenic medium, supporting osteocommitment at the expense of adipocommitment, a situation confirmed by the downregulation of the specific transcription factor PPARγ.
In the context of tissue engineering and regenerative medicine, it is increasingly recognized that it is important for different cell types the copresence in a 3D environment to generate constructs with a greater functionality and engraftment capacity [20]. In particular, coculturing tissue-specific cells with endothelial cells has been proposed as a way to address one of the main limitations of tissue-engineered grafts, namely their rapid vascularization. In this study, we therefore aimed to use ASCs, a readily available source of cells, to generate a 3D construct that was both osteogenic and vasculogenic. Our results showed that ASCs expanded in 3D in the presence of osteogenic medium were capable of generating bone tissue. In parallel cultures containing vasculogenic factors as well, we demonstrated that ASCs also commit to mature endothelial cells, as confirmed by the expression of superficial markers such as CD31 or specific endothelial soluble factors such as von vW or VEGF.
Surprisingly, we found that osteogenic marker expression increased by about 25% when ASCs were committed to both cell types at the same time.
This confirms that we can reproduce in an in vitro model an important in vivo event: the osteogenic commitment is strongly associated with the presence of endothelial cells. Indeed, interactions between endothelial cells and osteoblasts play a key part in skeletal development and regeneration [21]. More recently, transplanted endothelial cells have been reported to enhance orthotopic bone formation by bone marrow stromal cells in vivo [16]. In our in vitro setting, we developed a novel model for studying the 3D interactions between endothelial and osteoblastic cells as well as a tool for investigating the mutual conditioning of the 2 cell populations, including a precise control of their capacity for self-renewal and differentiation, which requires the formation of an appropriate niche.
The same result was strongly evident also when in vivo calvarial defects were treated with HA granules embedded with ASCs. Here again, morphological and molecular analyses confirmed a great commitment to the osteogenic and vascular phenotypes by the adult stem cells.
With a view to use our tissue in humans, we need to perform detailed cytogenetic analyses to validate the safety of the cells obtained. It is well known since few years that cell-based products need to be tested for their safety before in vivo implantations. This safety could be ascertained by the validation of chromosomal stability. Chromosome analysis remains one of the most commonly performed diagnostic genetic tests, being offered for a wide variety of indications in oncology, gynecology, and pediatrics. At the cytological level, banded human chromosomes show a consistent and similar pattern in clinically healthy individuals. Hence, balanced and unbalanced chromosomal aberrations can serve as informative markers for a clinical phenotype such as the dangerous transformation of a normal genotype in a tumor one. The method most commonly used for this purpose is the analysis of MSC metaphases. In fact, Rubio and coworkers [22] demonstrated that transformed MSCs always showed chromosome alterations, such as trisomy, tetraploidy, and/or chromosome rearrangements. In view to test this stability, we performed an innovative molecular cytogenetics approach: array CGH. This chromosomal microarray analysis is a molecular–cytogenetic method for the analysis of copy number changes (gains/losses) in the DNA content of a given subject's DNA [23,24]. Chromosome analysis is one of the most commonly used diagnostic genetic tests, with indications in obstetrics, gynecology, pediatrics, and oncology. At cytological level, banded human chromosomes show a consistent and similar pattern in clinically healthy individuals, and so balanced and unbalanced chromosomal aberrations can serve as informative markers of clinical phenotypes. The method relies on the development of a monolayer cell culture that can be analyzed under the microscope after treatment with a mitotic fuse blocker. With the advent and application of array-based comparative genomic hybridization (array CGH), by which the genome extracted from cells can be analyzed at a significantly higher resolution than was previously possible, scientists can now screen all the DNA content even if cells have been cultured in 3D. This tool thus enables us to establish whether prolonged in vitro cultures can give rise to chromosomal anomalies that might be implicated in the etiology of diseases or disorders.
In our study, we had the ability to test whether with our strategies ASCs, expanded in vitro in 3D feature and in presence of differentiative factors, were able to maintain the chromosomal stability and their safety when implanted in vivo. In Fig. 6a, CGH array for 2 different specimens (one from a male and one from a female lipoaspirated) is reported, and it is clear that no DNA alteration occurred. This confirms that with our techniques the cells were able to differentiate, maintaining a genomic stability.
In conclusion, we summarize that to obtain a successful tissue-engineered bone graft we could combine the following components:(a) Cells: ASCs could be easily obtained from patients, easily expanded, and committed in 2 cell lines inside the same scaffold; the osteogenic commitment was strongly improved when it occurred at the same time with the vasculogenic commitment; the angiogenic event in vivo was increased, ensuring the long-term survival of cells once in vivo implanted and then the engraftment of the scaffold.(b) Scaffolds: HA-based scaffolds in both granules and cubes were confirmed as the gold standard for tissue-engineered bone products.(c) Genetical tools: CGH array could be considered as perfect tools to validate the chromosomal stability of cells cultured on 3D scaffolds and then as a test to ascertain the safety of the cellular products.
Footnotes
Acknowledgments
This research was supported by AIRC, Telethon, COFIN, and Italian Ministry of Health (to P.P.).
Author Disclosure Statement
The authors declare that there is no competing interest.