
Abstract
Tendons and ligaments (T/L) are dense connective tissues of mesodermal origin. During embryonic development, the tendon-specific cells descend from a sub-set of mesenchymal progenitors condensed in the syndetome, a dorsolateral domain of the sclerotome. These cells are defined by the expression of the transcription factor scleraxis (Scx), which regulates tendon formation and several other characteristic genes, such as collagen type I, decorin, fibromodulin, and tenomodulin (Tnmd). In contrast to other mesenchymal progenitors, the genealogy and biology of the tenogenic lineage is not yet fully understood due to the lack of simple and efficient protocols enabling generation of progenitors in vitro. Here, we investigated whether the expression of Scx can lead to the direct commitment of mesenchymal stem cells (MSCs) into tendon progenitors. First, MSC derived from human bone marrow (hMSC) were lentivirally transduced with FLAG-Scx cDNA to establish 2 clonal cell lines, hMSC-Scx and hMSC-Mock. Subsequent to Scx transduction, hMSC underwent cell morphology change and had significantly reduced proliferation and clonogenicity. Gene expression analysis demonstrated that collagen type I and several T/L-related proteoglycans were upregulated in hMSC-Scx cells. When stimulated toward 3 different mesenchymal lineages, hMSC-Scx cells failed to differentiate into chondrocytes and osteoblasts, whereas adipogenic differentiation still occurred. Lastly, we detected a remarkable upregulation of the T/L differentiation gene Tnmd in hMSC-Scx. From these results, we conclude that Scx delivery results in the direct programming of hMSC into tendon progenitors and that the newly generated hMSC-Scx cell line can be a powerful and useful tool in T/L research.
Introduction
Tendons and ligaments (T/L) connect and transmit force from muscle to bone and bone to bone, respectively. Both tissues are able to store elastic energy and withstand high-tensile forces, on which locomotion is entirely dependent [1]. T/L are predominantly composed of collagen type I fibrils organized in a highly hierarchical manner that is unique for the T/L. Other collagens (types III–VI, XI, XII, XIV, and XV) and various proteoglycans [decorin, cartilage oligomeric matrix protein (COMP), byglican, lumican, fibromodulin, tenascin-C, etc.] are building the remaining T/L substance [2]. The cellular content of T/L is dominated by tendon-specific fibroblasts named tenocytes. During embryonic development, the tendon progenitors derive from mesenchymal progenitors located in the syndetome, a sub-compartment of the sclerotome [3,4]. These cells are defined by the expression of the transcription factor scleraxis (Scx), a member of the basic helix-loop helix (bHLH) family [3,5]. To initiate transcription, Scx forms heterodimers with E12 or E47, which belong to the widely expressed class I bHLH proteins [6 –8]. So far, the known key targets of Scx-dependent transcription are the α1 and α2 chains of type I collagen, aggrecan, and tenomodulin (Tnmd) genes [9 –13]. Genetic ablation of Scx in mice results in severe T/L phenotype, ranging from a dramatic failure of tendon progenitor condensation and differentiation to the formation of small and poorly organized T/L [14]. Further, the molecular characterization of the Scx knockouts revealed a clear decrease in the levels of collagen I α1 gene and a complete loss of collagen XIV and Tnmd transcripts [14]. Tnmd is a transmembrane protein with a cleavable C-terminal cystein-rich domain and is highly expressed in T/L [15,16]. Mice deficient for Tnmd display decreased tenocyte proliferation and altered collagen fibril structure, thus suggesting that Tnmd is important for T/L maturation [17]. Hence, Tnmd is a terminal differentiation marker of the tendon cell lineage [11,17].
Mesenchymal stem cells (MSCs) are multipotent cells that give rise to tissues of mesodermal origin, such as adipocytes, chondrocytes, osteoblasts, skeletal myocytes, and visceral stromal cells during embryonic development [18,19]. In the adult organism, MSC reside in the bone marrow (BM-MSC) as well as in other tissue-specific niches such as adipose tissue, periosteum, tendon, muscle, and so on (reviewed in [20]). The BM-MSC are easily obtainable and can be expanded to large numbers on polystyrene dishes. Further, by using well-developed protocols, MSC can be stimulated in vitro and directly differentiated into adipocytes, chondrocytes, and osteoblasts. Therefore, these cells are currently considered a high-potential source for musculoskeletal regeneration [21,22].
In contrast to adipogenic, chondrogenic, and osteogenic differentiation, a simple and efficient protocol to generate tendon progenitors from MSC has not been reported. Therefore, the aim of this study was to recapitulate in vitro the process of T/L development, where the transition of mutipotent MSC to tendon progenitors is marked by Scx upregulation, thus allowing the establishement of a novel approach for generation of tendon progenitors. We hypothesized that introducing Scx expression in cultivated BM-MSC will result in a gene expression shift, reduced cell proliferation, and multipotentiality, thus eventually leading to induction of MSC commitment into the tenogenic lineage. For this purpose, we applied a lentiviral transfer of FLAG-Scx cDNA in BM-derived human MSC (hMSC) and characterized the cellular phenotype of the gene-targeted cells. (FLAG is an eight amino acid peptide tag.)
Materials and Methods
Cell culture
The well-established BM-hMSC cell line (SCP-1, hTERT-immortalized BM-derived MSC) described in [23] was used in the study. hMSC were maintained in Alpha minimum essential medium (MEM) GlutaMAX culture media (Gibco, Karslruhe, Germany) supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich, Munich, Germany) and 1% penicillin/streptomycin (PAA, Pasching, Austria). Cells were cultivated on polystyrene dishes in a humidified incubator at 5% CO2 and 37°C.
Cloning of FLAG-Scx and transduction of hMSC
The original FLAG-Scx plasmid was kindly provided by Prof. David Hall, NIH, Bethesda, MD [7]. FLAG-Scx cDNA was first subcloned into pENTR11 plasmid (Invitrogen, Carlsbad, CA) by EcoRI/NcoI digestion and then transferred into pLenti4/V5-DEST plasmid (containing CMV promoter and zeocin resistant gene, Invitrogen) by the LR clonase reaction. In parallel, a Mock pLenti4/V5-DEST plasmid (without the expression cassette) was created. The final plasmids (Fig. 1A) were verified by restriction digestion analysis and sequencing.
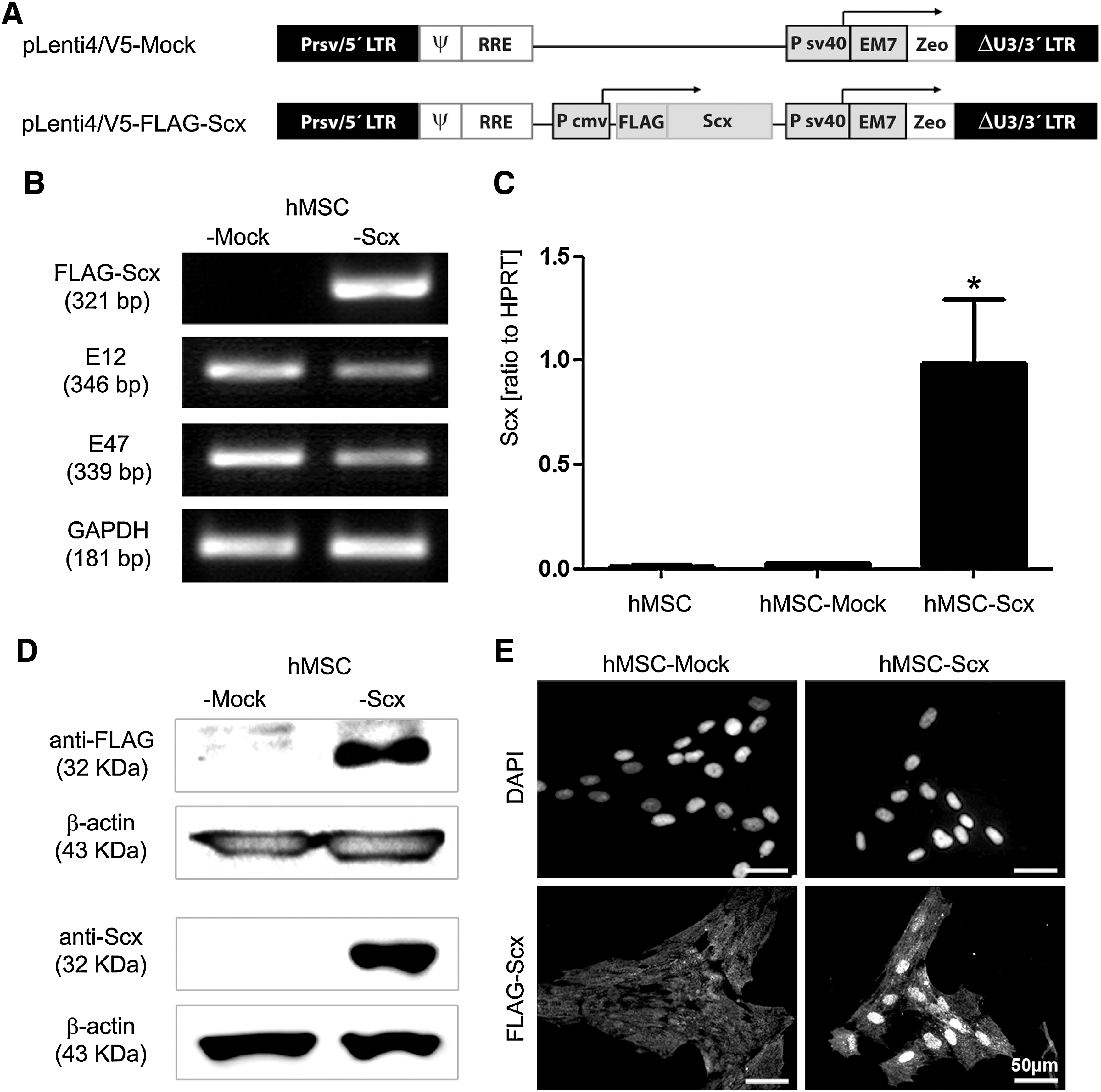
Lentiviral transduction of FLAG-Scx cDNA in hMSC and establishment of stable cell lines.
Lentivirus production and transduction of hMSC were accomplished as described in [23] with minor modifications. Briefly, pLenti4/V5-DEST-Mock or -FLAG-Scx were cotransfected with ViraPower packaging mix (Invitrogen) in 293FT cell line, and lentiviral supernatants were collected after 48 h. hMSC were incubated with a 1:1 mixture of viral supernatant and growth medium in the presence of 16 μg/mL polybrene (Sigma). After 48 h, cell selection with medium containing 50 μg/mL zeocin (Invitrogen) was carried out for 8 days. hMSC-Mock and hMSC-Scx in 6–10 consecutive passages were used for analyses.
Analysis of cell area
Cell morphology was monitored and imaged with an Axiocam ICc3 camera mounted on a phase-contrast microscope Axiovert 40 CFL (both Carl Zeiss, Göttingen, Germany). hMSC-Mock and hMSC-Scx cells were plated on polystyrene and after 24 h, cell areas were measured with the polygonal tool of Image J 1.38 software (
Self-renewal analysis
Long-term cell growth was evaluated by calculation of population doubling (PD) as described in [24]. Cumulative PD and PD time were determined from 6 consecutive passages.
For analysis of clonogenic potential, colony-forming unit (CFU) assay was applied. Briefly, cells were seeded in 10 cm Petri dishes with a density of 10 cells/cm2. After 12 days, formed colonies were visualized with 0.5% crystal violet/methanol staining. Single colonies with 1–8 mm diameter were counted and in the case of small cell clusters, a cluster of >25 cells was considered a colony. Finally, CFU efficiency was estimated with the formula CFU [%]=(number of colonies/number of plated cells)×100. CFU assays were performed for hMSC-Mock and hMSC-Scx in 2 different passages (6 and 10), and each assay contained triplicates.
Reverse transcriptase-polymerase chain reaction
Total RNA was extracted from hMSC-Mock and hMSC-Scx with RNeasy Mini Kit (Qiagen, Hilden, Germany), and 1 μg RNA was used for cDNA synthesis with AMV kit (Invitrogen) according to the manufacturer’ instructions. Semi-quantitative reverse transcriptase-polymerase chain reaction (RT-PCR) was performed with Taq DNA Polymerase (Invitrogen) in a PTC-200 Thermal Cycler (Bio-Rad Laboratories, Munich, Germany). The cDNA input was normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The PCR products were analyzed on 2% agarose gels. For lumican and alpha smooth muscle actin (α-SMA), PCR bands were densitometrically quantified by using the BioCapt software (Vilber Lourmat, Eberhardzell, Germany). Values were normalized to GAPDH, and results were reported as relative gene expression. The primer pairs and RT-PCR conditions used in this study are listed in Table 1. Quantitative RT-PCR was performed in a LightCycler 1.5 instrument equipped with LightCycler 3.5 software (Roche, Penzberg, Germany). LightCycler Fast Start DNA Master SYBR Green Kit (Roche) and primer kits for Scx, decorin, fibromodulin, sex determining region Y-box 9 (Sox9), runt-related transcription factor 2 (Runx2), transcriptional coactivator with PDZ-binding motif (TAZ), Tnmd, and hypoxanthine phosphoribosyltransferase (HPRT) designed by Search-LC (Heidelberg, Germany) were used. Relative gene expression was calculated as a ratio to HPRT. All RT-PCR results were reproduced at least twice independently.
Western blot analysis
For protein isolation, cells were lysed in radio-immuno-precipitation assay buffer (0.1% sodium dodecyl sulfate (SDS), 1% Na-deoxycholate, 1% Triton X-100, 50 mM Tris HCl pH 8.2, 150 mM NaCl, 10 mM ethylenediaminetetraacetic acid, and 20 mM NaF) supplemented with complete protease inhibitors (Roche). Total protein was quantified with Micro BCA protein assay kit (Pierce, Rockford, IL). Aliquots of 20–50 μg total protein were separated on 15% SDS-polyacrylamide gel electrophoresis gels and transferred onto polyvinylidene fluoride membranes (Roche). Membranes were blocked with 5% skimmed milk in Tris-buffered saline, 0.05% Tween-20 for 2 h at room temperature and then incubated with mouse anti-FLAG M2 (Sigma), rabbit anti-Scx (Abgent, San Diego, CA), or mouse anti-β actin (Santa Cruz, Heidelberg, Germany) primary antibody overnight at 4°C. Corresponding secondary horseradish peroxidase-conjugated antibodies (Rockland, Gilbertsville, PA) were applied for 1 h at room temperature. Proteins were visualized with ECL Plus Detection System (GE Healthcare, Buckinghamshire, UK) and detected on Lumi-films (Roche).
Enzyme-linked immunosorbent assay for collagen type I
hMSC-Mock and hMSC-Scx cells were plated in 6-well dishes at a density of 6×103 cells/cm2. After 3 days, the conditional media was collected, supplemented with cocktail protease inhibitors (Roche), and stored at −80°C. In parallel, the cell number was counted. Secreted collagen type I was detected by using Human Collagen Type I ELISA kit (Cosmo Bio, Tokyo, Japan) according to the manufacturer's instructions. Optical density was measured at 450 nm on a Multiscan FC microtiter-plate reader (Thermo Scientific, Vantaa, Finland). The collagen I concentration was calculated by using a collagen I standard curve and normalized to cell number. The assay was performed in triplicate and reproduced twice independently.
Hydroxyproline assay
hMSC-Mock and hMSC-Scx cells were plated in T-225 flasks (Nunc, Roskilde, Denmark) at a density of 4.5×103 cells/cm2. After 6 days, the conditioned medium was collected and concentrated with Amicon Ultra centrifugal filters (Millipore, Temecula, CA) to a final volume of 100 μL. In parallel, total amount of cells were counted and centrifuged. Cell pellets and concentrated conditioned media were hydrolyzed in 6N HCl for 15 h at 100°C in a termomixer Comfort (Eppendorf, Hamburg, Germany). Hydroxyproline content was measured with the Hydroxyproline Assay Kit (QuickZyme Biosciences, Leiden, The Netherlands) according to the manufacturer's instructions. Optical density was measured at 550 nm on a Multiscan FC microtiter-plate reader (Thermo Scientific). The hydroxyproline concentration was calculated by using a hydroxyproline standard curve and normalized to cell number. The assay was reproduced twice independently.
Luciferase assay
Original plasmids containing promoter-less firefly luciferase II (pGL4-Luc II) and Tnmd promoter (-769/+84F)-driven luciferase (pGL4-Tnmd-LucII) were subcloned in pENTR11. hMSC-Mock and hMSC-Scx (3×103 cells/cm2) were transfected with 2.5 μg plasmid DNA by using Opti-MEM media and Lipofectamine 2000 kit (Invitrogen). After 6 h, transfection media was replaced with complete growth media, and cells were maintained in culture for 2 days. Afterward, the luciferase activity was detected with Luciferase Assay E4030 kit (Promega, Madison, WI) according to the manufacturer's instructions. Briefly, cells were lysed in 1×lysis buffer, and 20 μL of the cell lysates were mixed with 100 μL of luciferase reagent and immediately measured on Safire II Luminometer (TECAN, Männedorf, Germany). Three independent transfections were performed and in each measurement, triplicates for hMSC-Mock and hMSC-Scx were used.
Cell differentiation
hMSC-Mock and hMSC-Scx were differentiated, twice independently, into 3 different mesodermal lineages. Adipogenic differentiation was performed as described in [25] by using Dulbecco's modified Eagle's medium (DMEM) high-glucose medium, 1 μM dexamethasone, 0.2 mM indomethacin, 0.1 mg/mL insulin, and 1 mM 3-isobutyl-1-methylxanthine (Sigma). Cells were stimulated for 21 days, and lipid vacuoles were visualized by Oil Red O staining by using a standard protocol. The extent of adipogenic differentiation was determined by AdipoRed assay (Lonza, Walkersville, MD), according to the manufacturer's instructions. Results are shown as relative fluorescence units. Chondrogenic differentiation was performed, with minor modifications, as described in [26]. Cells were seeded in V-bottom 96-well polypropylene plates, and pellets with 2.5×105 cells were formed by centrifugation. Differentiation medium composed of DMEM high glucose, 10 μM dexamethasone, 1 nM sodium-pyruvate, 0.195 mM L-ascorbic-acid, 1% insulin transferrin selenit (all Sigma), and 10 ng/mL TGF-β 1 (R&D Systems, Wiesbaden, Germany) was applied for 4 weeks. Pellets were fixed with 4% formaldehyde, paraffin embedded, cut in 7 μm slices, and stained for collagen type II. The area positive for collagen type II was then measured with the polygonal tool of Image J 1.38 software, and results were shown as a percentage of the total pellet area. Osteogenic differentiation was assessed according to [27]. Briefly, 4×104 cells/cm2 were seeded in 6-well plates and incubated in osteogenic medium (DMEM high glucose, 10% FBS, 10 mM β-glycerophosphate, 50 μM
Immunocytochemistry
hMSC-Mock and hMSC-Scx (1×104 cells/cm2) were grown on glass slides coated with 10 μg/mL collagen type I (Millipore) or fibronectin (BD Bioscience, Bedford, PA). Cells were fixed with 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, and blocked with 2% BSA. Before blocking, Image-iT FX Signal Enhancer (Invitrogen) was applied for 30 min. Primary antibodies for mouse anti-FLAG M2 (Sigma), rabbit anti-Tnmd [17], rat anti-CD44 (DSHB, Iowa, IA), or mouse anticollagen type I (Sigma) were applied overnight at 4°C. Next, corresponding secondary antibodies conjugated with Alexa Flour 488 or 546 and the nuclear dye 4′,6-diamidino-2-phenylindole (DAPI) (all Invitrogen) were used. For collagen type II immunostaining, chondrogenic pellet sections were deparaffinized and treated with 0.02 mg/mL proteinase (bacterial type XXIV, Sigma) and consecutively with 2 mg/mL hyaluronidase (Sigma) for 30 min at 37°C. After 2 h blocking with 5% normal horse serum, collagen type II antibody (DSHB) was applied overnight at 4°C. Next, secondary anti-mouse biotinylated antibody combined with ABC kit was used at room temperature, and a color product was developed with DAB kit (VectorLab, Burlingame, CA). Finally, sections were counterstained with hematoxylin. Negative controls were carried out by omitting the primary antibodies, and at least 2 independent stainings were performed. Photomicrographs were taken with an Axiocam MRm or Axiocam ICc3 camera on an Axioskope 2 microscope (Carl Zeiss).
Statistics
Quantitative data were analyzed with GraphPad Prism 5 software (GraphPad, La Jolla, CA). Bar charts show mean values and standard deviations. Unpaired t-test was used, and a P value of 0.05 was considered statistically significant.
Results
Endogenous and transgene expression of Scx in BM-derived hMSC
Using lentiviral expression system, FLAG-Scx cDNA was stably delivered into the well-established hMSC line SCP-1 (hTERT-immortalized hMSC, [23]). hMSC were infected in parallel with Mock- or FLAG-Scx-lentivirus (Fig. 1A) and by applying antibiotic selection, nontransduced cells were eliminated. Next, we obtained and expanded >20 single cell-derived clones from the heterogeneous hMSC-Mock and hMSC-Scx populations. Using RT-PCR, we screened for Scx expression nontransuduced hMSC; hMSC-Mock and hMSC-Scx heterogeneous cells; 3 control hMSC-Mock clones; and 7 hMSC-Scx clones (Supplementary Fig. S1; Supplementary Data are available online at
First, we analyzed these clones for the expression of E12 and E47, both of which are ubiquitously expressed and co-operate with Scx (Fig. 1B). As expected, we detected their expression in both hMSC-Scx and hMSC-Mock cell lines. Next, we confirmed the Scx transgene expression first, at the mRNA level by quantitative PCR (Fig. 1C) and second, at the protein level by Western blot (Fig. 1D). Lastly, by performing immunocytochemical analysis, we showed a nuclear localization of FLAG-Scx in the hMSC-Scx (Fig. 1E). In hMSC-Mock, we observed only a slight background signal. In conclusion, FLAG-Scx was successfully transduced in BM-derived hMSC, and 2 stable cell lines were generated—hMSC-Scx and hMSC-Mock (control). No endogenous Scx expression was detected in both hMSC and hMSC-Mock cells, whereas the transgene expression in hMSC-Scx was clearly demonstrated at the RNA and protein levels.
Scx expression leads to morphological changes and reduced self-renewal of the hMSC
hMSC-Mock and hMSC-Scx were first monitored for morphological differences by phase-contrast microscopy (Fig. 2A). hMSC-Mock exhibited bi-or 3-polar shape morphology, which was similar to the nontransduced hMSC (data not shown). In contrast, hMSC-Scx cells were more spread, having approximately 60% bigger cell area than the control cells (Fig. 2B). Next, we investigated whether Scx expression leads to alteration of hMSC self-renewal potential. We calculated and compared the cumulative PD of hMSC-Mock and hMSC-Scx for 3 weeks. In this period, hMSC-Mock reached 18 PD and had a PD time of 29±4.3 h, whereas hMSC-Scx doubled only 12 times, and their average PD time was increased to 40.5±5.3 h (Fig. 2C). The clonogenic ability of hMSC-Mock and hMSC-Scx cells was tested by performing CFU assay. At day 12, the colonies were visualized with crystal violet staining, and it became apparent that hMSC-Scx formed less colonies than hMSC-Mock (Fig. 2D). Calculating the CFU efficiency of both cell types revealed a significant 2.4-fold reduced clonogenicity of hMSC-Scx compared with that of hMSC-Mock cells (Fig. 2E).
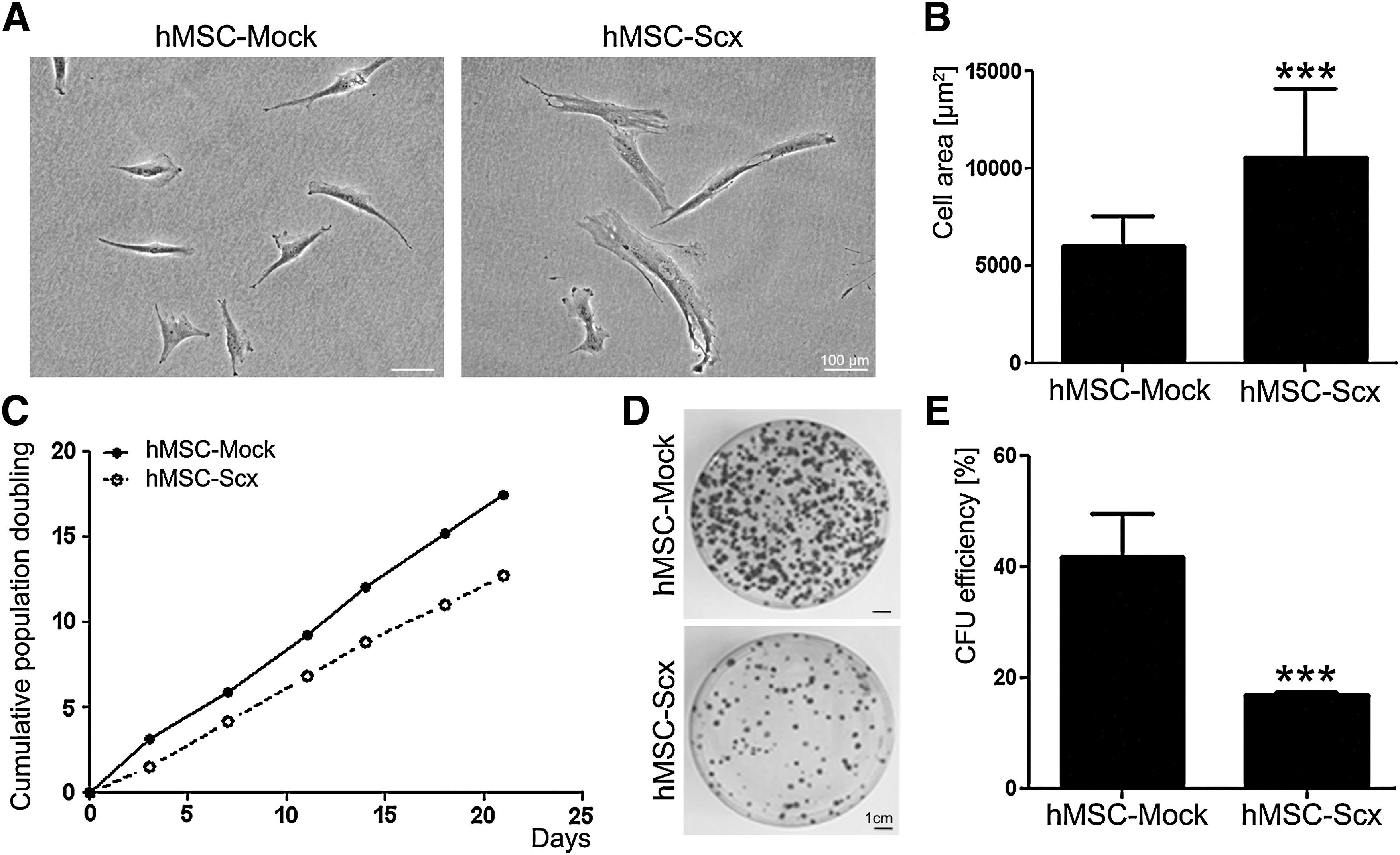
Analysis of cell morphology and self-renewal of the hMSC-Mock and hMSC-Scx cell lines.
hMSC undergo gene expression shift subsequent to the Scx expression
We expected gene expression changes to occur in hMSC after the ectopic expression of Scx and, therefore, we investigated the expression of several T/L- and mesenchyme- related genes. Figure 3A shows the gene expression analysis of collagens that are typical for T/L. No pronounced differences in the mRNA levels of collagen type I α1, III, XIV, and XV were detected by semi-quantitative RT-PCR in the 2 cell lines. Immunocytochemistry revealed intracellular production of the triple helical form of collagen I in each cell type (Fig. 3B). Next, to investigate for quantitative difference, total hydroxyproline (Fig. 3C) and secreted collagen I were measured (Fig. 3D). These analyses demonstrated a higher hydroxyproline content and collagen I secretion in hMSC-Scx cells compared with hMSC-Mock. In Fig. 3E and F, we screened the expression of several proteoglycans that are known to be highly expressed in T/L. We detected basal expression levels of all 6 proteoglycans in hMSC-Mock. In contrast, RT-PCR for 3 proteoglycans (decorin, fibromodulin, and lumican) showed an upregulation in hMSC transduced with Scx. In addition, we examined the expression of another characteristic gene for T/L-derived cells, α-SMA, which is a contractile isoform of actin [28]. The semi-quantitative PCR clearly showed that α-SMA is upregulated in hMSC-Scx cells. Finally, we studied the expression of 5 genes that are known to be crucial regulators of the chondrocyte, osteoblast, and adipocyte lineages. In Fig. 3G, the expression of 3 transcription factors, Sox9, Runx2, and TAZ, is shown. Sox9, an HMG box-containing transcription factor, is highly expressed in chondrocytes [29], and it showed a basal level of mRNA in hMSC-Mock cells. Interestingly, on Scx ectopic expression in hMSC, we observed almost a complete loss of Sox9 expression. Regarding the expression of Runx2, an osteoblast-specific transcription factor [30], and TAZ, a transcriptional modulator of the switch between osteoblasts and adipocytes [31], our RT-PCR analysis revealed no significant differences between hMSC-Mock and hMSC-Scx cells. We also analyzed the expression of peroxisome proliferator-activated receptor γ (PPAR-γ) and adipocyte protein 2 (aP2) genes, which are typical for adipocytes [32], but neither of the 2 cell lines showed expression for these genes (data not shown). Taken together, our results demonstrate that Scx ectopic expression results in a marked upregulation of T/L-related genes in hMSC.
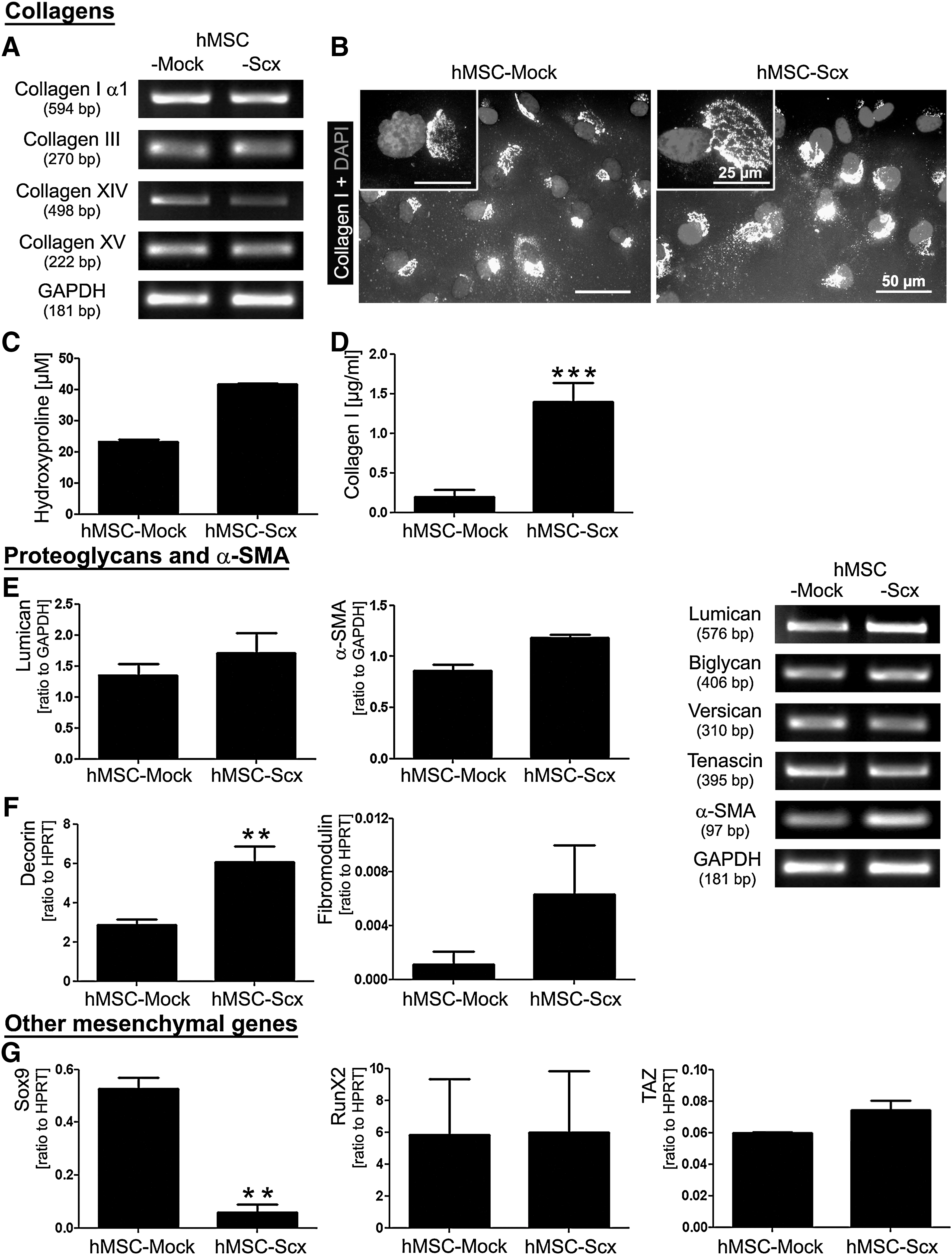
Expression screening for tenogenic and other mesenchymal lineage genes.
Scx expression in hMSC results in impaired multi-differentiation potential
To investigate whether hMSC multipotentiality is affected by the presence of Scx transgene, hMSC-Mock and hMSC-Scx cells were subjected to adipogenic, chondrogenic, and osteogenic differentiation protocols. The control, hMSC-Mock cells successfully differentiated into the 3 lineages, whereas hMSC-Scx cells were able to differentiate only into adipocytes. This was revealed by staining and quantification for accumulated lipid vacuoles (Fig. 4A) and RT-PCRI analysis as analysis for the expression of typical markers of adipogenesis, PPAR-γ and aP2 (Fig. 4B). With regard to the chondrogenic differentiation, hMSC-Scx pellets were clearly negative for the cartilage marker collagen type II (Fig. 4C). The impaired chondrogenic potential of hMSC-Scx cells was further confirmed by RT-PCR for additional chondrogenic markers such as aggrecan and COMP, which showed no upregulation in these cells (Fig. 4D). Finally, hMSC-Scx cells failed to produce mineralized matrix, which was monitored by Alizarin Red staining and quantification (Fig. 4E). Further, expression analysis of the osteogenic markers, Runx2 and bone sialo protein (BSP), revealed their upregulation only in stimulated hMSC-Mock cells (Fig. 4F). These findings clearly demonstrate that the typical multipotentiality of hMSC is hampered due to the Scx expression.
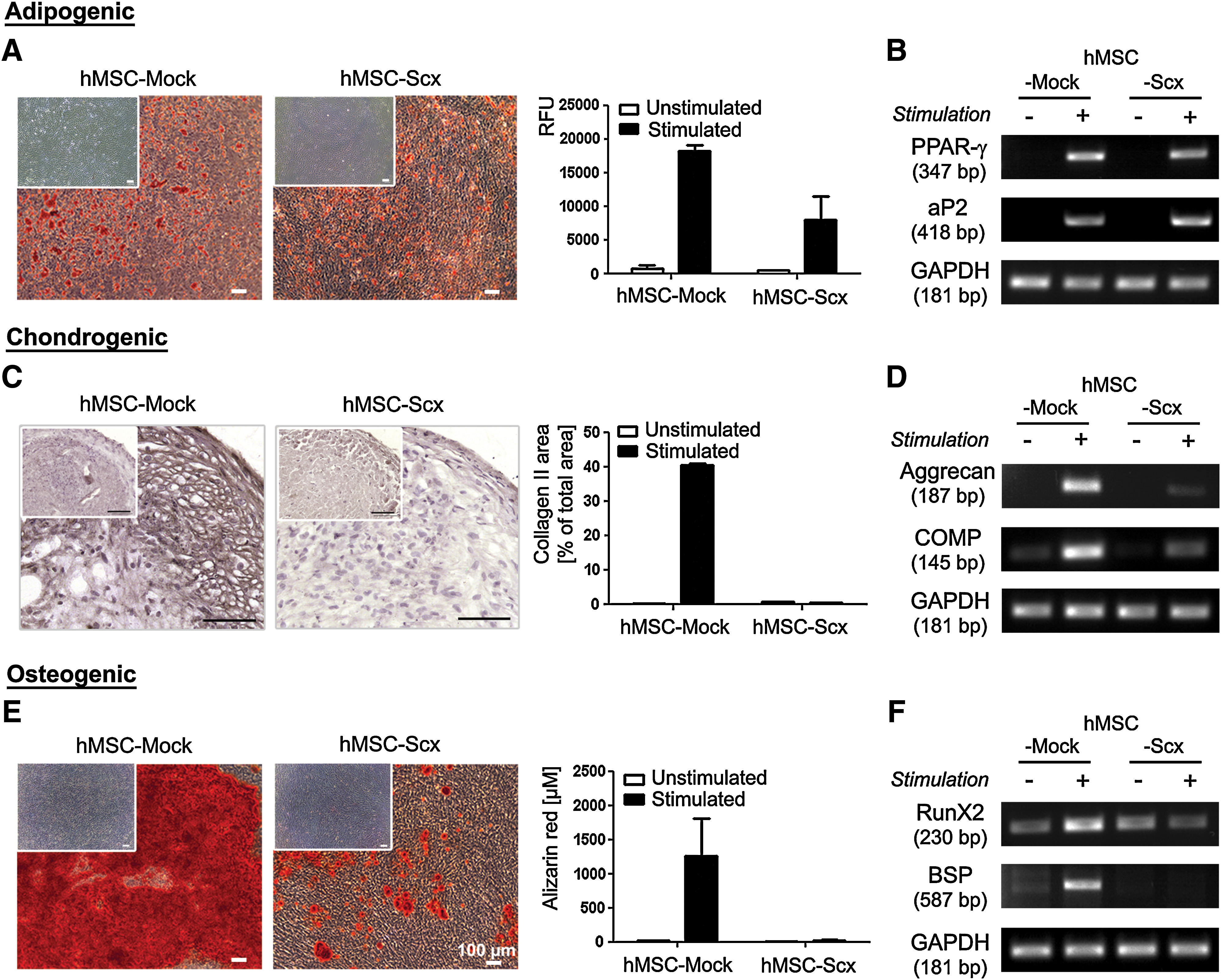
Investigation of hMSC-Mock and hMSC-Scx multipotentiality. Cells were differentiated toward 3 mesenchymal lineages.
Scx drives the expression of the T/L differentiation gene Tnmd in hMSC
To further verify that hMSC-Scx cells are tendon progenitors, we investigated whether these cells express the late tenogenic marker Tnmd. We transfected hMSC-Mock and hMSC-Scx cells with Tnmd promoter luciferase reporter construct (pENTR11-Tnmd-769/+84F-Luc II) and performed luciferase activity assay (Fig. 5A). In parallel, control cells were transfected with promoter-less luciferase plasmid (pENTR11-Luc II). Our results showed a significantly higher increase of luciferase activity (2.8-fold) in hMSC transduced with Scx. We then analyzed the expression of Tnmd transcript in hMSC-Mock and hMSC-Scx by quantitative RT-PCR (Fig. 5B). hMSC-Mock cells had almost undetectable Tnmd expression, whereas Tnmd mRNA levels in hMSC-Scx cells were significantly upregulated (14-fold). Lastly, we also performed double immunocytochemistry of Tnmd and CD44, a membrane marker to distinguish the cell boundaries (Fig. 5C). Consistent with the RT-PCR data, hMSC-Scx strongly expressed Tnmd protein; the cells were positively stained for both Tnmd and CD44, whereas hMSC-Mock cells were only CD44-positive. In conclusion, Scx expression in hMSC convincingly results in the upregulation of Tnmd.
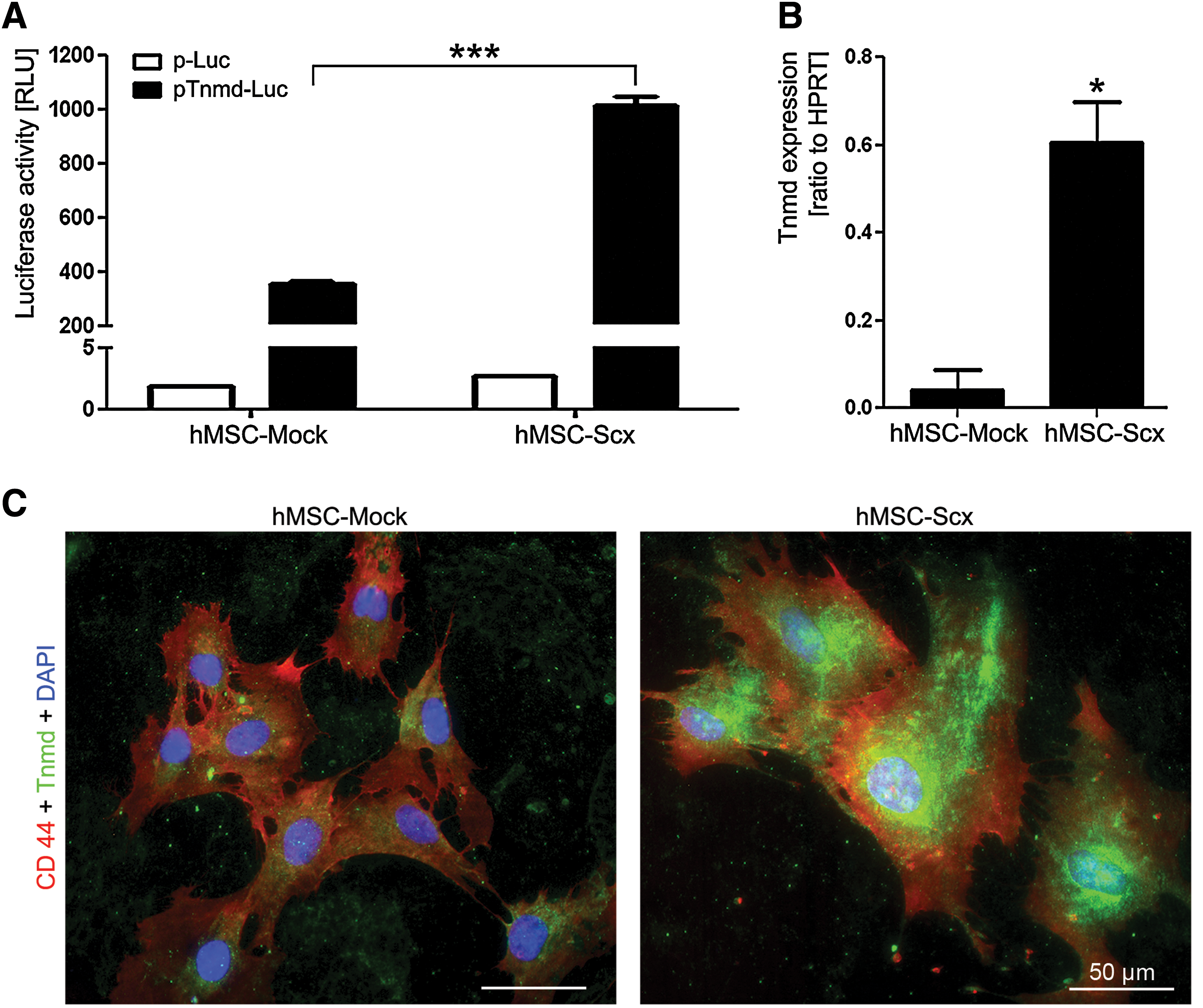
Expression analysis of the differentiation marker Tnmd in hMSC-Mock and hMSC-Scx cell lines.
Discussion
The developmental and cell biology of T/L still has to solve major challenges; for instance, the T/L field urgently requires medicaments that exclusively boost tenocyte functions as well as a higher optimization rate of T/L disease management. To allow long-term characterization of T/L-specific cells, here we aimed at establishing a protocol for the generation of tendon progenitors in vitro. We focused on the transcription factor Scx, which has been shown to be indispensible for T/L development and function [14]. As a source for primitive, undifferentiated cells, we took advantage of MSC from human BM that can be easily harvested from BM aspirates. It has been already reported that T/L tissues contain their own MSC-like cells, named tendon stem/progenitor cells (TSPC), which can as well be isolated and expanded in culture [33]. However, these cells are not as thoroughly characterized as their counterparts in the BM. In addition, the availability of T/L biopsies for TSPC isolation is clinically more limited compared with obtaining BM aspirates. First, we analyzed the BM-hMSC for endogenous Scx, E12, and E47 expression and demonstrated that these cells are Scx-negative but positive for the ubiquitously expressed E-proteins. This result confirmed a previous study which showed that Scx expression is one of the features distinguishing TSPC from BM-MSC [33]. Second, we established a stable ectopic expression of Scx in BM-hMSC. For this purpose, we implicated immortalized hMSC (SCP-1 cell line, [23]), which avoid senescence and allow for further long-term standardized research in vitro or in vivo. Next, by the means of lentiviral technology, we created a single-cell derived hMSC-Scx cell line in which every cell ectopically expresses Scx. Third, we investigated whether the generated hMSC-Scx cells exhibit a tenogenic phenotype and based on our findings, we concluded that the clonal hMSC-Scx cell line represents a homogeneous population of committed tendon progenitors (Fig. 6).
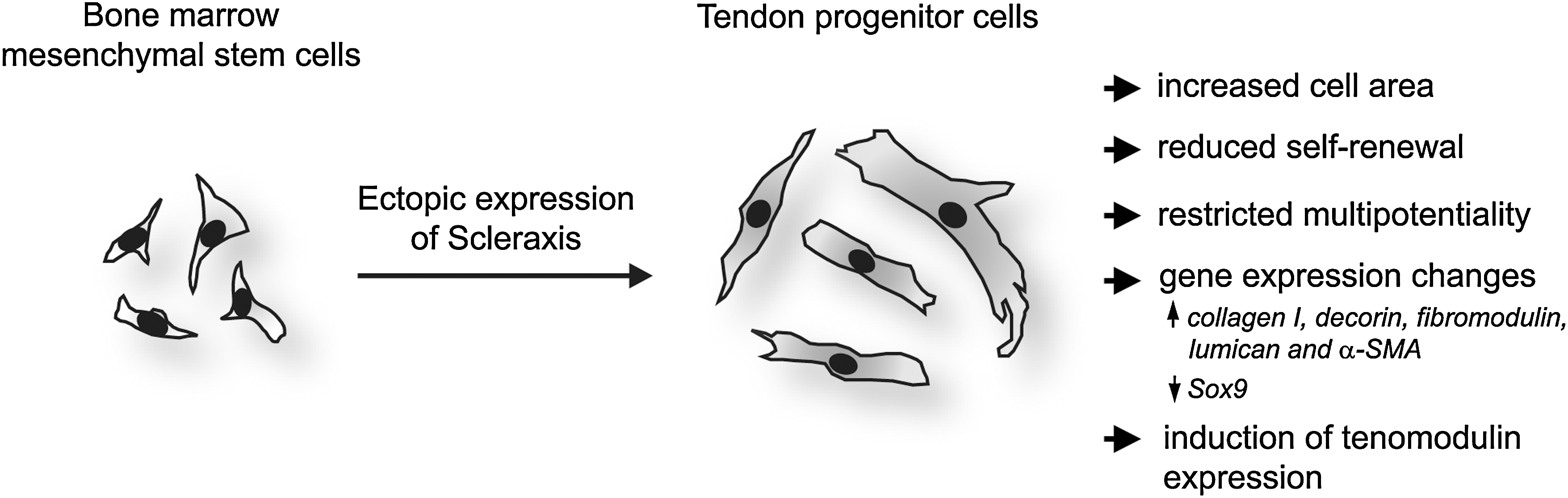
A schematic summary representing the experimental model and findings of the study. Scx cDNA was lentivirally delivered into human bone marrow-derived MSC. Subsequent to Scx expression, hMSC underwent several major changes that collectively suggest their conversion into tendon progenitors. Symbol:↑, upregulation;↓, downregulation.
The strategy to introduce transcription factors into multipotent MSC for the direct generation of various progenitor types has been already suggested as a straightforward therapeutic method in the field of stem cell-based regenerative medicine [34]. Moreover, this strategy is currently considered as an alternative and simpler approach than the generation of induced pluripotent stem cells (iPSC) in which terminally differentiated somatic cells are initially reprogrammed to the embryonic stem cell-like state and are then differentiated toward a desirable cell lineage [35,36]. Several studies have already applied the direct conversion approach on mesenchymal cells, such as fibroblasts and MSC. For example, consecutively to the genetic manipulation with single or multiple factors, these cells have been converted to cardiomyocytes, multilineage blood progenitors, and dopaminergic progenitors [37 –40]. Up to now, it has been recognized that proliferation and differentiation potential of MSC reduces in association with MSC conversion to progenitor states. After the ectopic expression of Scx in the BM-hMSC, we also observed such an inverse relation between cell lineage specification and self-renewal and multipotentiality. Here, we reported that hMSC-Scx cells had reduced self-renewal capability, as their proliferation rate and colony-forming ability were significantly lower than those of hMSC-Mock. Moreover, hMSC-Scx cells had a more restricted differentiation potential. In comparison to hMSC-Mock cells with the typical 3-lineage differentiation potential (adipogenic, chondrogenic and osteogenic), hMSC-Scx cells differentiated only toward the adipogenic lineage.
The lineage commitment of stem cells is accompanied by the activation and repression of various gene expression clusters. By performing expression screening focused on T/L-related genes and several other mesenchymal markers, we demonstrated that hMSC-Scx cells had undergone an expression shift compared with the control hMSC-Mock cells. Based on previous literature about Scx transcriptional activities, changes in collagen I, collagen XIV, and Tnmd expression were expected [10 –14]. Interestingly, we did not detect by semi-quantitative RT-PCR an augmentation in the collagen mRNA levels and since they are very complexly regulated at post-transcriptional and post-translational levels, we next performed quantitative protein analyses by comparing the hydroxyproline content and secreted collagen I of hMSC-Mock and hMSC-Scx. Our results clearly showed, consecutively to the Scx expression, a significantly higher amount of total hydroxyproline and increased collagen I secretion in hMSC-Scx cells. In addition, we also observed an upregulation in the expression levels of decorin, fibromodulin, lumican, and α-SMA. Hence, we suggest that these genes might be under a direct transcriptional control of Scx. In contrast to many common connective tissue collagens and proteoglycans, the Tnmd gene is defined as the best-known marker for tenogenic differentiation, because its expression is ultimately restricted to T/L tissues [11,15,16]. We not only demonstrated a significant enhancement of Tnmd gene transcription in hMSC-Scx cell line but also that every cell expresses Tnmd protein as revealed by immunocytochemistry.
Developmental studies have reported a coordinated expression of Scx and the chondrogenic transcription factor Sox9 to suggest an intertwined relation between cartilage and tendon progenitors [29,41]. Further, it was reported that Scx and E47 can directly cooperate with Sox9 and regulate its transcription [8]. Therefore, we analyzed whether Scx ectopic expression in BM-hMSC alters the expression of Sox9 and 2 additional transcription factors, Runx2 and TAZ, which are responsible for the osteoblast lineage and the osteoblast-to-adipocyte switch, respectively [30,31]. Quantitative RT-PCR revealed almost a complete loss of the basal Sox9 expression in undifferentiated hMSC-Scx cells. Regarding Runx2 and TAZ levels, no significant changes between hMSC-Scx and hMSC-Mock cells were detected. Nevertheless, as just mentioned, hMSC-Scx cells failed to differentiate toward both chondrogenic and osteogenic lineages. These results suggested that, once Scx expression is induced in hMSC, the Sox9 expression and transcriptional cascade are repressed, thus allowing the switch to chondrocyte phenotype to be constrained. However, further investigation is required to test whether Scx is a direct repressor of Sox9 and what exact mechanisms are restricting the hMSC-Scx adoption of osteogenic fate but allowing hMSC differentiation into adipocytes. Finally, it will be also important to study whether such juxtaposition between tendon and other mesenchymal progenitors exists in vivo.
The straight modification of MSCs into tendon progenitors has been suggested and explored by others as well [42]. Several studies applied a gene transfer of bone morphogenetic protein (BMP) 12 or both BMP12 and BMP13 into BM-MSC, and concluded that these growth factors induce a tenocyte or ligamentocyte-like phenotype [43 –45]. Hoffmann et al. 2006 [46] used a biologically active Smad8 variant that was coexpressed with BMP2 in a mouse MSC line. The engineered cells demonstrated not only a gene expression profile of tendon progenitors but were also capable of initially inducing T/L-like tissue formation and subsequently contributing to T/L regeneration in a rat Achilles tendon defect model. Interestingly, similar to our data, it was reported that the generated tendon progenitors did not possess osteogenic and chondrogenic differentiation capacity. The authors suggested a novel mechanism in which BMP2 activates the MSC but due to Smad8, the BMP2-dependent osteogenesis is inhibited and thereby, tenogenic differentiation is tolerated. The major difference between the studies just described and ours is that the conversion of hMSC to tendon progenitors relies on complex autocrine pathways in which numerous BMP downstream targets, with known but also unknown effects, will be activated. In contrast, our study is based on the introduction of a single key regulatory transcription factor that directly programs hMSC into tendon progenitors.
In the field of T/L biology, there is an impeding lack of specific markers distinguishing each step of the process of tendon differentiation from stem cells via progenitors to mature tenocytes. By comparing the behavior of our cells to previously described tendon-derived cells [33,47], we defined hMSC-Scx cells as tendon progenitors and not tendon stem cells because of their restricted self-renewal and multipotential. Moreover, we did not define hMSC-Scx as tenocytes, because tenocytes, similar to osteocytes or chondrocytes, truly exist only in vivo. It has been already reported that tenocytes require a 3-dimensional environment and mechanical stimuli to sustain their completely differentiated state [48]. Finally, to fully explore the potential of hMSC-Scx cells, a further in vivo investigation of their ability to form T/L-like tissue or to participate in T/L-repair will be necessary. A positive effect of Scx in T/L regeneration has been recently suggested in a rat model for rotator cuff healing in which rat MSC expressing transiently Scx were implicated [49]. Despite the several limitations and the lack of information on the cell phenotype, the study showed augmented biomechanical properties of the regenerates implanted with the Scx-transduced MSC. Currently, in vivo studies dealing with the hMSC-Scx cell line established by us are underway.
Taken together, after the Scx ectopic expression, hMSC underwent several major changes comprising of reduced self-renewal, restricted multipotentiality, and upregulation of T/L gene markers, which collectively suggest their successful conversion into tendon progenitors (Fig. 6). In future, this novel hMSC-Scx cell line can be used in multiple experimental set ups spanning from basic understanding of T/L molecular and cell biology to various applications in T/L tissue engineering.
Footnotes
Acknowledgments
This study was funded by the AO Research Fund of the AO foundation (Project S-07-18D). PA was supported by the Bavarian Research Foundation (Project DOK-100-08); DD and CP were supported by the German Research Foundation (DO 1414/1-1); and DD and JK were supported by the Bavarian Research Foundation (FORZEBRA, TP1/WP2). The authors thank professor David Hall (NIH, Bethesda, MD) for providing FLAG-Scx cDNA; Martina Burggraf for excellent technical assistance; professor Stefan Milz and Claudia Harbauer for their help in the histological laboratory; Dr. Wolfgang Böcker's group for virus production and infection; Dr. Attila Aszodi and Anna Maria Casalini for carefully reading the article; and professor Wolf Mutschler (Director of the Surgery Clinic-Downtown, LMU) for the support of the research laboratory.
Author Disclosure Statement
The authors have nothing to disclose.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.