
Abstract
Recent studies support cell-based therapies for cancer treatment. An advantageous cell type for such therapeutic schemes are the mesenchymal stem cells (MSCs) that can be easily propagated in culture, genetically modified to express therapeutic proteins, and exhibit an innate tropism to solid tumors in vivo. Recently, we successfully isolated and expanded MSCs from second-trimester amniotic fluid (AF-MSCs). The main characteristic of AF-MSCs is their efficient and rapid expansion in vitro. Herein, we investigated the AF-MSCs tropism and capability to transport interferon beta (IFNβ) to the region of neoplasia in a bladder tumor model. To this end, we used the T24M bladder cancer cell line, previously generated from our studies, and developed a disease progression model in immunosuppressed mice, that can recapitulate the molecular events of bladder carcinogenesis. Our results documented that AF-MSCs exhibited high motility, when migrated either to T24M cells or to T24M-conditioned medium, and we further identified and studied the secreted factors which may trigger these enhanced migratory properties. Further, lentivirus-transduced AF-MSCs, expressing green fluorescent protein (GFP) or IFNβ, were intravenously administered to T24M tumor-bearing animals at multiple doses to examine their therapeutic effect. GFP- and IFNβ-AF-MSCs successfully migrated and colonized at the tumor site. Notably, significant inhibition of tumor growth as well as prolonged survival of mice were observed in the presence of IFNβ-AF-MSCs. Collectively, these results document the great potential of AF-MSCs as anti-cancer vehicles, implemented by the targeting of the tumor site and further facilitated by their high proliferation rate and expansion efficiency in culture.
Introduction
B
Cell lines and animal models are valuable research tools to increase the knowledge related to the molecular mechanisms of neoplasia and develop potential therapeutic strategies [4]. In our previous studies, we developed the T24M cell line, an invasive and metastatic bladder cancer model, derived from the T24 cells after serial sub-culturing and subcutaneous injections in SCID mice [2,5,6]. Detailed cytogenetic and proteomic characterization of T24M cells, in comparison to their parental cell line, showed that these cells exhibited molecular characteristics frequently encountered in aggressive urothelial carcinomas. Further, ectopic implantation of T24M cells generated a reliable bladder cancer disease progression model for the development and evaluation of novel therapeutic approaches [2,5].
One possible therapeutic molecule for bladder cancer is interferon beta (IFNβ), a member of type I IFN family [7]. It has been shown that high concentrations of IFNβ repress malignant cell growth in vitro [8]. IFNβ suppresses tumor cell growth by induction of differentiation, S phase accumulation, and apoptosis [7,9,10]. Nevertheless, the therapeutic use of IFNβ in vivo is limited because of its high cytotoxicity when systemically administered at high doses [11,12]. Additionally, systemic IFNβ treatment for most solid tumors has limited effectiveness, as the short half life of this protein could not reach the desired levels to inhibit cell growth, suppress angiogenesis, and enhance host immune response [7,13]. Thus, cell-based therapies are recently employed as useful alternatives for IFN treatment for aggressive solid tumors.
Human mesenchymal stem cells (MSCs) are considered useful delivery vehicles for many types of solid tumors or gliomas [14 –20]. MSCs can be isolated from a wide variety of sources such as the most well-characterized bone marrow (BM), adipose tissue, umbilical cord, peripheral blood, fetal liver, and placenta [21 –25]. Several recent reports have shown the use of BM-MSCs as delivery vehicles for anti-cancer molecules into solid tumors in vivo, taking advantage of their unique feature to integrate into the tumor site [8,17,18,26,27]. The MSCs' innate ability to migrate to the tumor microenvironment is thought to take place due to the inflammatory signaling at the tumor site, resembling a wound, and, thus, providing a permissive environment for the engraftment of MSCs [17,19,28]. Another possible theory involves the cellular and molecular processes performed during tumor stroma development [29]. Stromatogenesis is a complex event including recruitment of local tissue fibroblasts and circulating MSCs into the tumor, which contribute to the stroma formation by differentiating into cells, such as fibrocytes that produce extracellular matrix components [29].
Recently, our group and others isolated MSCs from an alternative source, the second-trimester amniotic fluid (AF), which can be obtained during routine amniocentesis [30 –35]. We previously characterized these cells based on their phenotype, proliferation rate, and differentiation potential [33,35]. More importantly, we showed that these cells can differentiate in vitro into cell types derived from mesoderm (adipocytes and osteoblasts) and endoderm (hepatocytes) [33,35]. Further, AF-MSCs represent an advantageous mesenchymal cell type for transplantation, as they can be expanded in high numbers in culture and exhibit low expression of HLA-ABC and no HLA-DR antigens, reducing in this way the risk of cell rejection and the potential of graft-versus-host disease [30 –33,35].
In the current study, we aim at evaluating the migratory potential of AF-MSCs into tumor cells in vitro and in vivo and additionally determining their function as delivery vehicles for anti-tumor molecules, such as IFNβ, at the site of neoplasia [11,24 –26,36,37]. More specifically, herein, we report, for the first time, the utilization of a therapeutic strategy based on MSCs in the T24M bladder tumor model, and, more importantly, we introduced the AF-MSCs as carriers of IFNβ into tumor sites in vivo.
Materials and Methods
Isolation and culture of MSCs from human AF
Cultured AF-MSCs were isolated from 6 human AF samples, collected during scheduled normal pregnancy amniocenteses between the 15th and 18th week of gestation, as previously described [33,35]. All second-trimester AF samples were obtained following a written informed consent, approved by the Ethical Committee of the Alexandra Hospital and the Bioethics Committee of the University of Athens, School of Medicine. Using a 22G needle and under ultrasonographic control, 10–15 mL of AF was aspirated under aseptic conditions. The procedure-related spontaneous abortion rate ranges from 0.06% to 0.5% [38,39]. Each sample was centrifuged at 1,300 rpm for 10 min. The cell pellet was resuspended in Dulbecco's modified Eagle's medium (DMEM; Gibco-BRL) supplemented with 20% (vol/vol) fetal bovine serum (FBS) (Gibco-BRL) in a 25 cm2 tissue culture-treated flask and incubated at 37°C in a 5% humidified CO2 chamber for ∼8–12 days, until the first colonies appeared. Colonies were then selected, and spindle-shaped AF-MSC colonies were subcultured [35].
Antibodies, flow cytometry analysis (FACS), and differentiation assays
Details are provided in the Supplementary Data; Supplementary Data are available online at
Cell lines
Details are provided in the Supplementary Data.
Lentiviral vector generation and production
The vesicular stomatitis virus G protein pseudotyped self-inactivating HIV-1-based lentiviral vector pCCLsin.PPT.hPGK.eGFP.WPRE [35,40,41] with the woodchuck hepatitis virus posttranscriptional regulatory element (WPRE) was used. This vector contains the enhanced green fluorescent protein (eGFP) reporter gene driven by the human phospho-glycerate kinase gene (hPGK) promoter. For the production of the lentiviral vector expressing the human IFNβ gene, the open reading frame of the human IFNβ was amplified by polymerase chain reaction (PCR) from the ORF-hIFNb v.11 plasmid (InvivoGen). We cloned the IFNβ gene into the pCCLsin.PPT.hPGK.WPRE plasmid in place of the GFP gene, using a set of Forward 5′- CTGAGATCACCGGTCAACATGACCAACA-′3 and Reverse 5′- GTCGACCTCAGTTTCGGAGGTAACCTGTA-′3 primers.
Virus was produced by transient transfection of 293T cells as previously described [42] and collected by ultracentrifugation using an Ultracentrifuge Discovery 100 Sorvall (Thermo Fisher Scientific Inc.). The concentrated virus was resuspended in PBS supplemented with 0.5% bovine serum albumin (Sigma-Aldrich). The GFP lentiviral titers were determined by infection of HT1080 cells with serial dilutions of the concentrated viral stock. GFP fluorescent cells were identified by fluorescent microscopy and FACS analysis. Titers ranged from 5×108–3×109 infectious units (IU)/mL.
For the determination of the IFNβ lentiviral titers, the biological titration method was performed by using GFP lentivirus FACS titration as standard assay and real-time PCR analysis. HT1080 cells were independently infected with serial 10-fold dilutions of GFP and IFNβ concentrated viral stocks, and genomic DNA was isolated by using Proteinase K (Sigma-Aldrich) treatment, followed by phenol-chloroform extraction. HT1080 cells transduced with GFP lentivirus were simultaneously subjected to FACS titration and real-time PCR analyses. The sample giving 10% of GFP positive cells corresponded to a value of 0.1 copy of virus sequences per cell, and a regression curve was created. The regression curve was then used to calculate IFNβ lentivirus copy number after real-time PCR analysis [43]. Real-time PCR analysis was carried out by using SYBR Green master mix (Roche Applied Sciences) and the following primers, designed on the WPRE sequence of the lentiviral vectors: Forward WPRE 5′-TTCTCCTCCTTGTATAAATCCTGGTT-3′ and Reverse WPRE 5′-CGCCACGTTGCCTGACA-3′, according to manufacturer's protocol. IFNβ lentiviral titers ranged from 6×107–108 IU/mL. The reaction was normalized to RnaseP internal control (Rnase P, No.4316844; ABI). For the AF-MSCs transduction, ∼5×104/well AF-MSCs were seeded in a 6-well plate, and virus was added in a multiplicity of infection (MOI) of 1 to 60.
ELISA
To quantitate the production of IFNβ by the transduced AF-MSCs, cells were incubated with the IFNβ lentivirus for 24 h with MOI of 10, 20, or 30, respectively. Four days later, the conditioned media (CM) were collected, and ELISA assay was performed for the detection of human IFNβ (PBL; Biomedical Laboratories), in duplicate, according to manufacturer's protocol. The absorbance was determined by a microplate reader at 450 nm (ELX 800; Biotek Instruments Inc). Statistical analysis was performed by unpaired Student's t-test method. The systemic IFNβ serum levels within groups in mice were determined by ELISA, as just described.
Transwell migration assay
To examine the biological effect of T24M cells on the migration properties of AF-MSCs or IFNβ-AF-MSCs, a transwell migration assay was established. In fact, 293T and T24M cells were cultured for 24 h in DMEM (0.5% FBS) and were then transferred at 105/600 μL density to the bottom well of a transwell plate with 8 μm pore size (Corning-Costar) for 8 h, to allow the cells to adhere. For the preparation of the CM from T24M cells, the cells were cultured in DMEM (0.5% FBS) for 8 h, and the supernatant was collected, centrifuged, filtered, and added to the bottom well of the transwell plate. Then, AF-MSCs or IFNβ-AF-MSCs, also cultured in DMEM (0.5% FBS) for 24 h, were transferred at 4×104/100 μL density to the insert of the transwell plate. AF-MSCs or IFNβ-AF-MSCs were allowed to migrate for 16 h across the pore membrane, toward T24M cells, 293T cells, CM from T24M cells, or DMEM supplemented with 0.5% FBS as a control.
After that, the nonmigrated cells were removed from the top of the insert with a wet cotton swab. The migrated cells at the bottom of the insert were then fixed with 4% paraformaldehyde (Sigma-Aldrich) and stained using the Ral Kit (Ral Reactif) according to manufacturer's instructions. Migration was quantified by counting the stained nuclei that passed through the filter. Photographs from the stained nuclei were taken from a minimum of 10 fields of view (20×) for each membrane, using a Leica CTR MIC microscope, and then the nuclei were counted by using the Image J 1.38× software. Three independent experiments were performed, each including 3 replicates, and the mean of each experiment was calculated. Statistical analysis was performed by using the Student's t-test.
The effects of inteleukin-8 (IL-8) and vascular endothelial growth factor (VEGF) cytokines in the migration properties of AF-MSCs and IFNβ-AF-MSCs were studied by treating T24M cells or CM derived from T24M cells with IL-8 (Peprotech House; 5 μg/mL) or VEGF neutralizing antibodies (Peprotech; 5 μg/mL). Appropriate isotype antibodies were used as negative controls. Recombinant IL-8 (Peprotech) or VEGF (Peprotech) were added, both at 500 ng/mL, in DMEM supplemented with 0.5% FBS.
Proteome array of T24M-CM
CM was prepared by collecting medium containing 0.5% FBS after 24 h culture of T24M cells or 293T cells. The CM was concentrated ∼10-fold, by using ultra filtration units (Millipore) with a 3-kDa cut-off. Then, CM was analyzed for specific proteins by using proteome profiler array for angiogenesis growth factors (Catalog # ARY007; R&D Systems Inc.) according to manufacturer's instructions. Quantitation of the detected spots was performed by using the Quantity One Software 4.4.1 (Bio Rad Laboratories Inc). Values were normalized to positive controls. Data are presented as the mean±SD for 3 independent experiments.
Proliferation of T24M cells in the presence of recombinant IFNβ
T24M cells were plated at a density of 1.5×104/mL in a 96-well plate and were cultured for 5 days in the presence of DMEM (10% FBS) supplemented with gradually increasing concentrations of recombinant IFNβ (Peprotech) from 0 to 1.25×105 pg/mL, in 5 replicates for each concentration. Medium was changed daily, and after 5 days, CellTiter 96 AQueous One Solution cell proliferation assay (MTS) (Promega Ltd) was performed. The absorbance was recorded at 490 nm by using a microplate reader (ELX 800; Biotek Instruments Inc). Results were expressed as the percentage of proliferation increase, calculated using the following formula: [(ODday5−ODday0)/(ODcontrol−ODday0)x100], where ODcontrol corresponded to the absorbance measured in non-IFNβ treated cells on day 5. All assays were performed in triplicate, and the mean of each experiment was calculated. Statistical analysis was performed by using the Student's t-test.
Proliferation of T24M cells in the presence of CM from IFNβ-AF-MSCs
AF-MSCs were transduced with IFNβ lentivirus with MOI of 10, 20, or 30. Four and 10 days post transduction, CM were collected from 2.5×104 cells/mL, filtered, and stored in −80°C until use. T24M cells were seeded at a density of 1.5×104/mL in a 96-well plate in 5 replicates and incubated either with the respective CM or DMEM (10% FBS) at 37°C. CM was changed every day. After 3 days, MTS assay (Promega Ltd) was performed, and the absorbance was recorded at 490 nm by using an ELISA plate reader. Results were expressed as the percentage of proliferation increase, calculated using the following formula: [(ODday3−ODday0)/(ODcontrol−ODday0)x100], where ODcontrol corresponded to the absorbance measured at day 3 for the wells incubated with DMEM (10% FBS). The experiment was performed in triplicate, and the mean of each experiment was calculated. Statistical analysis was performed by using the Student's t-test.
Effect of IFNβ on the viability of AF-MSCs
The proliferation rate of AF-MSCs transduced with IFNβ lentivirus with MOI of 10, 20, 30, and 60 was estimated by an MTS assay, as just described, for a period of 4 and 10 days post transduction.
Animals and cell administration
NOD-SCID immunodeficient mice were housed and maintained at the Animal Facility of the Biomedical Research Foundation of the Academy of Athens. The procedures for the care and treatment of animals were performed according to the institutional guidelines that follow the guidelines of the Association for Assessment and Accreditation of Laboratory Animal Care, the recommendations of the Federation of European Laboratory Animal Science Associations, and the National Institute of Health. For the establishment of tumors, T24M cells were subcutaneously administered in 200 μL of PBS into the tail base of each mouse. Ten days later, tumors were established, and 1×106 AF-MSCs (n=7) or AF-MSCs transduced with GFP (MOI 60, n=7) or IFNβ lentivirus (MOI 10, n=7) were intravenously (i.v.) administered into the lateral tail vein in a volume of 200 μL. The MSC injections were performed in 3 weekly doses. Tumor bearing mice without treatment were used as controls (n=7). One week after the last cell dose, animals were sacrificed, and lung, spleen, and liver tissues were collected. Tumors were also collected and measured by caliper.
Immunohistochemistry, immunofluorescence, and TUNEL assay
Details are provided in the Supplementary Data.
Determination of animal survival and tumor measurements
For the determination of the IFNβ effect on the animal survival, mice were treated with AF-MSCs (n=12), GFP-AF-MSCs (n=12), or IFNβ-AF-MSCs (n=12), as previously described. Tumor bearing mice without treatment were used as controls (n=10). Animal survival was measured, starting from the day of the T24M cell injection until death, or until the presence of a tumor diameter>17 mm, tumor ulceration, or bleeding, where in those cases mice had been sacrificed earlier. The difference in survival was determined by Kaplan–Meier's log rank test. The tumor growth was monitored by measuring 2 perpendicular axes every 7 days, using a caliper, from the beginning of the cell treatment.
Results
Characterization of lentiviral transduced AF-MSCs
Initially, cell pellets from 6 second trimester AF samples were plated according to previous protocols, and the first plastic adherent colonies of AF-MSCs began to appear 8–12 days after the initial plating [33,35]. AF-MSCs were isolated and further expanded up to 40–50 passages to date with normal karyotype and high proliferation capacity [33,35]. AF-MSCs expressed high levels of CD73, CD90, CD105, and CD166 markers, whereas hematopoietic molecules such as CD45 were not detected [33,35], as analyzed by flow cytometry (Supplementary Fig. S1A). Further, AF-MSCs, when cultured under appropriate differentiation inducing conditions, were successful in forming adipocytes, osteocytes (mesoderm-derived), and hepatocytes (endoderm-derived) cells in vitro (Supplementary Fig. S1B).
We further investigated whether AF-MSCs can be efficiently transduced with lentiviral vectors for further in vivo experiments. Cells were transduced with GFP lentivirus with MOI of 60, which led to a 92%–100% efficiency of infection, as determined by FACS analysis (Fig. 1A, B). In general, an increase in fluorescence of GFP positive AF-MSCs was shown, when the cells were transduced with rising doses of GFP lentivirus (MOI from 10 to 100) ranging from 45% to 100% [35]. The expression levels of MSC markers such as CD90, CD105, CD166, and CD73 in GFP-AF-MSCs remained the same after transduction (Fig. 1C). We observed that the expression of GFP remained stable after cycles of freezing and thawing [35]. GFP transduced AF-MSCs exhibited similar proliferation capacity as AF-MSCs and could successfully differentiate into functional adipocytes, osteocytes, and hepatocytes in vitro [35].
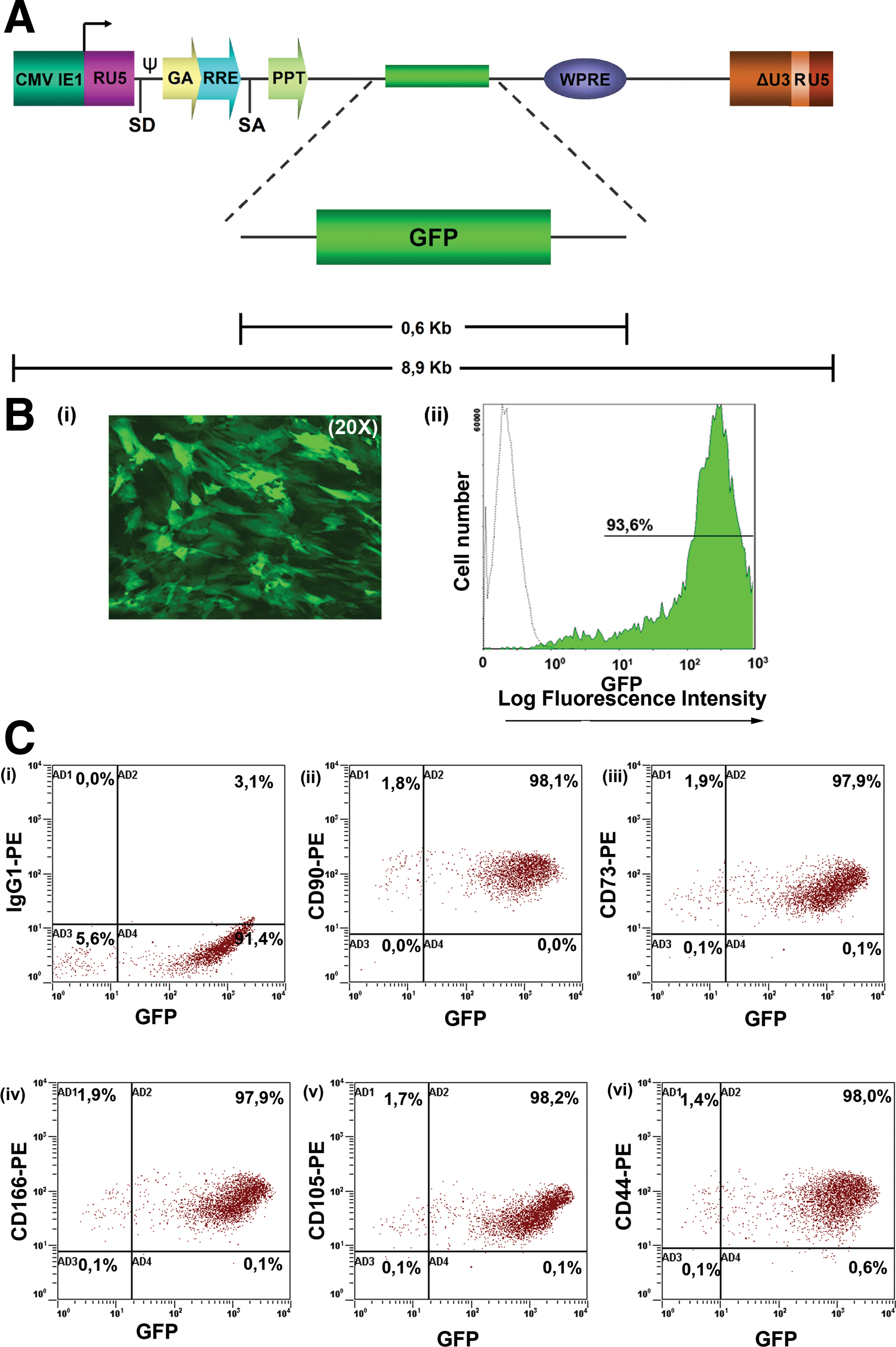
GFP lentiviral production and GFP-AF-MSCs marker expression.
For assessing the therapeutic effect of secreted IFNβ from AF-MSCs, we transduced these cells with a lentivirus carrying the human IFNβ coding sequence (Fig. 2A), at multiple MOIs. Lentiviral integration at different MOIs was estimated by real-time PCR for WPRE viral sequence in IFNβ-AF-MSCs (Fig. 2B). Concentration of the secreted IFNβ from AF-MSCs was determined by ELISA at 1,016 pg/mL (MOI 10), 4 days after transduction, which corresponded to an approximate production of 406.3×10−4pg of IFNβ per cell (Fig. 2C). As expected, the IFNβ concentration from IFNβ-AF-MSCs was increased in a MOI dose-dependent manner. IFNβ-AF-MSCs expressed CD90, CD105, CD166, and CD73 antigens at the same levels after transduction (data not shown).
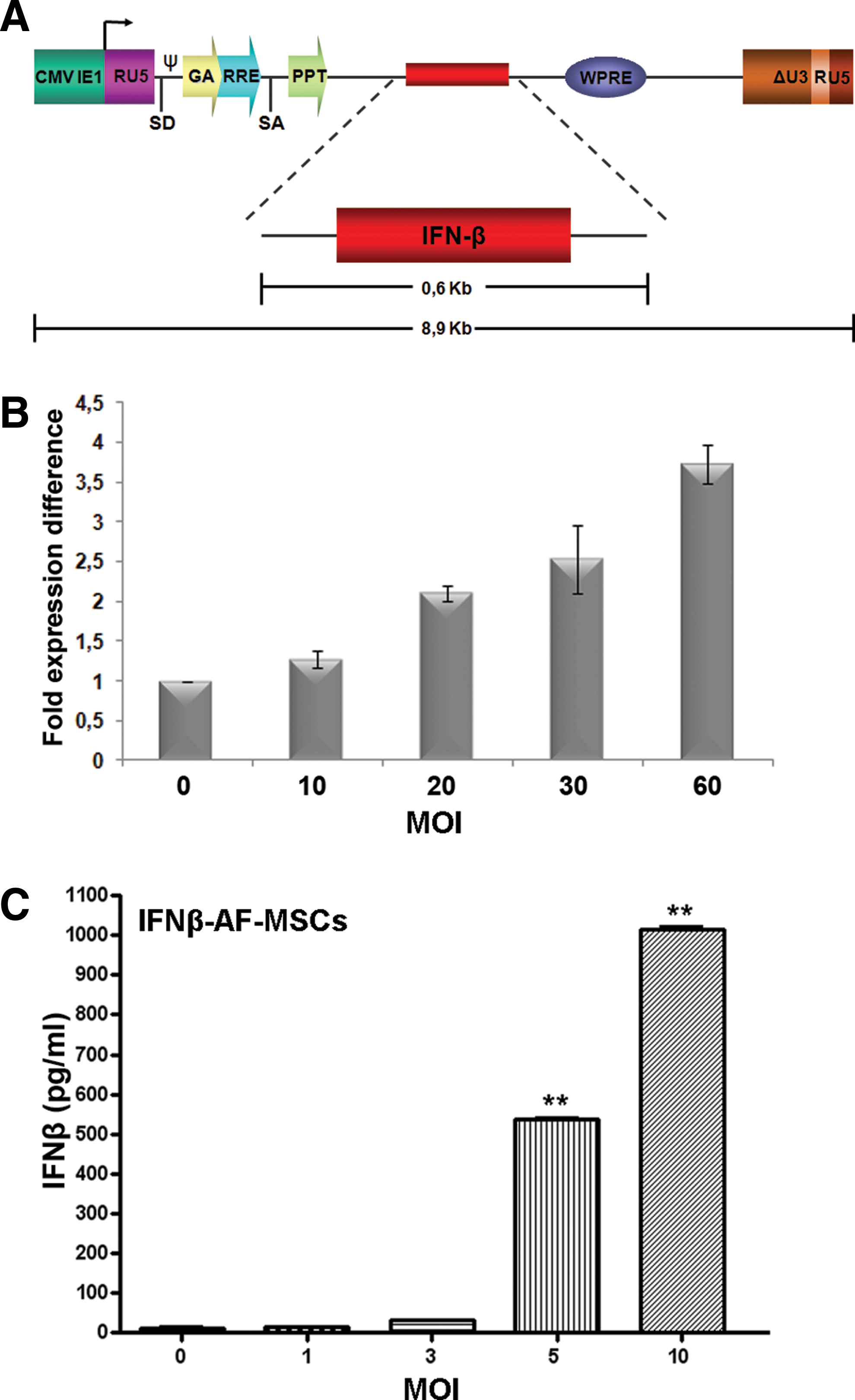
IFNβ production by AF-MSCs.
Migratory capacity of AF-MSCs toward T24M cells in vitro
We then evaluated the migratory capacity of AF-MSCs toward the bladder cancer cell line T24M in vitro (Fig. 3). Although AF-MSCs did not migrate spontaneously toward the control medium or the nontumorigenic cell line 293T, they exhibited increased motility toward T24M cells (62-fold increase, P<0.01, Student's t-test) using an in vitro transwell system. AF-MSCs were also found to exhibit significantly greater migratory capacity toward CM derived from T24M cells (68-fold increase, P<0.01, Student's t-test), compared with control medium.

Migratory capacity of AF-MSCs in vitro.
Cytokines and growth factors triggering the AF-MSCs migration toward T24M cells
Numerous cytokines, growth factors, and their receptors have been shown to affect stem cell migration. To determine the molecular mediators responsible for the enhanced migration of AF-MSCs toward bladder cancer cells, we examined the CM, derived from T24M cells, by using proteome array specific for angiogenic cytokines and growth factors (Fig. 4). DMEM supplemented with 0.5% FBS was used as a negative control (data not shown).

T24M CM proteome levels of cytokines.
A number of such cytokines were found to be secreted by T24M cells, as identified by the cytokine array. For negative control, we used CM from 293T cells, a cell line that does not induce specific AF-MSCs migration. For instance, VEGF, was highly expressed by T24M cells and represents an MSC-attracting factor [44]. In addition, urokinase plasminogen activator (uPA), secreted by T24M cells, and its receptor, uPAR, are involved in chemotaxis and cell guidance during normal development and are up-regulated in invasive tumors. Evidence has been provided that uPA and uPAR, in malignant solid tumors, augment neural and MSC tropism [45].
Other highly expressed cytokines in the CM of T24M cells that are correlated with MSCs recruitment to the tumor area were IL-8 [44], platelet-derived growth factor BB [46], transforming growth factor beta-1 (TGF-β1) [44,47], and monocyte chemotactic protein-1 [45,48].
More importantly, VEGF [44,47], IL-8 [44], angiogenin [49], and TGF-β1 [44] are all key mediators in the complex process of tumor angiogenesis. This could indicate that the same pathways are used for both the induction of angiogenesis and for MSC recruitment by cancer cells.
The role of VEGF and IL-8 in migration of AF-MSCs and IFNβ-AF-MSCs
To delineate the role of highly secreted factors from T24M cells, such as VEGF and IL-8, in AF-MSCs and IFNβ-AF-MSCs migration, we performed an in vitro transwell migration assay, using neutralizing antibodies for the respective cytokines, as well as recombinant molecules. In greater detail, AF-MSCs and IFNβ-AF-MSCs exhibited similar migration potential toward T24M cells or T24M-CM, when tested in an in vitro transwell migration system (Fig. 5). Further, we showed that in the presence of IL-8 neutralizing antibody in CM derived from T24M cells (Fig. 5A), the migratory capacity of AF-MSCs and IFNβ-AF-MSCs was reduced at 37.91%±8.6% and 64.27±7.82, respectively. In addition, when we added recombinant IL-8 to DMEM supplemented with 0.5% FBS, a statistically significant increase in the migration capacity of both cell types was observed (Fig. 5A). A similar effect in reduction of the migration capacity of both AF-MSCs (63.95%±7.09%) and IFNβ-AF-MSCs (69.8%±9.7%) was observed in the presence of VEGF neutralizing antibody in CM derived from T24M cells (Fig. 5A). However, recombinant VEGF in DMEM supplemented with 0.5% FBS did not induce the migration potential of these cells (Fig. 5A).

The role of IL-8 and VEGF in the migratory capacity of AF-MSCs and IFNβ-AF-MSCs in vitro.
When we tested the migration potential of these cells toward T24M cells, we observed a statistically significant reduction in migration of both AF-MSCs (67.53%±12.38%) and IFNβ-AF-MSCs (87.29%±5.81%), only in the presence of IL-8 neutralizing antibody (Fig. 5B).
In vitro effect of recombinant IFNβ on the proliferation of T24M cells
We examined the effect of recombinant IFNβ on the proliferation of T24M cells, and we showed that recombinant IFNβ inhibited the growth of T24M cells in a concentration-dependent manner (Fig. 6A). More importantly, concentrations ≥25×103 pg/mL of recombinant IFNβ resulted in a statistically significant decrease in the proliferation of T24M cells (P<0.01, Student's t-test).

Biological activity of recombinant and secreted IFNβ.
Biological activity of the secreted human IFNβ on T24M cells and AF-MSCs
The biological effect of secreted IFNβ on T24M cell proliferation was analyzed. CM collected from IFNβ-AF-MSCs (MOI 10, 20, and 30), 4 days post transduction, resulted, for example, in a 66.55%±4.3% (P<0.01, Student's t-test) inhibition of tumor cell growth for MOI 10 [Fig. 6B (i)]. Additionally, CM collected from IFNβ-AF-MSCs (MOI 10, 20, and 30) 10 days post transduction had similar effects on the T24M proliferation [74.87%±6.56% (P<0.01, Student's t-test), for MOI 10], thus showing that IFNβ expression remained stable in time [Fig. 6B (ii)]. These results indicated that IFNβ-AF-MSCs secrete biologically active soluble IFNβ.
Moreover, the described analyses indicated that IFNβ secretion from AF-MSCs resulted in similar inhibition of T24M proliferation, when cells were transduced with MOI greater than 10.
We further tested the effect of IFNβ production on the carrier cells, the AF-MSCs. For this reason, we performed the proliferation assay of AF-MSCs transduced with IFNβ lentivirus at MOIs of 10, 20, 30, and 60 [Fig. 6C (i)]. The IFNβ-AF-MSCs growth rate was evaluated 4 and 10 days post transduction by MTS assay, thus showing a nonsignificant decrease in cell proliferation at an MOI of 10, 4 days after transduction. However, an MOI of 60 resulted in an ∼75% decrease in cell viability at the same time point [Fig. 6C (ii)]. Thus, an MOI of 10 was selected for further experimental procedures, as it resulted in T24M cell proliferation inhibition and at the same time had no negative effect on the AF-MSCs viability.
Tumor tropism and engraftment of transplanted AF-MSCs
To confirm that AF-MSCs engrafted in tumors in vivo, we utilized T24M tumor bearing mice and applied a previously established therapeutic scheme [8,18] including 3 weekly doses of AF-MSCs, GFP-AF-MSCs, or IFNβ-AF-MSCs. In all cases, T24M tumors were palpable (∼2–3 mm3), 10 days after the T24M cell injection, when the MSCs treatment had been initiated. We further traced these cells 7 days after the last injection, by immunohistochemical staining of tumors and other mouse organs, by using anti-GFP or anti-CD90 antibodies. Numerous GFP and CD90 positive cells were located mostly in the periphery of the tumor area; however, some also infiltrated into the tumor (Fig. 7A). These results suggested that AF-MSCs survived in the animals for at least 1 week after transplantation and possibly formed colonies which were incorporated into the tumor structure. Additionally, very rare GFP positive cells were found in the lung, whereas they were absent from any other type of organs examined (Fig. 7B). The present data implied that the tumor microenvironment plays a critical role in the successful engraftment and proliferation of MSCs in vivo. To exclude the possibility that any apoptotic effects observed in tumors of mice treated with GFP, transduced AF-MSCs were induced by the GFP expression, we performed double staining analysis for TUNEL and GFP. The results showed no co-localization of the TUNEL and GFP stain, implying that the GFP positive cells were not apoptotic after in vivo transplantation (Supplementary Fig. S2).

Tumor progression in the presence of AF-MSCs, GFP-AF-MSCs, or IFNβ-AF-MSCs.
The systemic levels of IFNβ in mouse serum after therapy
To analyze the systemic levels of IFNβ after treatment, serum samples were collected 7 days after the last cell injection from T24M tumor bearing mice (control); T24M tumor bearing mice treated with AF-MSCs, GFP-AF-MSCs, or IFNβ-AF-MSCs; and NOD/SCID mice without tumors (normal). The concentrations of IFNβ were measured by ELISA, and the results showed no significant difference in systemic IFNβ serum levels within groups (Supplementary Fig. S3), suggesting that the IFNβ serum levels were not altered by the AF-MSCs local production of IFNβ at the site of the tumor.
Effect of IFNβ-AF-MSCs on tumor volume and animal survival
We further investigated whether AF-MSCs, GFP-AF-MSCs, or IFNβ-AF-MSCs could have an effect on T24M tumor growth during the 3 weeks of treatment. AF-MSCs and GFP-AF-MSCs resulted in no alteration of the tumor diameter, whereas IFNβ-AF-MSCs treatment exhibited a statistically significant retardation of 27.24%±2.25% in tumor growth (P<0.05, Student's t-test) (Fig. 7C).
Further, to determine whether IFNβ-AF-MSCs could provide therapeutic benefits in vivo for a longer period of time, T24M tumor bearing mice were treated with AF-MSCs, GFP-AF-MSCs, or IFNβ-AF-MSCs in 3 weekly doses, as just described and followed until death. Tumor volume was estimated by measuring tumor diameters on a weekly basis after T24M cells administration. As shown in Fig. 8A, the therapeutic approach using the IFNβ-AF-MSCs significantly suppressed tumor growth (482.69±116.28 mm3), when compared with untreated animals (1842.9±413.825 mm3) (P<0.01, Student's t-test), 59 days after the T24M cell injection. In contrast, there was no significant difference between the untreated group and the group that received treatment with AF-MSCs (1318.22±302.14 mm3) or GFP-AF-MSCs (1163.37±266.11 mm3) throughout the experiment.

Therapeutic efficacy of IFNβ-AF-MSCs. T24M tumor-bearing mice received i.v. AF-MSCs (n=12), GFP-AF-MSCs (MOI 60, n=12), or IFNβ-AF-MSCs (MOI 10, n=12) in 3 weekly doses.
To assess whether the inhibition of tumor growth, observed after treatment with IFNβ-AF-MSCs, was also associated with the prolonged survival of the animals, Kaplan–Meier survival analysis was performed (Fig. 8B). Among mice bearing T24M tumors, those treated with IFNβ-AF-MSCs lived significantly longer [median survival time: 75 days (P<0.01, log rank test)] than the mice treated with AF-MSCs [median survival time: 57.5 days (P=0.855, log rank test)] or GFP-AF-MSCs [median survival time: 55 days (P=0.9143, log rank test)], compared with the untreated mice (median survival time: 57.5 days).
More importantly, apoptosis of tumor cells was induced in higher levels with IFNβ-AF-MSCs (3.3%±0.73%), compared with AF-MSCs (1.1%±0.32%) or GFP-AF-MSCs (1.1%±0.45%) treatment, as determined by TUNEL assay (Fig. 9). Moreover, IFNβ-AF-MSCs treatment resulted in reducing the vascularization of the tumors, as shown by immunohistochemistry for mouse CD31 in T24M tumor sections, compared with GFP-AF-MSCs treated or nontreated animals (Supplementary Fig. S4A). In addition, we examined the infiltration of NK1.1 cells in the tumors after injection of IFNβ-AF-MSCs, AF-MSCs, or GFP-AF-MSCs. No significant differences between different groups were observed (Supplementary Fig. 4B).
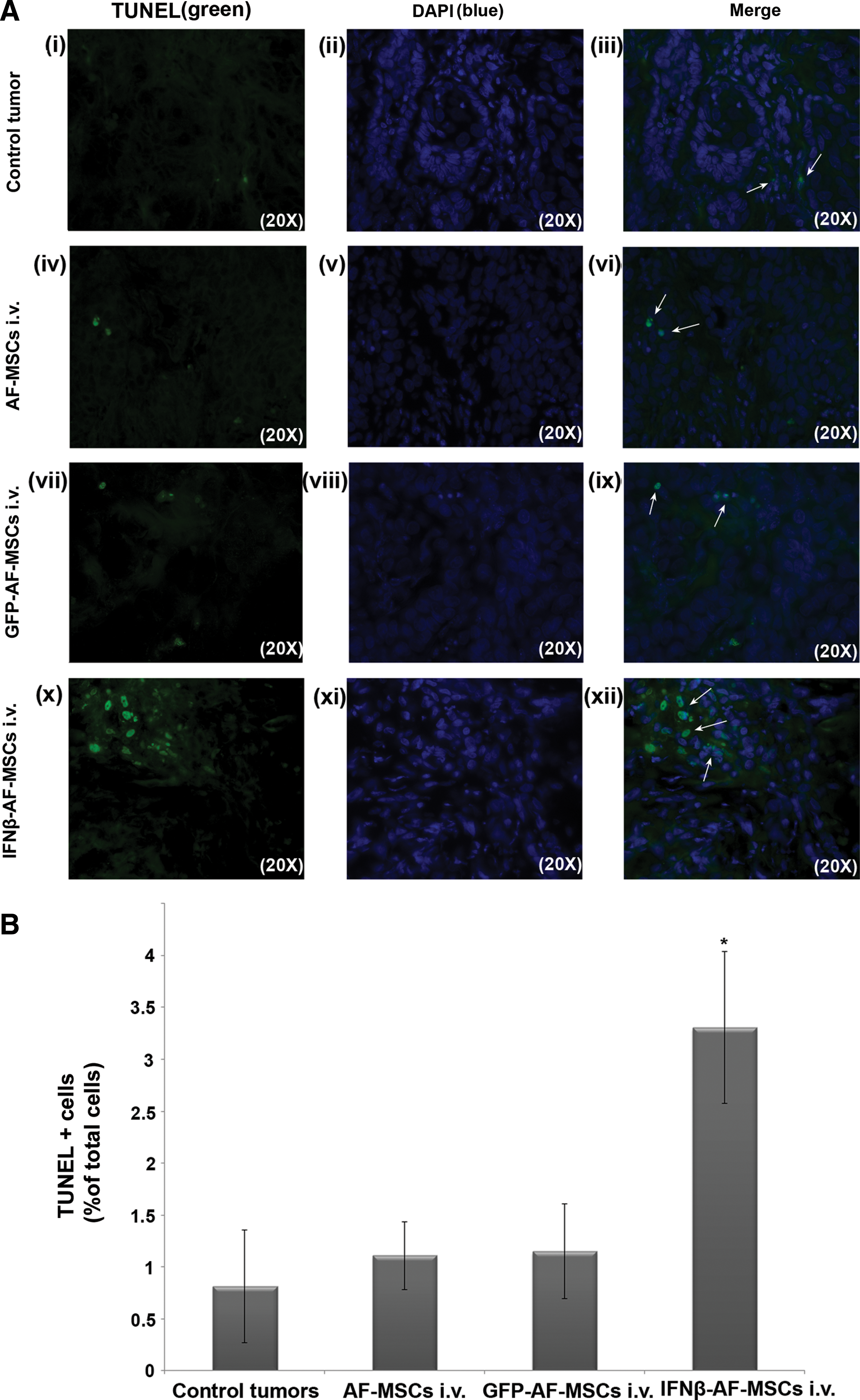
Determination of apoptosis in tumors by TUNEL assay.
Moreover, the health of the mice was monitored daily, and when necessary, mice were sacrificed according to humane end-point guidelines. The majority of the untreated and GFP-AF-MSCs treated animals exhibited macroscopic changes, such as ruffled fur, loss of weight, and pathological signs, such as splenomegaly and ulcerations. In IFNβ-AF-MSCs treated mice, the respective macroscopic symptoms were observed later and, in a lesser extent, whereas splenomegaly was absent.
Discussion
Cell culture and animal models are invaluable research tools that are utilized to increase our knowledge on the molecular mechanisms of neoplasia, to identify and characterize diagnostic and prognostic markers, and to evaluate potential drugs and therapeutic targets [4]. Here, we employed the T24M model for bladder cancer [2,5], to evaluate for the first time the effectiveness of novel stem cell therapies for solid tumors, based on the use of AF-MSCs, as valuable delivery vehicles for established anti-tumor agents. The proof of principle and the efficiency of this approach, to act locally at the tumor microenvironment and inhibit malignant cell growth in vivo, were shown in other types of cancers using MSCs derived from adult sources, such as BM [8,18,50,51]. Specifically, homing of MSCs after systemic or local administration has been studied in a variety of tumor animal models, such as lung metastasis [8,52], melanoma [27], brain glioma [17,19] models, and others. Collectively, these studies supported the notion that exogenously administrated BM-MSCs preferentially engrafted at the tumor site by contributing to the population of the stromal fibroblasts, thus forming the tumor's microenvironment [28,29]. This specific tropism may allow the development of efficient and rational therapeutic approaches based on the local production of biological agents by gene-manipulated MSCs. Along these lines, there is a general consensus that MSCs have a tremendous potential in cancer therapeutics [18,19,29,36,53].
In such cases, large numbers of MSCs are required. However, the frequency and expansion capacity of adult MSCs, such as those derived from BM, may decrease with age. Thus, recent studies were focused on fetal sources of MSCs with high proliferative activity, such as the AF [30 –35]. Given these unique characteristics, AF-MSCs may be considered as a powerful tool for gene therapy and tissue repair in clinical approaches. Further, AF-MSCs exhibited a chromosomal stability with no karyotypic abnormalities, even in high passages, with long telomeres and no tumorogenic effect in vivo [30,34,35,54].
In this study, we showed for the first time that both AF-MSCs and IFNβ-AF-MSCs efficiently migrated toward T24M cells or T24M-CM in vitro. We further analyzed by cytokine array the molecular mediators expressed by T24M cells, such as VEGF, IL-8, TGF-β1, and others, that could trigger the migration of AF-MSCs toward cancer cells. Additionally, we performed functional assays demonstrating that IL-8 and VEGF molecules play a chemoattractant role and promote the migration of AF-MSCs toward T24M cells or T24M-CM in vitro.
More importantly, we used GFP transduced AF-MSCs in a mouse bladder tumor model to evaluate their in vivo migratory properties. The localization of AF-MSCs at the tumor area suggested that these cells, similar to BM-MSCs [27 –29], can reach the extravascular space and contribute to the development of tumor connective stroma. However, the migratory properties of AF-MSCs in vivo need to be further evaluated for the possible signaling mechanisms involved.
Human IFNβ has been thoroughly studied in cancer cell lines and animal models and is found to exhibit anti-tumor effects [17,18,55]. IFNβ is a factor with known immuno-enhancing activities that plays an important function in protecting the host from viral infections [56]. Additionally, it has been shown that this cytokine has anti-tumor activity, thus showing potential clinical benefits in the treatment of certain types of cancer [11]. High concentrations of IFNβ are required to obtain therapeutic responses in patients with cancer. Nevertheless, the therapeutic use of IFNβ is limited because of its high cytotoxicity [11,37,57]. Therefore, IFNβ therapy should target the tumor tissue to avoid undesired side effects to the neighboring tissues. For this reason, we developed an efficient lentiviral system for generating AF-MSCs stably expressing IFNβ, and we initially verified their inhibitory effects on the proliferation of T24M cells, in vitro. We further analyzed the clinical efficacy of recombinant AF-MSCs in vivo by introducing them into mice modeling the evolution of aggressive bladder tumor. These data showed that i.v. injected IFNβ-AF-MSCs were distributed through the circulation to the tumor site, where they engrafted and inhibited malignant cell growth through a paracrine mechanism of locally produced IFNβ. Moreover, systemically administered IFNβ-AF-MSCs not only reduced the tumor volume, but also prolonged the survival of the T24M tumor bearing animals. Further, absence of significant increase in the infiltration of NK1.1 cells in the tumors treated with IFNβ-AF-MSCs suggested that the anti-tumor effect might be related to a potential decrease of angiogenesis and/or induction of tumor cell apoptosis [27].
In conclusion, the current study demonstrated, for the first time, the therapeutic efficacy of MSCs derived from AF against bladder tumor xenograft in vivo and in vitro. AF-MSCs can be utilized as delivery vehicles for anti-cancer agents, such as IFNβ, at the tumor site. These data showed that local IFNβ production, by the engrafted AF-MSCs in the tumor area, could efficiently inhibit malignant cell growth, without increasing the systemic levels of IFNβ in serum. However, recent studies from our group were also focused on the identification of newly discovered secreted factors with tumor modulating activity, such as secreted protein acidic and rich in cysteine (SPARC) [5]. Potential novel therapeutic targets for bladder cancer were identified by comparing the secretome of lineage-related bladder cancer cell lines differing in metastatic potential such the T24 and T24M cells [5]. Based on this analysis, a number of novel factors can be further selected as targets for MSC therapy.
Collectively, these data support that AF-MSCs have a great potential as vehicles for anti-cancer agents at the tumor site due to their apparent homing specificity, the high proliferation rate, and the lack of ethical concerns associated with human embryo research [30 –33,35]. Their evaluation in the treatment of bladder cancer represents an additional novelty of our study. Our current studies include further evaluation of the use of AF-MSCs in the treatment of metastatic bladder cancer using respective models and various alternative therapeutic schemes, including assessment of the stem cell dose range.
Footnotes
Acknowledgments
This research was supported by Grant PENED No. 03ED 652 from the Greek Secretariat of Research and Technology and the European Union to N.P.A. Partial support was provided by Iaso Hospital, Athens, Greece. The authors would like to thank professor Luigi Naldini for the generous donation of the pCCLsin.PPT.hPGK.GFP plasmids, Drs. G. Vassilopoulos and E. Siapati for kindly providing the NOD/SCID mice, and professor Thalia Papayannopoulou for useful and productive scientific advice and guidance.
Author Disclosure Statement
The authors indicate that no potential conflicts of interest exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.