
Abstract
Microarray analyses of transcriptomes have been used to characterize mesenchymal stem cells (MSCs) of various origins. MicroRNAs (miRNAs) are short, nonprotein-coding RNAs involved in post-transcriptional gene inhibition in a variety of tissues, including cancer cells and MSCs. This study has integrated the use of miRNA and mRNA expression profiles to analyze human MSCs derived from Wharton's jelly (WJ) of the umbilical cord, milk teeth (MT), and adult wisdom teeth (AT). Because both miRNA and mRNA expression in MT and AT MSCs were so similar, they were combined together as tooth MSCs for comparison with WJ MSCs. Twenty-five genes that were up-regulated in tooth MSCs and 41 genes that were up-regulated in WJ MSCs were identified by cross-correlating miRNA and mRNA profiles. Functional network analysis show that tooth MSCs signature genes, represented by SATB2 and TNFRSF11B, are involved in ossification, bone development, and actin cytoskeleton organization. In addition, 2 upregulated genes of tooth MSCs—NEDD4 and EMP1—have been shown to be involved in neuroectodermal differentiation. The signature genes of WJ MSCs, represented by KAL1 and PAPPA, are involved in tissue development, regulation of cell differentiation, and bone morphogenetic protein signaling pathways. In conclusion, the combined interrogation of miRNA and mRNA expression profiles in this study proved useful in extracting reliable results from a genome-wide comparison of multiple types of MSCs. Subsequent functional network analysis provided further functional insights about these MSCs.
Introduction
Wharton's Jelly (WJ) is the connective tissue that surrounds the 2 umbilical arteries and 1 vein, making up the major mass of the human umbilical cord, which averages 40 g [8,9]. The abundant amount of WJ makes it an attractive source of MSCs [10,11]. Since the dental sources for MSCs include dental pulp, exfoliated deciduous teeth, periodontal ligaments, apical papilla, and dental follicles [12], 5 types of dental MSCs have been isolated and characterized. Dental pulp stem cells are considered a heterogeneous population of MSCs, because dental pulp contains both mesenchymal and ectodermic components [13]. Therefore, tooth MSCs have the potential for a wide array of clinical applications, including a cell-based therapy for Parkinson's disease [7].
Microarray analyses of transcriptomes have been used to characterize MSCs of various origins [11,14]. MicroRNAs (miRNAs) are short, nonprotein-coding RNAs involved in post-transcriptional gene inhibition in a diverse variety of tissues [15], including cancer cells [16 –18] and MSCs [19 –23]. In this study, we integrated miRNA and mRNA expression profiles to analyze human MSCs derived from WJ of the umbilical cord, milk teeth (MT), and adult wisdom teeth (AT). Because each miRNA often exerts mild regulation on hundreds of gene targets and each gene is regulated by multiple miRNAs [24], our rationale in this study was that the expression of the genes cotargeted by multiple miRNAs would be more inhibited.
Materials and Methods
Approval of the study
This study was approved by the institute review board of Chang Gung Memorial Hospital (IRB#92-176). MSCs derived from WJ of the umbilical cord (WJ, n=5), MT (n=5), and AT (n=5) were provided by Taiwan BIONET Corp. (
Isolation and culture of umbilical cord derived MSCs
The umbilical cords were first disinfected with 75% ethanol, and then transferred to phosphate-buffered saline (PBS), where the remaining blood was washed away. After removal of the blood vessels, the tissue was sliced into very small pieces and treated with 0.05% trypsin (Invitrogen) and 300 U of collagenase (Stem Cell Technologies) in alpha-minimum essential medium (MEM) (Invitrogen) for 1 h at 37°C. The cell suspension was filtered through a 75-μm mesh and centrifuged at 400 g. The cells were re-suspended and cultured in alpha-MEM supplemented with 10% fetal bovine serum (FBS) (Hyclone) in a 37°C incubator with 5% CO2 over night. After the non-adherent cells were removed, culture medium was changed every 3–4 days. Subculture was performed at a plating density of 3–6×103 cells/cm2.
Isolation and culture of dental pulp stem cells
Teeth were first disinfected with 75% ethanol and then washed with PBS. Wisdom teeth were cracked open, and pieces were moved to a dish of alpha-MEM. MT were disinfected and placed directly into dishes of alpha-minimum essential medium (MEM) to keep the tissue wet. The pulp tissue was extracted from the tooth by a barbed broach and cut into small pieces with a surgical knife when necessary. The tissue was collected by centrifugation, washed 3 times with culture medium, and cultured in alpha-MEM supplemented with 10% FBS (Hyclone) in a 37°C incubator with 5% CO2. Tissue pieces were removed once cell colonies appeared. Subculture was performed to achieve the cell number needed for the experiments and performed at a plating density of 3–6×103 cells/cm2. The culture medium was changed every 3 days.
Characterization of MSCs
Human WJ MSCs and dental pulp stem cells were characterized by flow cytometry using fluorescein isothiocyanate-conjugated or phythoerythrin-conjugated antibodies (BD Biosciences/Southern Biotech). For analysis, the cells were detached with trypsin/ethylenediaminetetraacetic acid (EDTA) in PBS, washed with PBS, and incubated for 30 min with its respective antibody. In each case, 1×104 events were acquired and analyzed using the Cell Quest software (Becton-Dickinson).
Calculation of cell doubling times
To assess the population doubling time, cells were seeded at 2.5×105 cells per culture dish and counted at 70%–80% of confluency. Population doubling time was calculated using the formula dt=t×ln2/ln(Ct/C0), in which dt=doubling time, t=time between cell count Ct and C0 (in hours), C0=initial cell count, Ct=cell count after time t.
In vitro differentiation of MSCs
WJ MSCs, MT MSCs and AT MSCs were seeded at a density of 1×105 per well (6-well plate) overnight. Osteogenic differentiation was induced using the STEMPRO® Osteogenesis Differentiation Kit (Invitrogen). Culture medium was changed twice a week over a period of 14–21 days. To visualize osteogenic differentiation, cells were stained for alkaline phosphatase using 5-bromo-4-chloro-3-indolyl-phosphate (BCIP)/nitroblue tetrazolium (NBT) substrate (Sigma). Adipogenic differentiation was induced using STEMPRO® Adipogenesis differentiation Kit (Invitrogen). Induction medium was changed every 3–4 days, and cells were cultured for at least 21 days. To demonstrate the presence of adipocyte-like cells, cells were fixed with 10% neutral buffered 3.7% formalin for 10 min, and the cytoplasmic inclusions of neutral lipids were stained with oil-red O (3 mg oil red/mL 60% isopropanol) for 15 min. For chondrogenic differentiation, micromass culture was used by seeding 10-μL droplets of cell suspension (2×105 cells/droplet) at the center of the culture dish. The cell droplets were incubated in a 37°C incubator with 5% CO2 for 2 h to allow cell adherence on a small surface area. Chondrocyte induction medium, STEMPRO® Chondrogenesis Differentiation Kit (Invitrogen) was then added. Induction medium was changed every 3–4 days over a period of 14 days. Cell pellets were stained with toluidine blue.
RNA preparation and microarray analysis
At 90% confluence, MSCs derived from WJ, MT, and AT at passages 2–3 were briefly rinsed with ice-cold PBS and lysed in TRIZOL reagent (Invitrogen,
Real-time quantitative polymerase chain reaction analysis
Real-time quantitative polymerase chain reaction (RT-Q-PCR) was carried out with the ABI 7900 HT RT-PCR system (Applied Biosystems) according to the manufacturer's protocol. miRNA quantification was performed using the SYBRgreen reagent (Applied Biosystems) as previously reported [17]. The conditions for Q-PCR were: 95°C for 10 min, then 40 cycles of 95°C for 15 s, 63°C for 32 s, followed by a final dissociation stage. Endpoint reaction products were analyzed on a 10% polyacrylamide gel stained with ethidium bromide to discriminate the correct amplification product (57–60 bp) from the potential primer dimers (<44 bp).
Data analysis
Procedures for the integrated analyses of miRNA and mRNA profiles have been previously reported [17]. The 3 steps were: (1) miRNA target prediction using TargetScan algorithm, (2) data compilation to identify target genes coregulated by multiple miRNAs, and (3) pathway enrichment analysis of coregulated targeted genes [15]. Statistical analyses included the Mann-Whitney test, principal component analysis (PCA), Pearson's correlation analysis, and hierarchical clustering, that were all performed using the Partek Genomics Suite (version 6.3). Pathway enrichment analysis was performed using the MetaCore database and algorithm (GeneGO). Procedures of functional network analyses based on the MetaCore database were identical to those previously reported [14,26 –28].
Results
Characteristics of MSCs from different origins
The MSCs isolated from human umbilical cord, dental pulp of milk and wisdom teeth all fulfilled the criteria of MSCs set by the International Society of Cellular Therapy [29]. Using flow cytometry, cell surface markers analysis of the MSCs from these 3 origins showed they were positive for CD73 (NT5E), CD90 (Thy-1) and CD105 (endoglin), CD13 (alanine aminopeptidase), CD29 (integrin β1), CD44 (HA receptor), HLA-A, B, and C (class I). These cells were negative for CD45, CD34 (both hematopoietic markers), CD14 (co-receptor of TLR4 and MD-2) and HLA-DR (human leukocyte differentiation antigen class II), CD31 (endothelial cell marker), and CD133 (hematopoietic stem cell marker) (Fig. 1). Cells from both sources (WJ and teeth) initially grew slowly but increased their proliferation rates after the first subculture. At passages 2–3, the average doubling times for WJ MSCs and tooth MSCs were not significantly different, 35.8 h and 42.3 h, respectively. In vitro differentiation analysis of these spindle-shaped MSCs confirmed that all the isolated MSCs from different origins exhibited the capacity to differentiate into various cell types, such as osteoblasts, chondrocytes, and adipocytes (Fig. 2). The capacity to differentiate, however, seemed to vary between the 3 different types of MSCs. Osteogenic differentiation was equally successful. Adipogenic and chondrogenic differentiation was more pronounced in WJ MSCs and MT MSCs than the differentiation seen in AT MSCs.
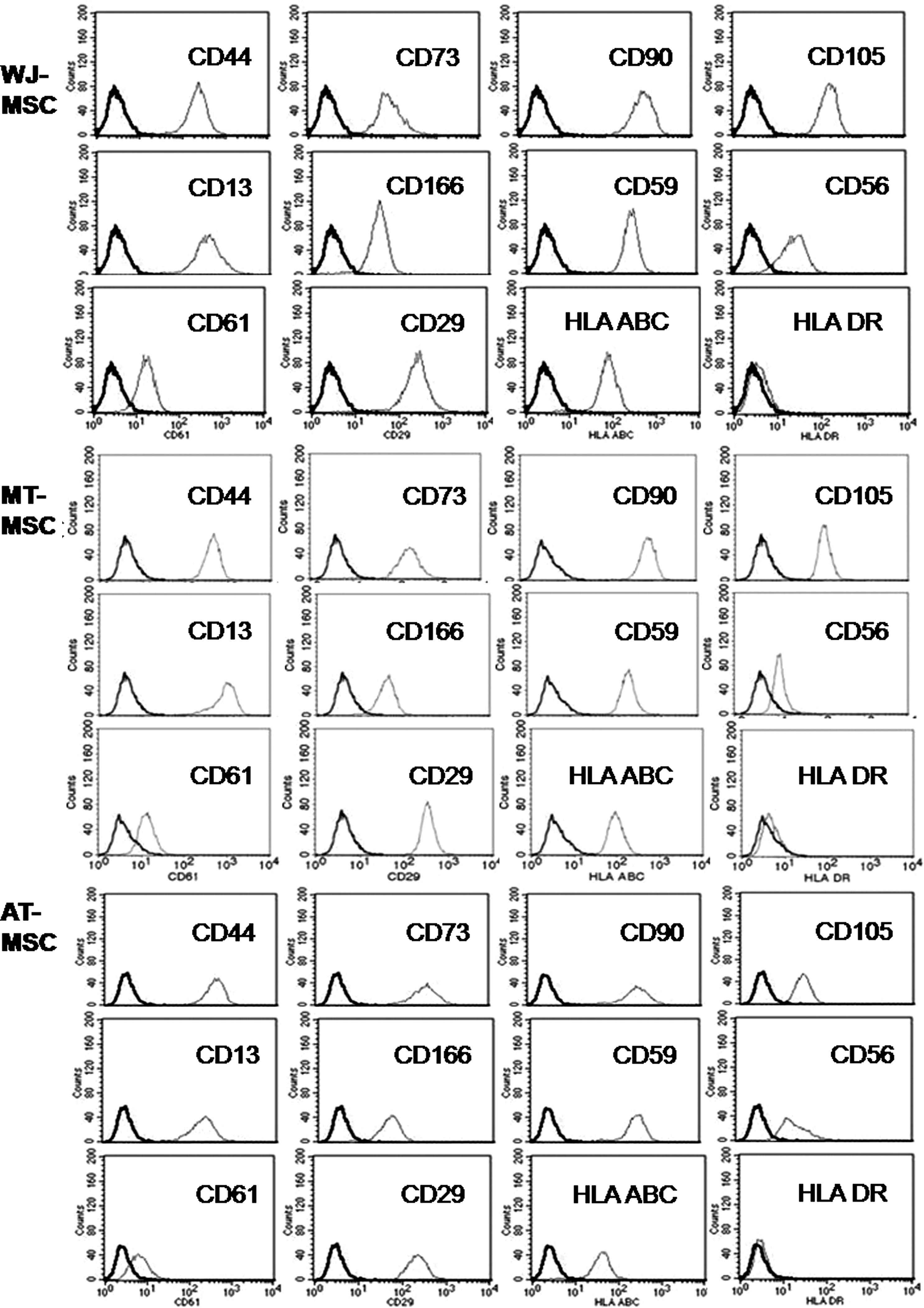
Cell surface markers of 3 types of mesenchymal stem cells (MSCs). WJ stands for Wharton's jelly of the umbilical cord, MT for milk teeth, and AT for adult wisdom teeth. In each panel, the first (left side, dark lined) peak depicts the cell population stained with an appropriate control isotypical antibody.
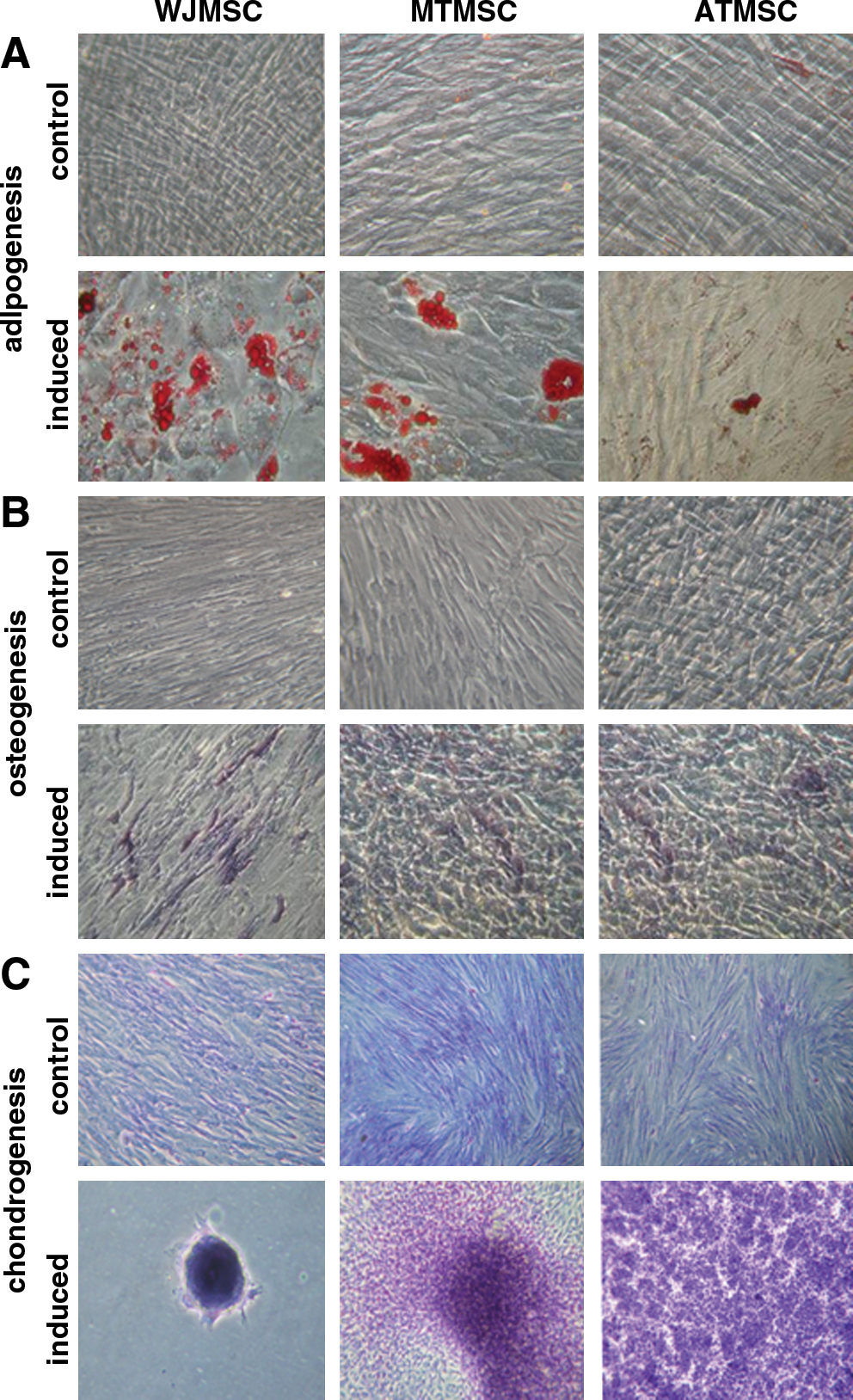
Differentiation potentials of 3 types of MSCs.
Transcriptomic similarity between milk tooth MSCs and adult tooth MSCs
PCA of mRNA transcriptomes indicated 5 AT MSCs and 5 MT MSCs clustered together, while the 5 WJ MSCs were in another cluster (Fig. 3A). Similar trends of miRNA profiles in these 3 MSCs were identified by both PCA (Fig. 3B) and hierarchical clustering analysis (Fig. 3C). Therefore, in the following analyses, we calculated AT MSCs and MT MSCs together as the tooth MSCs group, while WJ MSCs were calculated separately.
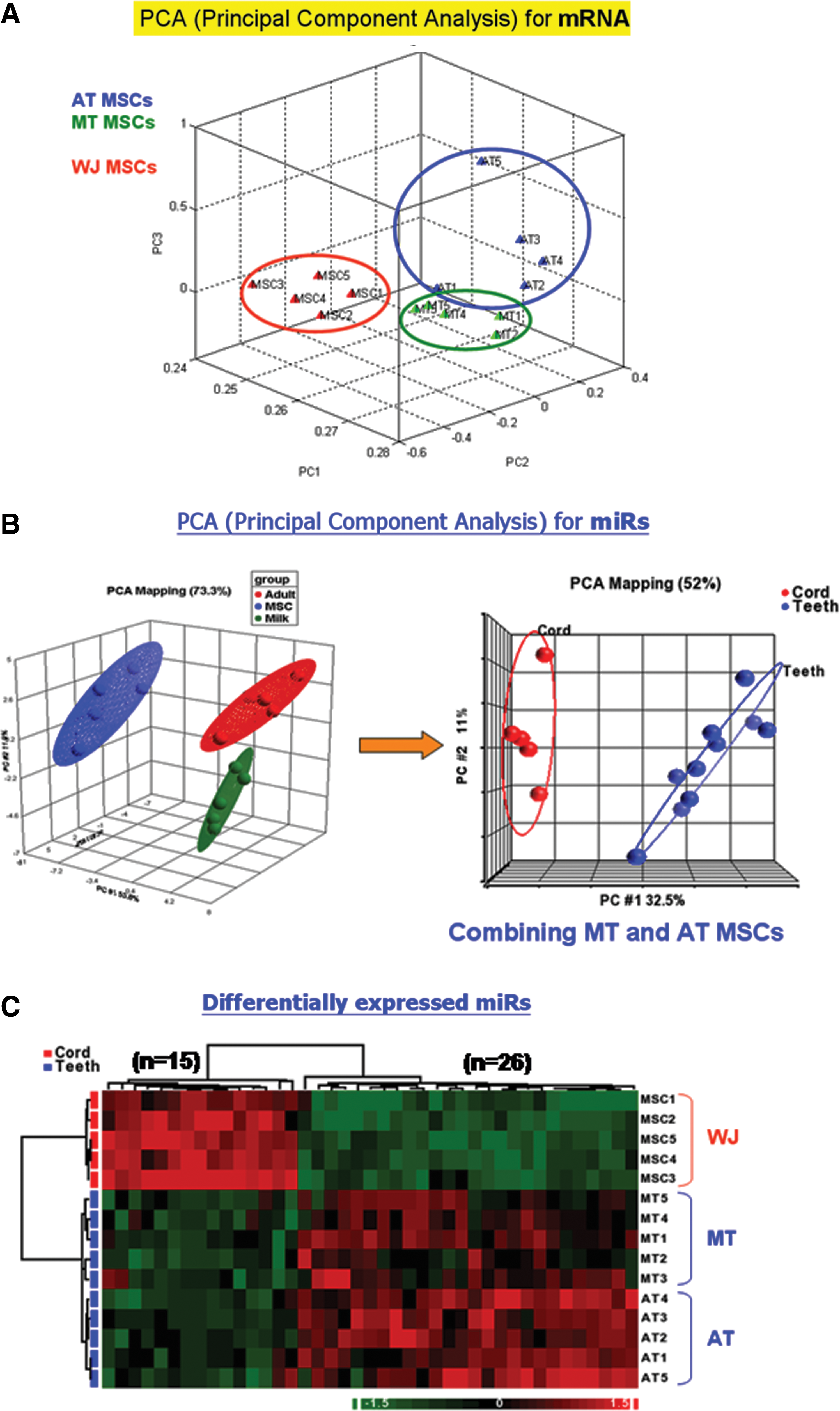
Analysis of expression profiles of 3 types of MSCs.
Identification of target genes coregulated by multiple miRNAs and validating these genes with the results of mRNA microarray analysis
Fifteen miRNAs were down-modulated in tooth MSCs and 26 miRNAs were down-modulated in WJ MSCs (Fig. 3C and Supplementary Tables S1 and S2; Supplementary Data are available online at
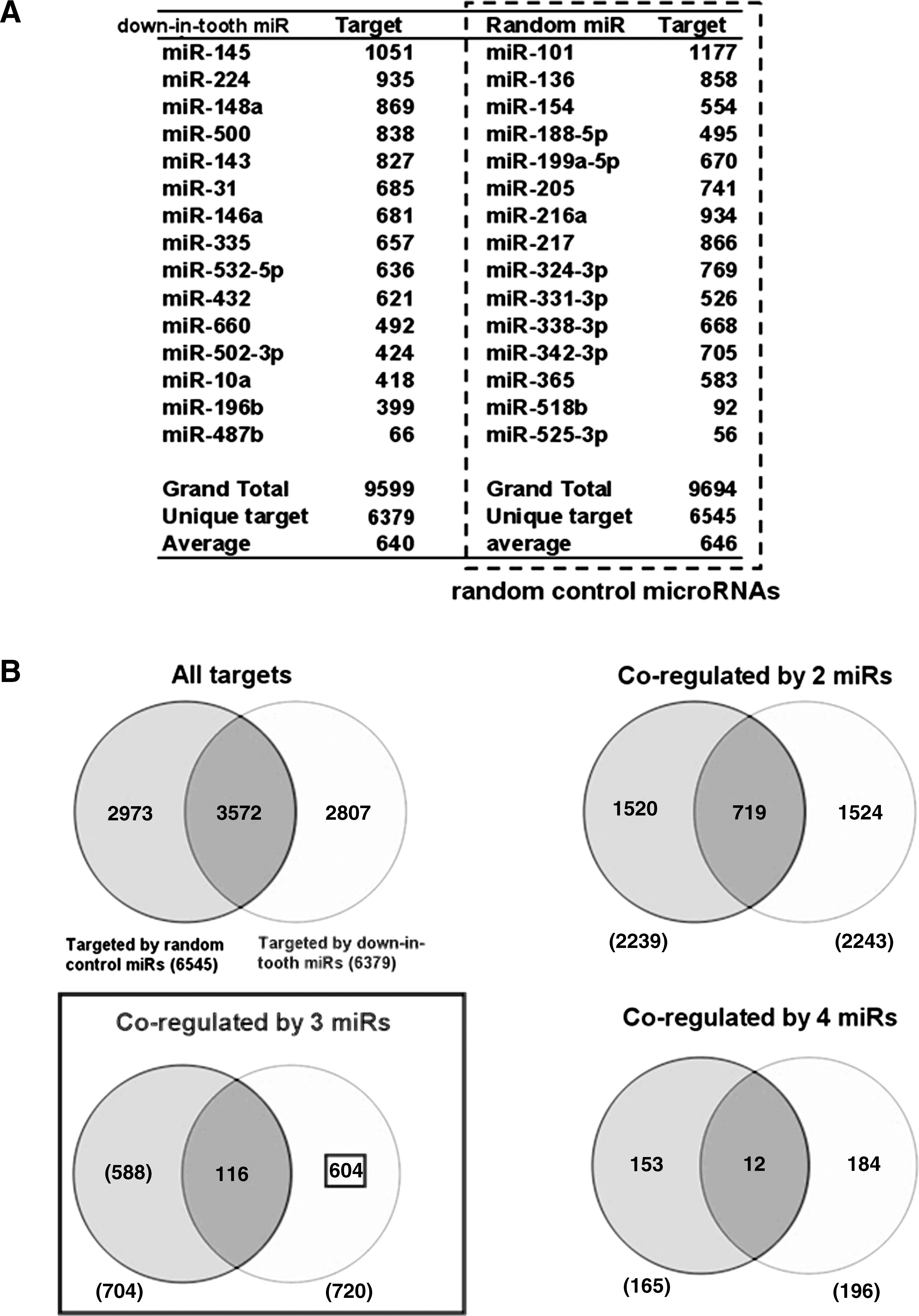
Selection of a control set of miRNAs to increase the specificity of targeted gene identification for the 15 down-in-tooth MSC miRNAs.
The numbers of total targets predicted for the 15 down-in-tooth MSC miRNAs and the 15 random control miRNAs were 6,379 and 6,545, respectively (Fig. 4B). As previously described in detail [17], we selected the dataset of the 3 coregulating miRNAs in which the fraction of overlapping targets (116 genes) decreased to <20% of the predicted targets by both down-in-tooth MSC miRNAs (116/720) and random control miRNAs (116/704) (Fig. 4B). The list of targets coregulated by 4 miRNAs was not selected because such high stringency resulted in far fewer genes. The 588 targeted genes (Fig. 4B) were further validated by the results of Affymetrix microarrays, finally resulting in 25 genes that were up-regulated in tooth MSCs (Supplementary Table S3). Using identical analytical procedures on the 26 down-in-WJ MSC miRNAs in combination with mRNA results of Affymetrix microarray, we identified a total of 41 genes that were up-regulated in WJ MSCs (Supplementary Table S4).
Functional network analyses of differentially expressed genes
To annotate gene functions at the network level, the genes that were up-regulated in tooth MSCs (Supplementary Table S3) were extrapolated using an intersection algorithm of MetaCore (Fig. 5). Both P values for the resulting 2 networks were 2.88E-14, indicating that the probabilities of assembly from random sets of gene nodes were very low [30]. The top 12 biological processes associated with the genes of these networks are summarized in Supplementary Tables S5 and S6. The 11 seed genes that were up-regulated in tooth MSCs (Table 1) were involved in anatomical structure development, ossification, bone development, regulation of apoptosis, and actin cytoskeleton organization. These biological processes were compatible with the functions of tooth MSCs.
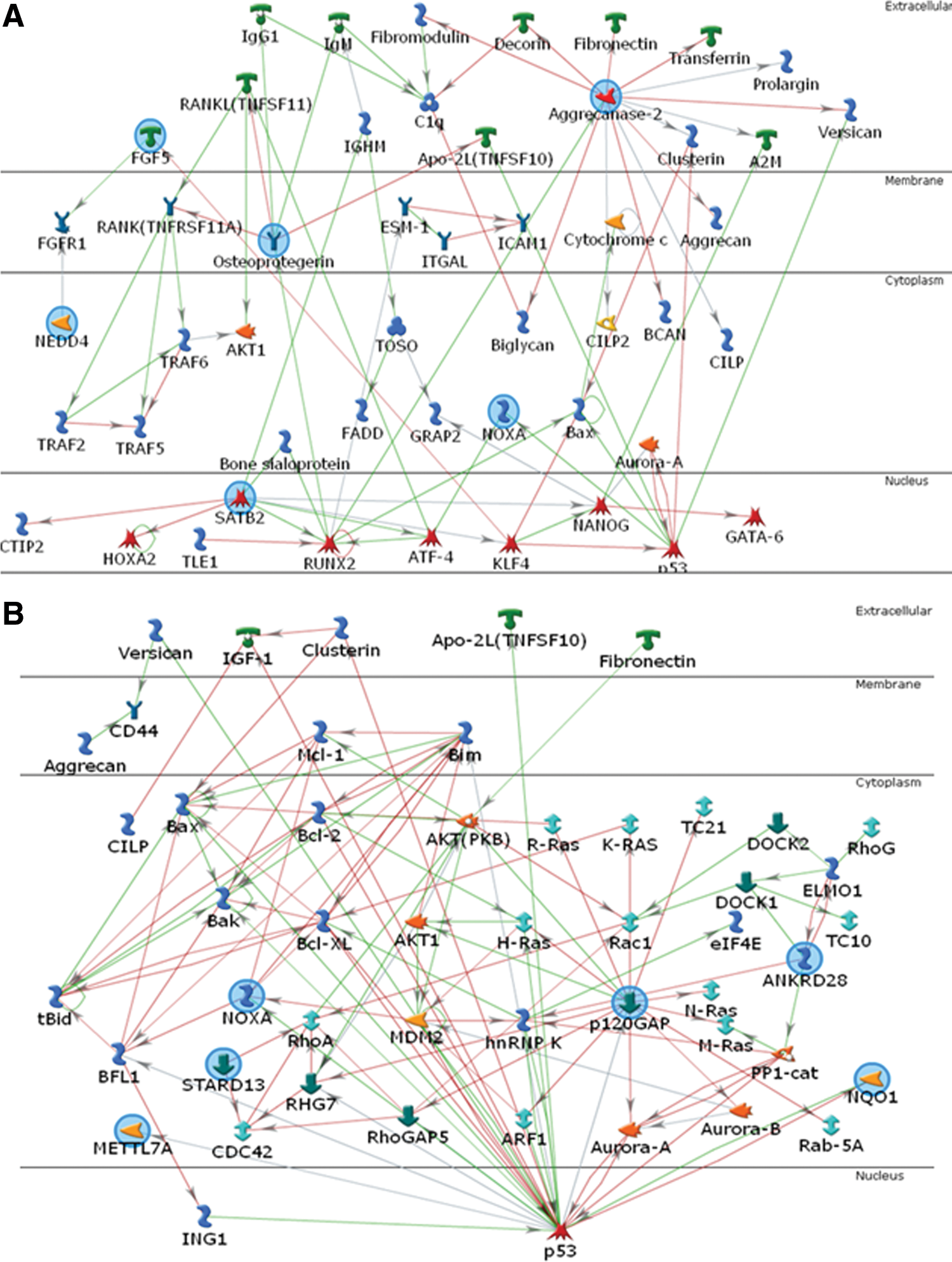
Functional network analyses of the genes up-regulated in tooth MSCs. The genes (blue circles) that were up-regulated in tooth MSCs were extrapolated into the network using the MetaCore program. The related biological processes in the network
Similarly, the genes that were up-regulated in WJ MSCs (Supplementary Table S4) were extrapolated using an intersection algorithm of MetaCore (Fig. 6). The P values for the resulting 2 networks were 1.78E-17 (Fig. 6A) and 5.56E-15 (Fig. 6B), indicating that the probabilities of assembly from random sets of gene nodes were very low [30]. The top 12 biological processes associated with genes in these networks are summarized in Supplementary Tables S7 and S8. The 13 seed genes that were up-regulated in WJ MSCs (Table 2) were involved in tissue development, regulation of cell differentiation, bone morphogenetic protein (BMP) signaling pathways, multicellular organismal process, response to hormone stimulus, and response to wounding. These biological processes are consistent with the versatile functions of WJ MSCs.
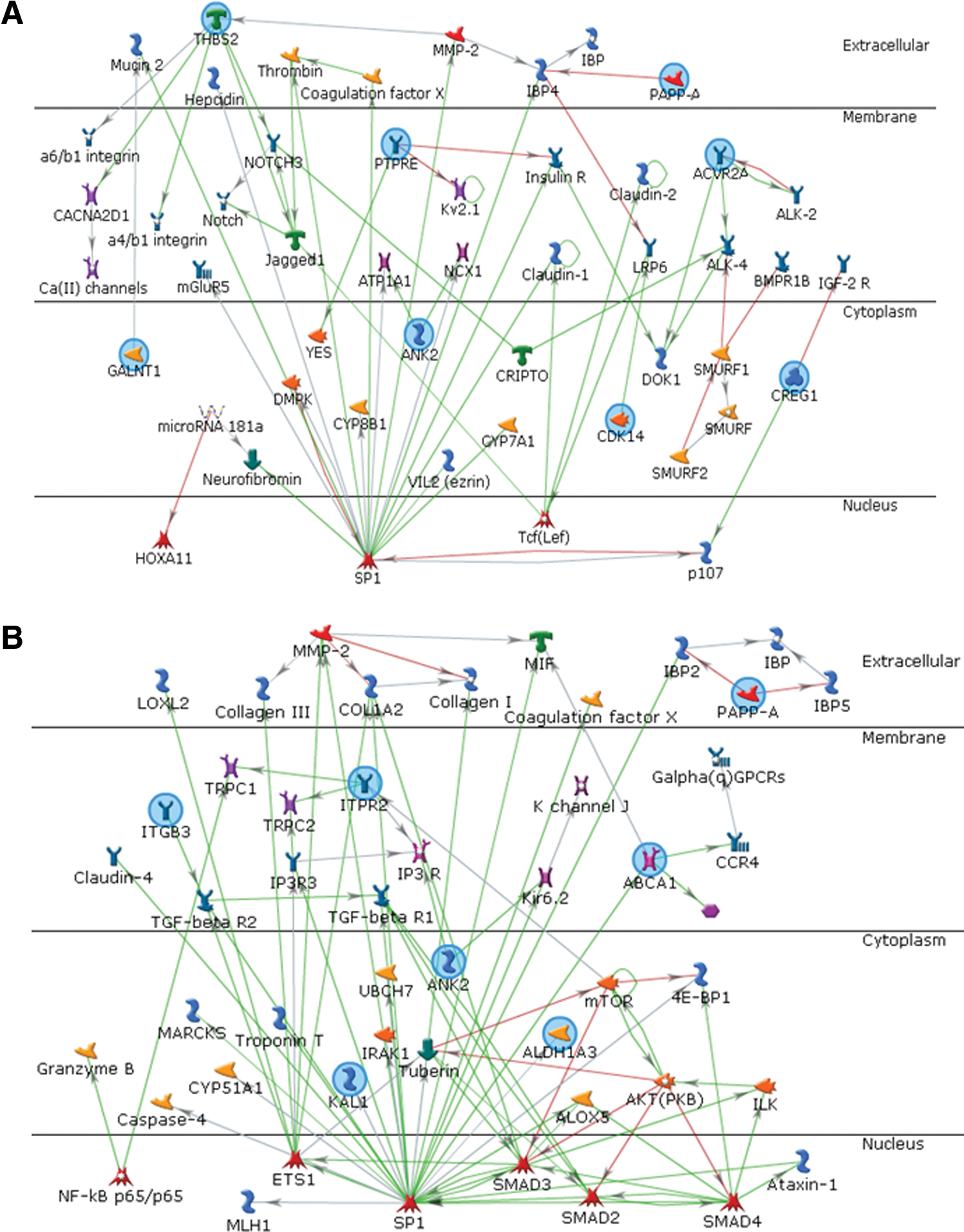
Functional network analyses of the genes up-regulated in WJ MSCs. The genes (blue circles) that were up-regulated in WJ MSCs were extrapolated into the network using the MetaCore program. The related biological processes in the network
Discussion
miRNAs are negative regulators that suppress protein-coding genes by recognizing and attaching to a specific sequence on the transcript of their cognate targets. Recent studies clearly demonstrated that the majority (>80%) of miRNA affect their cognate targets at the transcript levels by decreasing the stability of mRNA [31]. Although computational target prediction has been widely used to identify the potential targets for miRNA, the false positive rate in target prediction is relatively high. Genome-wide transcriptomics profiling using microarrays offers a convenient tool to resolve this problem as miRNA targets can be easily and accurately recognized using computational target prediction on the inverse expression profile of miRNAs and mRNAs. Such approach has been successfully used to study the pathogenesis of bladder cancer [32] and nasopharyngeal carcinoma [17].
Microarray analysis for gene expression has been used to characterize intrinsic properties of various MSCs [11,14,33]. miRNAs have recently been shown to play critical roles in dictating the differentiation of MSCs [19,34 –36]. However, only few studies systematically investigated the regulation of miRNAs on mRNA expression profiles in MSCs [22,37,38]. Both WJ MSCs [39 –43] and tooth MSCs [12,44,45] were shown to have profound potential for tissue engineering and cell-based therapy, but the roles of miRNAs have not been reported in these 2 types of MSCs. To our knowledge, this study is the first one to probe the roles of miRNAs in WJ MSCs or tooth MSCs by cross-correlating miRNA and mRNA expression profiles in these 2 types of MSCs. Cross-correlation of these 2 profiles led to identification of genes highly representative of the functions of each type of MSCs.
Dental pulp stem cells contain both mesenchymal and ectodermal components [13], suggesting that the heterogeneity of tooth MSCs may account for their neuroectodermal differentiation capacity. For instance, milk tooth stem cells were recently shown to differentiate into dopaminergic neuron-like cells [46]. In our study, the 2 genes that were up-regulated in tooth MSCs are involved in neuroectodermal differentiation. NEDD4 (Table 1) contains binding sites for 5 miRNAs down-modulated in teeth MSCs, including miR-10a, miR-224, miR-31, miR-660, and miR-143 (Supplementary Table S1). NEDD4 was shown to form a signaling complex with RAP2A and TNIK and regulate neuronal dendrite extension and arborization during development [47]. Similarly, EMP1 (Supplementary Table S3) contains putative binding sites for 4 miRNAs down-modulated in tooth MSCs, including miR-148, miR-31, miR-143, and miR-500 (Supplementary Table S1). During neurogenesis in the prenatal mouse brain, EMP1 was shown to be highly expressed in immature neurons and is assumed to be important in neuroectodermal differentiation [48].
In the list of signature genes of tooth MSCs (Table 1), SATB2 and TNFRSF11B were the top-ranked, and both have been linked to ossification and bone metabolism. SATB2 encodes a nuclear matrix protein, that represses the expression of HOXA1 and activates osteoblast-specific genes [49]. SATB2 was recently shown to synergize with RUNX2 to promote bone formation, acting as a molecular modulator in skeletal development and osteoblast differentiation [50]. Mice that have had Satb2 homozygously knocked out, exhibit craniofacial abnormalities and defects in osteoblast differentiation [49]. TNFRSF11B (osteoprotegerin) is a key regulator in the RANK/RANKL system that regulates osteoclastogenesis and bone metabolism. TNFRSF11B acts as a decoy receptor by binding to RANKL and preventing RANK signaling, thereby blocking osteoclast activation [51]. In addition, TNFRSF11B has been proposed as a link that accounts for the comorbidity of bone and vascular diseases in patients with chronic renal failure [52].
KAL1 and PAPPA were the top ranked in the list of WJ MSCs genes (Table 2), and both are known for playing an important role in maintaining the “stemness” of these cells. KAL1 was one of the genes up-regulated in cells that were immortalized by increasing telomerase expression [53]. KAL1 was shown to interact with EGFR, thereby impairing the migration of oligodendrocyte precursors, holding these neuron progenitor cells in a less differentiated status [54]. PAPPA is a metalloproteinase that controls the tissue availability of insulin-like growth factor. BMP2 was shown to up-regulate PAPPA expression, and both were involved in the matrix mineralization of MSCs [55]. The immune modulating effects of PAPPA revealed in knockout mice [56] may provide an insight to understanding the mechanism of how WJ MSCs prevent graft-versus-host disease [41]. The role of PAPPA in regulating the differentiation of myoblasts [57] may account for the long-term survival of WJ MSCs in dermal tissue engineering [42].
MSCs have been shown to exert therapeutic effects by secreting trophic factors [58,59]. Since cells may use RNAs, including miRNAs, to mediate intercellular communications [60,61], MSCs were recently shown to secrete microparticles that are enriched in pre-miRNAs [23]. This study is somewhat limited since miRNAs in the exosome of MSCs have not been analyzed. Future analyses of the miRNAs in the exosomes may help us predict the therapeutic effects of various MSCs on target tissues. Results of this study, however, provide groundwork for future research.
This study is further limited since miRNA targets were not validated at the protein level. Nevertheless, some of the miRNA-target gene pairs discovered by cross-correlation in our analysis have been experimentally validated using reporter assays and Western blot analyses by other researchers. For instance, SATB2 was shown to be directly regulated by miR-31 in cancer-associated fibroblasts [62] while GALNT1 was found to be targeted by miR-129-5p in bladder cancer [63]. Similarly, CCND2, DICER1, ITGB3, CDKAL1, and EDEM3 have been validated as the targets of several members of let-7 family miRNAs [64 –68].
In conclusion, the combined interrogation of miRNA and mRNA expression profiles in this study proved useful in extracting reliable results from the genome-wide comparison of multiple types of MSCs. Subsequent functional network analyses of these results provided further insights into the potential functions of miRNAs in regulating different types of MSCs, such as on bone metabolism and neuronal development by tooth MSCs and on immunomodulation by WJ MSCs.
Footnotes
Acknowledgment
The authors thank Bioinformatics Core Facility of Chang Gung University for the access of MetaCore database and algorithm, Shih-Chi Yeh (Chang Gung University) for technical assistance, and Dr. Shihyee Mimi Wang (White Memorial Medical Center, Los Angeles, CA) for English editing.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.