
Abstract
Adipose-derived stromal/mesenchymal stem cells (ASC) have gained interest as promising tools for delivering cancer therapy. Adipose tissue can be obtained readily in amounts sufficient for ASC isolation, which can be expanded rapidly, allowing its use at low passage numbers, and can be transduced by viral and nonviral means. Our goal was to examine the potential of ASC to deliver cytokine gene therapies melanoma differentiation associated gene-7 (MDA-7) or pigment epithelial-derived factor (PEDF) to cancer cells. These novel cytokines are a potent proapoptotic and an antiangiogenesis mediator, respectively, with potential as antitumor agents. Expression of cytokine therapies did not adversely affect ASC biology, and these cells were still able to differentiate and retain normal viability. The ASC cytokine therapies were efficient in reducing tumor cell growth in coculture and also in suppressing in vitro angiogenesis phenotypes. We also observed that ASC retained their innate ability to migrate toward tumor cells in coculture, and this ability could be blocked by inhibition of CXCR4 signaling. The ASC were found to be nontumorigenic in vitro using a soft agar assay, as well as in vivo, utilizing 2 prostate cancer xenograft models. The ASC-MDA7 only reduced tumor growth in the TRAMP-C2-Ras (TC2Ras) prostate cancer model. The ASC-PEDF, however, reduced growth in both the TC2Ras and the PC3 highly aggressive prostate cancer models, and it was able to completely prevent prostate tumor establishment in vivo. In conclusion, ASC expressing PEDF and MDA7 could effectively reduce prostate tumor growth in vivo, suggesting ASC-cytokine therapies might have translational applications, especially the PEDF modality.
Introduction
A
In the present study, our goal was to examine the potential of ASC to deliver cytokine gene therapies melanoma differentiation associated gene-7 (MDA-7) or pigment epithelial-derived factor (PEDF) to prostate and breast tumor cells. These molecules are potent proapoptotic and antiangiogenesis mediators with high promise as therapies for reducing tumor growth. MDA-7, also known as interleukin-24, is a tumor suppressor gene associated with inducing differentiation and apoptosis and reducing cellular proliferation. The MDA-7 expression is substantially reduced in malignant breast tissue, and low transcript levels are significantly associated with unfavorable pathological parameters and poor prognosis. Multiple studies have demonstrated that re-expression of MDA-7 in a wide variety of tumor cells (including prostate and breast cancer) causes their growth arrest and rapid cell death with no toxicity to normal cells [12,13]. Phase I clinical trial data have shown that a recombinant adenovirus (Ad) expressing MDA-7 was safe and had measurable tumoricidal effects in over 40% of patients, strongly arguing that delivery of this cytokine could have significant therapeutic value (reviewed in Ref. [14]).
PEDF is the pigment epithelium-derived factor and is significantly downregulated in patients with prostate cancer [15]. PEDF is an endogenous secreted monomeric glycoprotein of 50 kD and one of the most potent inhibitors of angiogenesis known, thus capable of playing an essential role in preventing tumor progression. PEDF is a key inhibitor of stromal vasculature and epithelial tissue growth in mouse prostate and pancreas [16], and gene disruption results in abnormal development of these organs due to vascular defects. Interestingly, in the same report, prostate cancer xenografts treated with rPEDF had significantly reduced growth, therefore implicating PEDF in preventing prostate cancer progression. Other studies also showed that dramatic reductions in PEDF levels correlate with enhanced tumor growth and angiogenesis during progression of breast, prostate, pancreatic, hepatocellular carcinomas, glioblastoma, and Wilms' tumor. Ad.PEDF has showed promising antitumor efficacy in prostate, brain, and pancreatic tumors by stimulating differentiation to a less malignant phenotype; in tumor and endothelial cells by promoting apoptosis, preventing cell migration by downregulating matrix metalloproteinase-2 and−9, and by inhibiting neovessel sprouting through induction of the Fas/FasL death pathway in endothelial cells [17,18]. Clinically, PEDF has been tested in macular degeneration Phase I trials without toxicity, and with promising suggestion of antiangiogenic activity starting at moderate doses (108 pfu) [19]. We propose that a less immunogenic cell-based PEDF therapeutic could even surpass the Ad efficacy reported in the clinic, as a strong immunologic response is typically mounted against the virus after the first round of vector administration. Therefore, it is critical to develop new delivery vehicles for gene therapy that could allow for more sustained expression of cytokine gene therapies over time, thus helping increase antitumor efficacy.
In summary, we present the potential of novel cytokine gene therapies delivered by adipose-derived MSCs in several prostate and breast tumor cell lines and in vivo in 2 prostate tumor models. We propose that the ASC therapies examined are potent proapoptotic and antiangiogenesis mediators and, thus, hold high promise as antitumor therapies, and may become strong candidates for translational applications.
Materials and Methods
Cell culture, viral transductions, and transfections
Human prostate and breast cancer cell lines have been obtained from ATCC or as described [19]. Mesenchymal cells of human or mouse origin of low passage numbers (P1–4) were used in all experiments. Human ASC were isolated from lipoaspirate tissue donated by patients undergoing elective procedures with informed consent under a protocol reviewed and approved by the Institutional Review Board, Pennington Biomedical Research Center. Human mesenchymal cells (STEMPRO Stem Cell Kit) were obtained and cultured according to manufacturer's protocols (Invitrogen). Mouse MSCs were isolated and cultured as previously described [20]. Lentiviruses for expressing human therapeutic Lentiviral constructs cDNAs were constructed in the chimeric CMV-HIV 5′LTR and vector backbone plasmid (pCCL) backbone containing a cytomegalovirus (CMV) promoter in frame with an internal ribosome entry sequence (IRES) and the reporter gene for green fluorescent protein (GFP) (Lv. CMV.ires.GFP). Lentiviruses were produced and tittered as described [21]. In more detail, 293T cells were transfected with calcium chloride (0.6M CaCl2) and 3 plasmids containing sequences coding for the lentiviral vector genome (pccL-CMV-PEDF-ires-GFP or pccL-CMV-ires-GFP), viral packaging proteins (pdelta8.2R), or viral VSVG (pVSVG) envelope from the vesicular stomatits virus. The transient transfection procedure for viral production is performed to maintain safe handling of the virus and avoid generation of self-replicating particles. Lentiviruses are produced and released into the media supernatants, which were collected at 48, 72, and 96 h post-transfection, filtered using a 2 μm membrane, and stored at −80°C. For determining viral concentration (titer), 200 μL of either pure supernatant (1×), or dilutions of 1:10 or 1:100 were used to infect 293T cells (5×104) seeded in 24-well plates. Infection was performed in the presence of the transduction enhancing agent Polybrene at 8 μg/mL for cells assayed for percentage of cells expressing GFP by flow cytometry 48 h after infection. For infecting experimental cells, multiplicity of infection (MOI) of 1 lentiviruses was used to transduce ASC cells.
Cell viability, migration, colony formation, tube formation, differentiation, and soft agar assays
For cell viability, either a Cell Counting Kit 8 (CCK8) assay or a crystal violet assay was used. For CCK8, manufacturer's instructions were followed (Dojindo). For the crystal violet assay, monolayers were washed in 1× Dulbecco’s phosphate buffered saline (DPBS), fixed in 10% buffered formalin, and stained with 0.5% Crystal violet in water, washed 3×, the dye solubilized in 250 μL 33% glacial acetic acid and 1/30 dilution sample read at absorbance 550 using a plate reader (Biorad). The migration assay consisted of 24-well plate inserts with 8 μ pores in the membrane (BD Falcon). ASC were plated in the top chamber at a density of 2×104 cells in a volume of 500 μL of stem cell media (MesenPRO; Invitrogen) containing 0.5% fetal bovine serum (FBS). The bottom chamber was filled with 500 μL of RPMI1640 media containing 10% FBS (control) or same media plus 105 tumor cell lines (prostate: TRAMP-C2, RM1, PC3, CWR22Rv1, breast: MCF7, MB-MDA-361, MB-MDA-231). The ASC were allowed to migrate to the bottom of the transwell for 18–24 h. At the end of the assay, media were aspirated from the lower chamber, and migrating ASC cells at the bottom of the transwell insert were fixed in 10% Formalin in 1×DPBS for 20–30 min. The media was then aspirated from the top chamber, cells atop the membrane were removed with a cotton swab, and the membrane was excised from the chamber using a No. 11 scalpel blade and mounted on a slide under a coverslip using Vectashield Hardmount with DAPI (Vector Labs). Each membrane was quantified by capturing digital images of at least 5 independent fields under a 10×lens on a fluorescent microscope and determining the percentage total area covered by cells in the field using NIH ImageJ software. The 5 fields were combined to gain a value for the total membrane coverage. Each experiment was performed in triplicate, and the average of it is shown. For blocking the CXCR4 signaling pathway, we used istoype control or anti-CXCR4 antibody (BD Biosciences), and migration assays were performed as just described. For apoptosis assays, we used a luminescent CaspGLO assay (Promega) that enables sensitive detection of Caspase 3/7 expression in a 96-well format, following manufacturer's protocols.
For examining angiogenesis potential of human umbilical vein endothelial cells (HUVEC2; BD Biosciences) growing in culture conditioned media (CCM), an Endothelial Tube Formation Assay (CellBiolabs, Inc.) was used, according to the manufacturer's protocols. After staining of the tubes formed with the provided fluorescein dye, the extent of tube formation (as assessed by the average tube length) was quantified using NIH ImageJ software. For preparing CCM, we plated 5×105 ASC in a 6-well format in 2.5 mL/well MesenPRO media (Invitrogen), a media that has low FBS (2%). When ∼50%–70% confluent, we changed the media to fresh MesenPRO and collected the resulting CCM 24 h later. For colony formation assay, 250 cells were seeded in a 12-well format and allowed to form colonies over a period of 14 days, after which wells were fixed in 10% buffered formalin and stained using 0.5% crystal violet in water. Colonies were counted using an NIH ImageJ program. For the differentiation assays, 1.5×105 cells were seeded in a 12-well format in triplicate and cultured with differentiation supplements as per manufacturer's instructions using Mesenchymal Stem Cell Osteogenesis or Mesenchymal Adipogenesis Kits (Millipore). On day 21 postseeding and culturing in differentiation supplements, cells were stained for either Alizarin S Red or Oil Red O stains (Millipore). For soft agar assay, we used a colorimetric Cytoselect cell transformation assay (CellBiolabs, Inc.) in a 96-well format. Cells were seeded at 5×105 cells/mL in a 96-well format in triplicate in soft agar solutions as per manufacturer's protocols and assayed for colony formation using a colorimetric assay (A570) on day 8.
Real-time quantitative reverse transcriptase-polymerase chain reaction analyses
Total RNA from 5 to 10×105 cell lines was extracted using a SurePrep kit (Fisher Scientific). Two micrograms of total RNA was reverse transcribed using TaqMan kit (Applied Biosystems). One microliter template cDNA was used in a real-time quantitative polymerase chain reaction (qPCR) reaction with 2×Sybr green master mix (Applied Biosystems), and 10 μM each forward and reverse primers for experimental or control housekeeping genes. Reactions were run on an ABI7300 cycler (Applied Biosystems), using 40 repeats of 95°C/15 s; 56°C/30 s, 72°C/30 s, and analyzed using ABI7300 software. All samples were normalized to the internal beta-actin control and expressed as change in Ct relative to beta-actin.
In vivo assays
All murine studies were performed using a protocol reviewed and approved by the Institutional Animal Care and Use Committee, LSU School of Veterinary Medicine, and were conducted under veterinary supervision. We utilized human ASC stably transduced with MOI=1 of lentiviruses as just described at passage 3–4 at a 20% ratio to human PC3 prostate tumor cells (ie, 1 ASC: 5 PC3). About 2×105 human ASCs (transduced with either GFP, MDA7, or PEDF lentiviruses) were commixed with 106 PC3 human prostate cancer cells in a final volume of 100 μL in 1×DPBS and subcutaneously implanted into Balb/c nu/nu male recipients (Taconic) in a 3:1 ratio with Matrigel™. Control groups received human ASC or PC3 cells alone. For the mouse ASC in vivo experiments, we used mouse ASC isolated from male C57/BL6 inguinal fat and expanded to passage 2–3 transfected with plasmids containing either “no cDNA” (empty vector control), mouse PEDF cDNA, or mouse mda-7 cDNA. About 4×105 mouse ASCs were commixed with 2×106 TRAMP-C2-Ras (TC2Ras) mouse prostate cancer cells in a final volume of 100 μL in 1×DPBS and subcutaneously implanted into C57/BL6 male recipients. Tumor volumes (mm3) were monitored using vernier caliper measurements and plotted as a function of time.
Statistical analysis
Assays were performed in triplicate, and values were provided as mean±standard error of the mean or 95% confidence interval. Comparisons were performed using an unpaired t-test, and P<0.05 was considered to indicate a significant difference.
Results
Expression of PEDF and MDA7 cytokine gene therapies do not adversely affect ASC biology
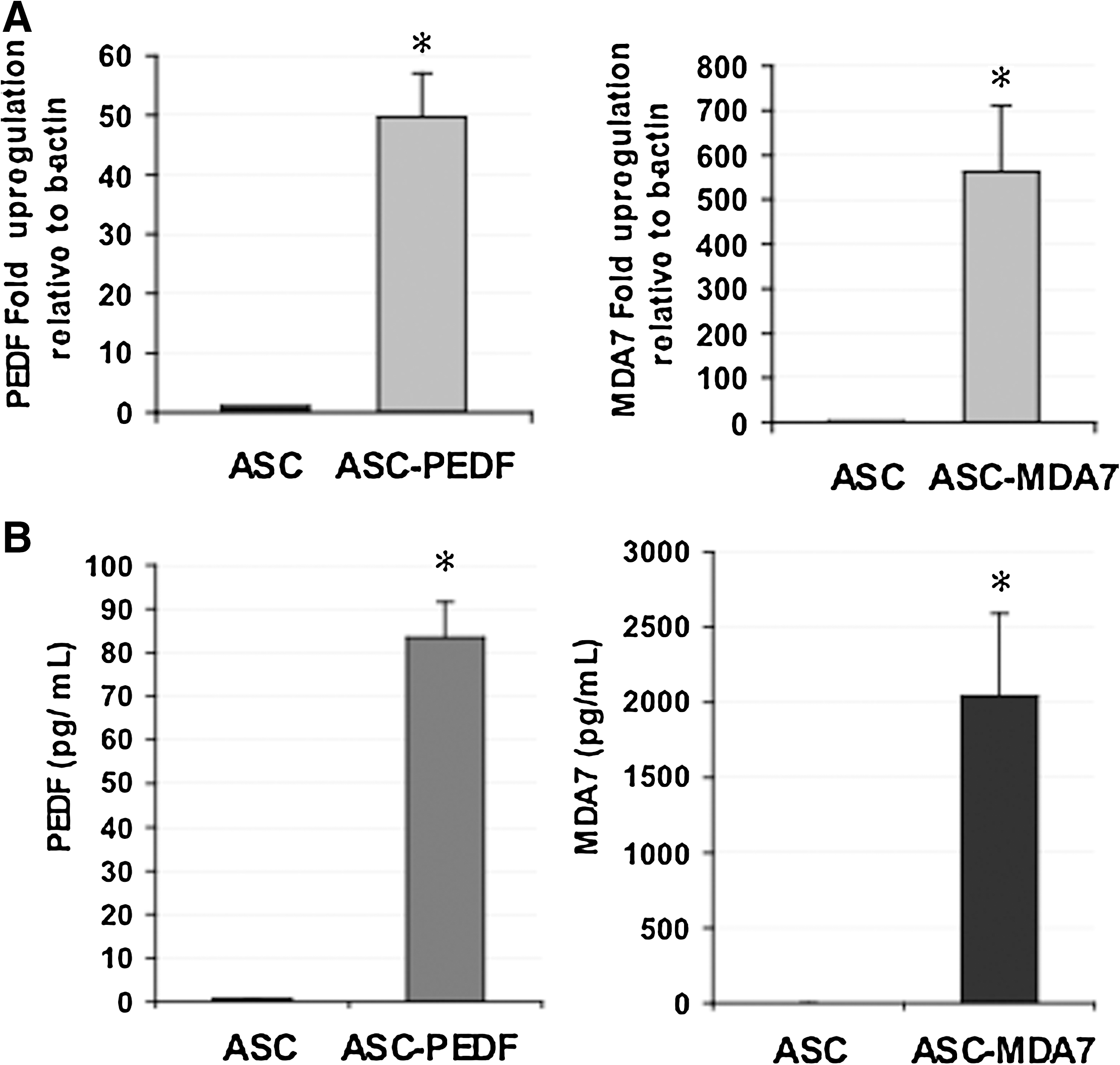
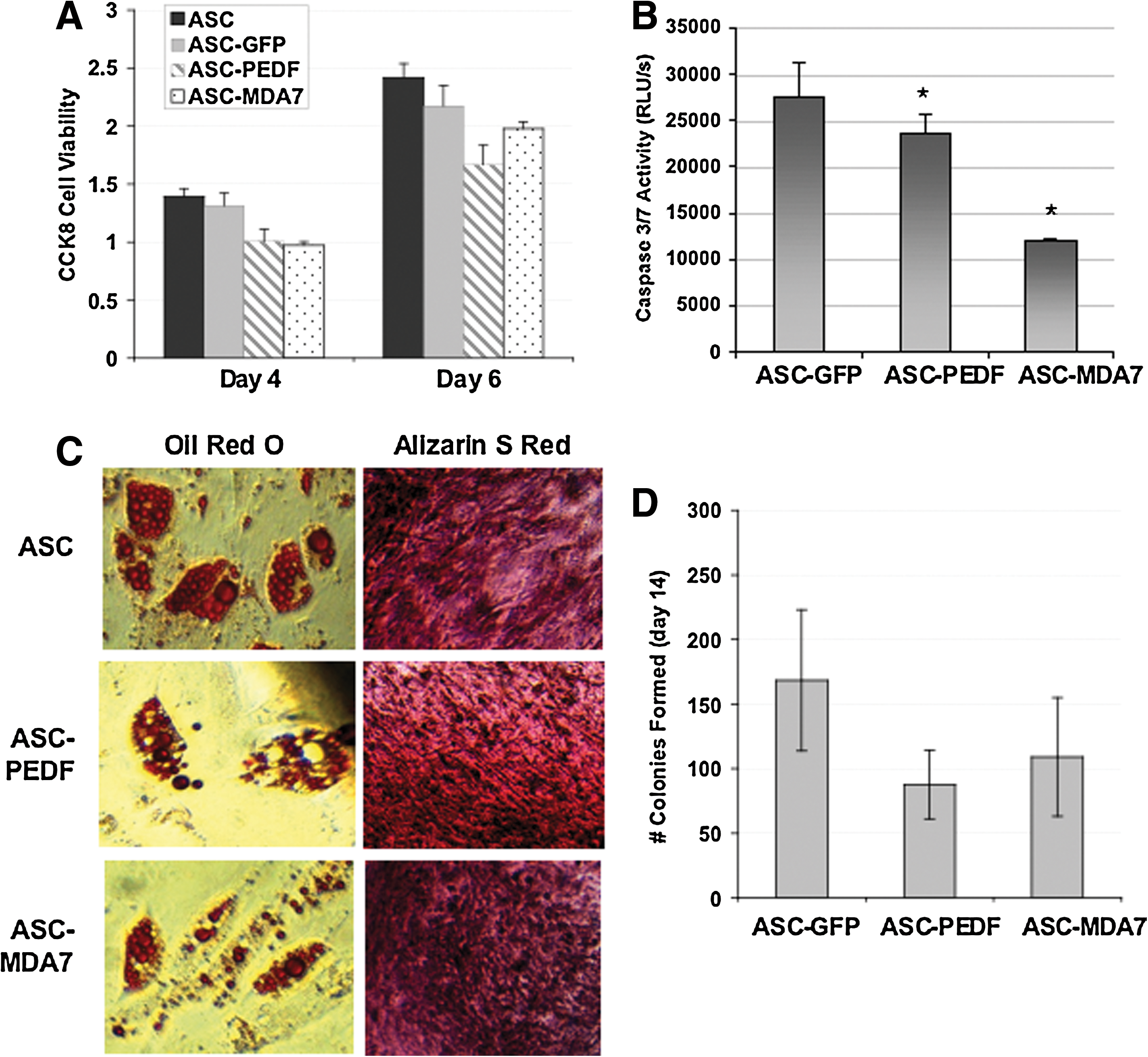
We examined the gene expression levels of PEDF or MDA7 in human MSCs (ASC) after transduction with lentiviruses at MOI=1 by real-time qPCR (Fig. 1A). The PEDF cDNA expression was upregulated by ∼50-fold, whereas MDA7 cDNA was upregulated ∼550-fold over control ASC (P<0.001). We also examined the presence of secreted proteins in the CCM from ASC cells expressing either PEDF or MDA7 by ELISA (Fig. 1B). Both PEDF and MDA7 were detected at levels significantly higher than those present in control (ASC with lentivirus control) (P<0.0002). Due to these high upregulated levels, we sought to determine whether expression of therapeutic cytokines in ASC might adversely affect ASC biological properties. First, cell viability was not adversely affected in ASC transduced by lentivirus (ASC-GFP) as compared with wild-type ASC (ASC) (Fig. 2A, P=0.4). Second, cell viability was also unaltered for ASC transduced by lentivirus and expressing either PEDF (ASC-PEDF) or MDA7 (ASC-MDA7) as compared with ASC-GFP control (Fig. 2A) (P=0.3). Third, the expression of PEDF or MDA7 in ASC did not appear to induce death of ASC, and Casp3/7 levels were actually reduced in ASC-PEDF and ASC-MDA7 (P<0.04). Additionally, cytokine-modified ASCs still could differentiate into the adipocytic and osteoblastic lineages (Fig. 2C) in a manner similar to control ASC-GFP. Finally, cytokine-modified ASCs also retained their ability to form equivalent numbers of colonies in a colony formation assay at day 14 in culture (Fig. 2D). Although the number of colonies was not significantly different than control ASC-GFP (P=0.1), there was, however, a trend in this assay toward fewer colonies for cytokine-expressing ASC (Fig. 2D).
The effect of expressing cytokine gene therapy on the differentiation and colony formation of adipose-derived mesenchymal stem cells.
Cytokine gene therapy expression does not appear to alter the differentiation and colony formation or cell viability of adipose-derived mesenchymal stem cells.
ASC cytokine therapies reduce tumor cell growth and angiogenesis modulation phenotypes
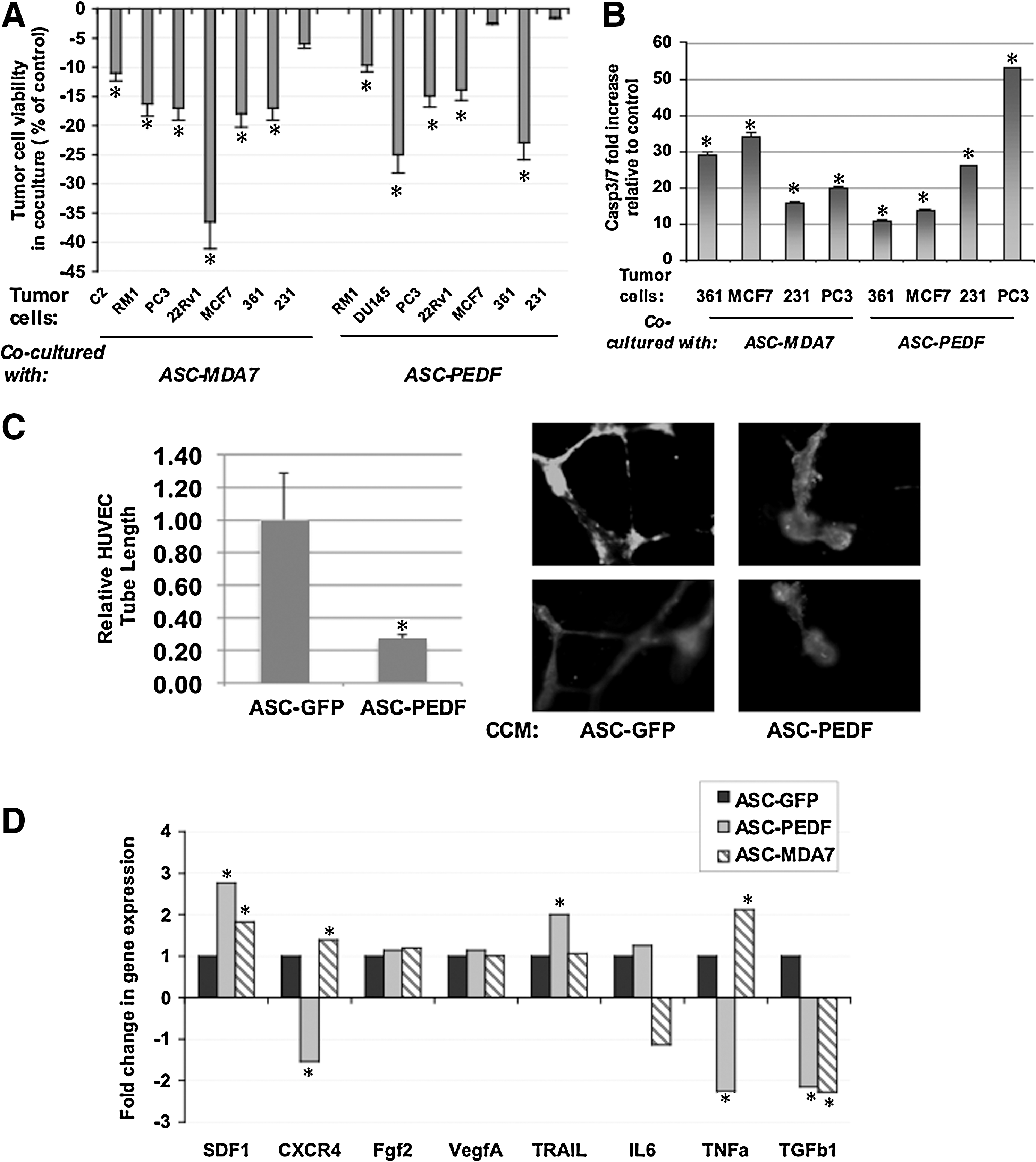
We examined the effect of coculturing ASC expressing cytokines as compared with ASC control on the viability of prostate and breast cancer cells in vitro (Fig. 3A). Coculture with ASC-MDA7 resulted in the greatest reduction in CWR22Rv1 (22Rv1) cell viability, and it reduced viability of all other prostate and breast cell lines (P<0.04). Coculture of tumor cells with ASC-PEDF resulted in the greatest reduction in DU145 and MDA-MB361 (MDA231) viability, and reduced viability of all other cell lines (P<0.02) except for MCF7 and MDA231, which presented a trend of modest reduction. This result suggests that ASC-cytokine therapies could be effective against a range of heterogeneous cell lines of prostate or breast tumor origin. Given the known activity of MDA7 as a proapoptotic agent, we examined the potential of ASC-MDA7 or ASC-PEDF in inducing apoptosis in cocultured cancer cells MDA 231, MDA361, MCF7, and PC3. We observed a significant (P<0.01) upregulation in Caspase 3/7 activity in cancer cells when cocultured with ASC-MDA7 or ASC-PEDF as compared with ASC control (Fig. 3B). For ASC-PEDF, we assessed the ability of cultured conditioned media (CCM) collected from ASC-PEDF cells cultured for 48 h to impact HUVEC endothelial tube formation in vitro in a tube formation assay. The CCM from ASC-PEDF cells dramatically reduced the relative HUVEC tube length in culture by ∼80% (Fig. 3C).
The effect of cytokine gene therapy expressed from ASC on tumor cell phenotype in coculture.
We also examined changes in expression of genes closely related to cellular migration, angiogenesis, and survival between ASC-GFP (control) and cytokine-modified ASC to help ascertain the potential effect of cytokine mRNA expression on ASC biology using real-time qPCR (Fig. 3D). Both ASC-PEDF and ASC-MDA7 significantly upregulated SDF1 and downregulated TGFb1 expression (P<0.003). Unique to ASC-PEDF was downregulation of CXCR4 and TNF and upregulation of TRAIL. Unique to ASC-MDA7 was upregulation of TNF. These changes suggest that ASC expressing PEDF or MDA7 can alter their gene expression profile toward that of enhanced migration likely by utilizing the SDF-1/CXCR4 signaling pathway, and may be acting to enhance apoptosis or reduce survival either through upregulation of TRAIL or TNF or through modification of survival signals (IL6, FGF2).
ASC migration toward cancer cells is not adversely affected by cytokine therapies and can be blocked by inhibition of CXCR4 signaling
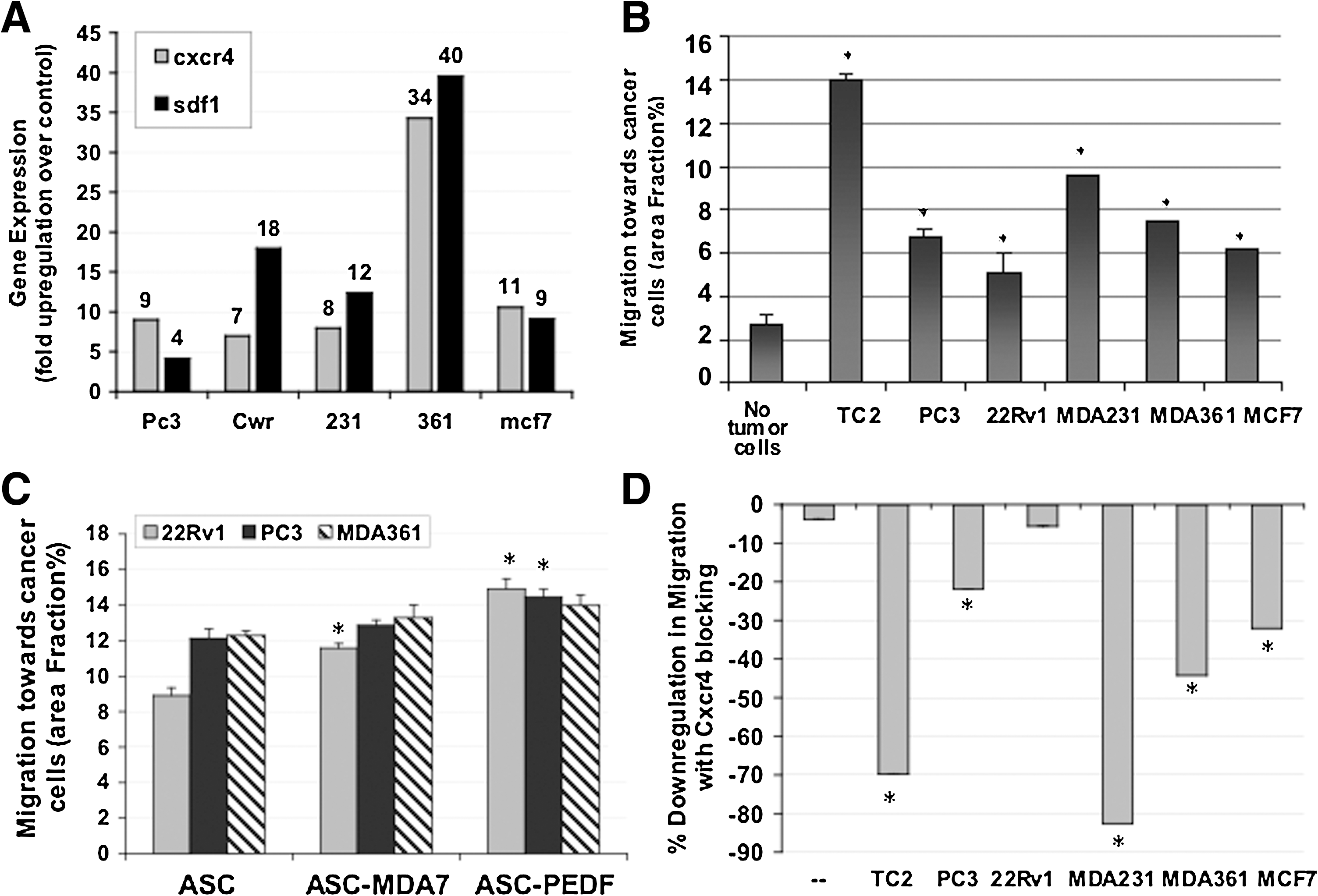
We examined the basal expression of CXCR4 and SDF1 in cancer cell lines by real-time quantitative PCR (qPCR) (Fig. 4A). Both genes were upregulated in cancer cell lines, with CXCR4 expression ∼7–35-fold and SDF1 ∼4–40-fold higher in cancer cell lines as compared with a control normal cell line (RWPE1). Then, using an assay whereby cells seeded atop a transwell insert are allowed to migrate toward cancer cells seeded at the bottom of a 24-well plate, we assessed the ability of ASC cells to migrate toward prostate or breast cancer cells in coculture, as assessed by the percentage of the membrane covered by migrating cells after 24 h (area fraction %). Interestingly, wild-type ASC cells appear to have an innate ability to preferentially migrate toward all tumor cells examined as compared with the “no tumor cell” control (Fig. 4B). A promising finding was that cytokine expression in gene-modified ASC did not appear to adversely impact their ability to migrate toward cancer cells in culture (Fig. 4C). In fact, MDA7 expression in ASC appeared to enhance their migratory ability toward 22Rv1 prostate tumor cells (P<0.02), whereas PEDF expression in ASC appeared to enhance their migratory ability toward 22Rv1 and PC3 prostate tumor cells (P<0.03). There was a trend in enhanced ASC-PEDF migration also toward MDA361 breast tumor cells, but this was not significant. Therefore, the expression of cytokine-encoding genes in ASC should not adversely affect their migration toward tumor cells, and might actually enhance tumor-homing ability, a property that is attractive for potential future systemic administration of ASC as therapy.
Migratory ability of ASC toward tumor cells.
Given the effect of cytokine expression on modulating SDF1/CXCR4 pathway gene expression changes in ASC (Fig. 4D), we examined one potential mechanism for the migration of ASC toward cancer cells. We tested whether migration of ASC toward tumor cells could be reduced by blockage of the CXCR4 signaling pathway in coculture. Presented is the percent downregulation in migration when an anti-CXCR4 antibody was used as compared with an isotype control (Fig. 4D). All CXCR4-blocked samples displayed a significant downregulation in migration toward cancer cells (P<0.05), except for the 22Rv1 prostate tumor line. This suggests the CXCR4 pathway is a key signaling pathway involved in the migration of ASC toward tumor cells, except for 22Rv1, for which another pathway(s) may be favored in impacting migration.
ASC are nontumorigenic in vitro and in vivo
The standard oncogenic transformation test assay for cell lines is usually performed in soft agar rather than in monolayers because of the higher transformation efficiency of cells in soft agar. We examined the potential of ASC to form colonies in soft agar, which indicated that ASC of either mouse or human origin are unable to form colonies, and instead remain as single cells in vitro (Fig. 5A). Controls used were negative control (no cells), which did not form any colonies, and a positive control (22Rv1 cells), which formed many colonies (Fig. 5A). The cells were recovered from the agar using a quantitative soft agar colorimetric kit (Cellbiolabs, Inc.), which confirmed that only the positive control sample produced a signal significantly higher than that of negative control (P<0.001; Fig. 5B). This result suggested that ASC are nontumorigenic.
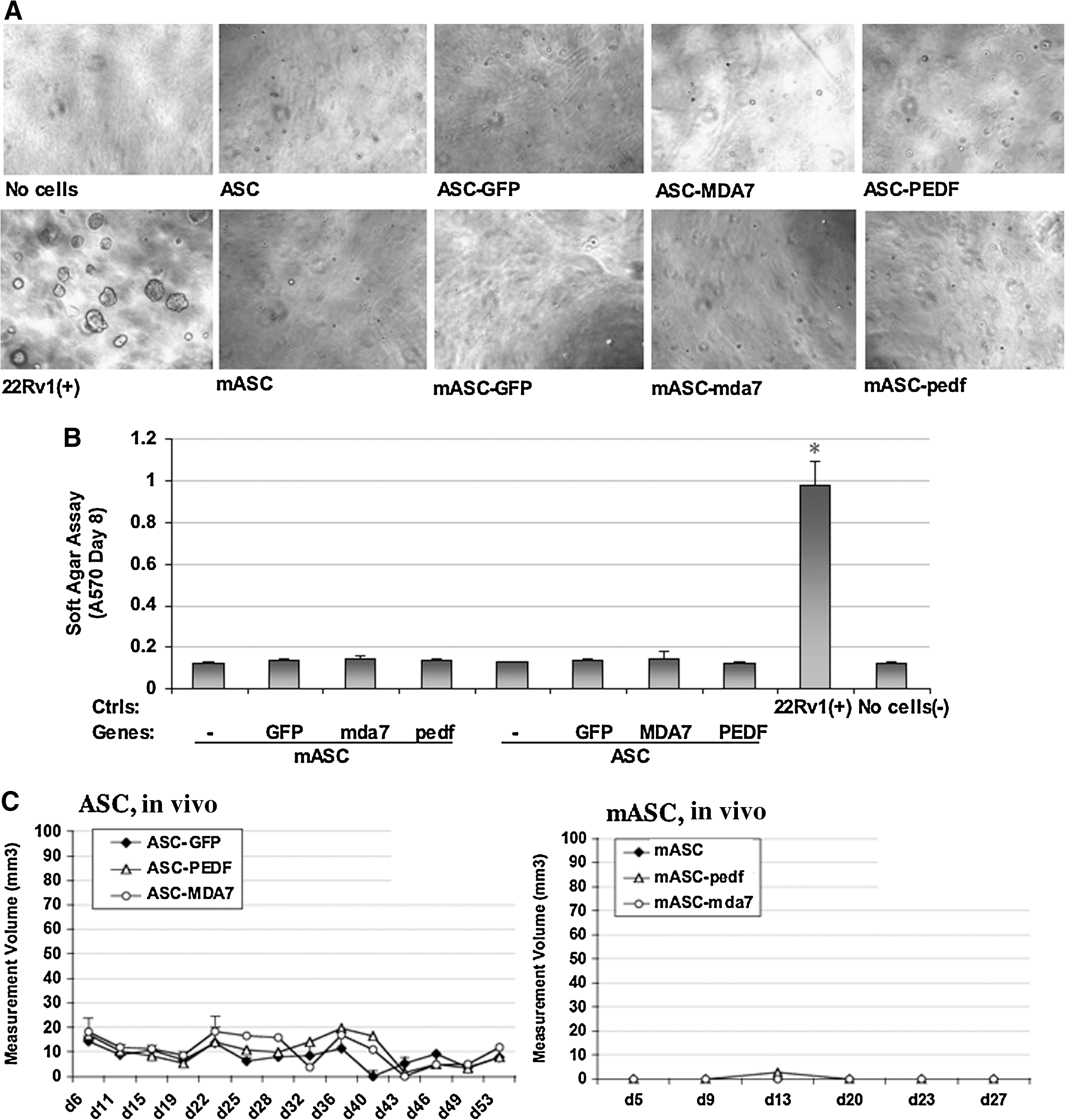
ASCs are nontumorigenic in vitro and in vivo
We confirmed this finding in vivo by assessing whether ASC implanted subcutaneously could form detectable tumors in either C57/BL6 (for mouse ASC or mouse ASC) or Balb/c nu/nu male mice (for human ASC) (Fig. 5C). Human ASCs (control GFP or cytokine-modified) formed a very small swelling of in average ∼10 mm3 that appeared to remain of a stable size throughout the experiment and rather appears to develop locally into adipose tissue. Typical tumor cell lines instead develop tumors within a ∼100–2,000 mm3 range. Mouse ASC did not appear to form any swelling or growth in vivo.
ASC can deliver cytokine gene therapies to reduce the growth rate of human or mouse prostate tumors in vivo
We examined the ability of ASC expressing either control (GFP or empty vector) or cytokine therapies (PEDF or MDA7) to reduce the growth of prostate tumors in vivo. For this purpose, we used 2 xenograft models of prostate tumorigenesis, the human prostate cancer PC3 model (implanted in nu/nu), and our mouse prostate cancer TRAMP-C2-Ras model (implanted in C57/BL6). Our goal was to show that both stably transduced ASC (lentivirus) and transiently transfected mouse ASC could act to reduce tumor growth rate in vivo, therefore illustrating that a variety of transduction means could be feasible and applicable for translation of ASC therapies. In the nu/nu model, we observed that ASC-PEDF commixed with PC3 prostate tumor cells completely inhibited the establishment of the tumor (Fig. 6A). The PC3+ASC-GFP did not significantly alter the growth of PC3 cells in vivo, whereas ASC-MDA7 appeared to adversely affect growth of PC3 cells as a trend, although this effect was not significant compared with the effect of control ASC-GFP (P=0.2; Fig. 6A). In the immunocompetent mouse model, the mouse ASC therapy with the best antitumor effect was once again, mouse ASC-PEDF, although mouse ASC-MDA7 also exhibited an effect in reducing the growth rate of TC2Ras cells (Fig. 6B). In conclusion, both human and mouse ASC expressing PEDF and mouse ASC expressing MDA7 effectively reduced prostate tumor growth in vivo, suggesting ASC-cytokine therapies might be of interest for translational applications.
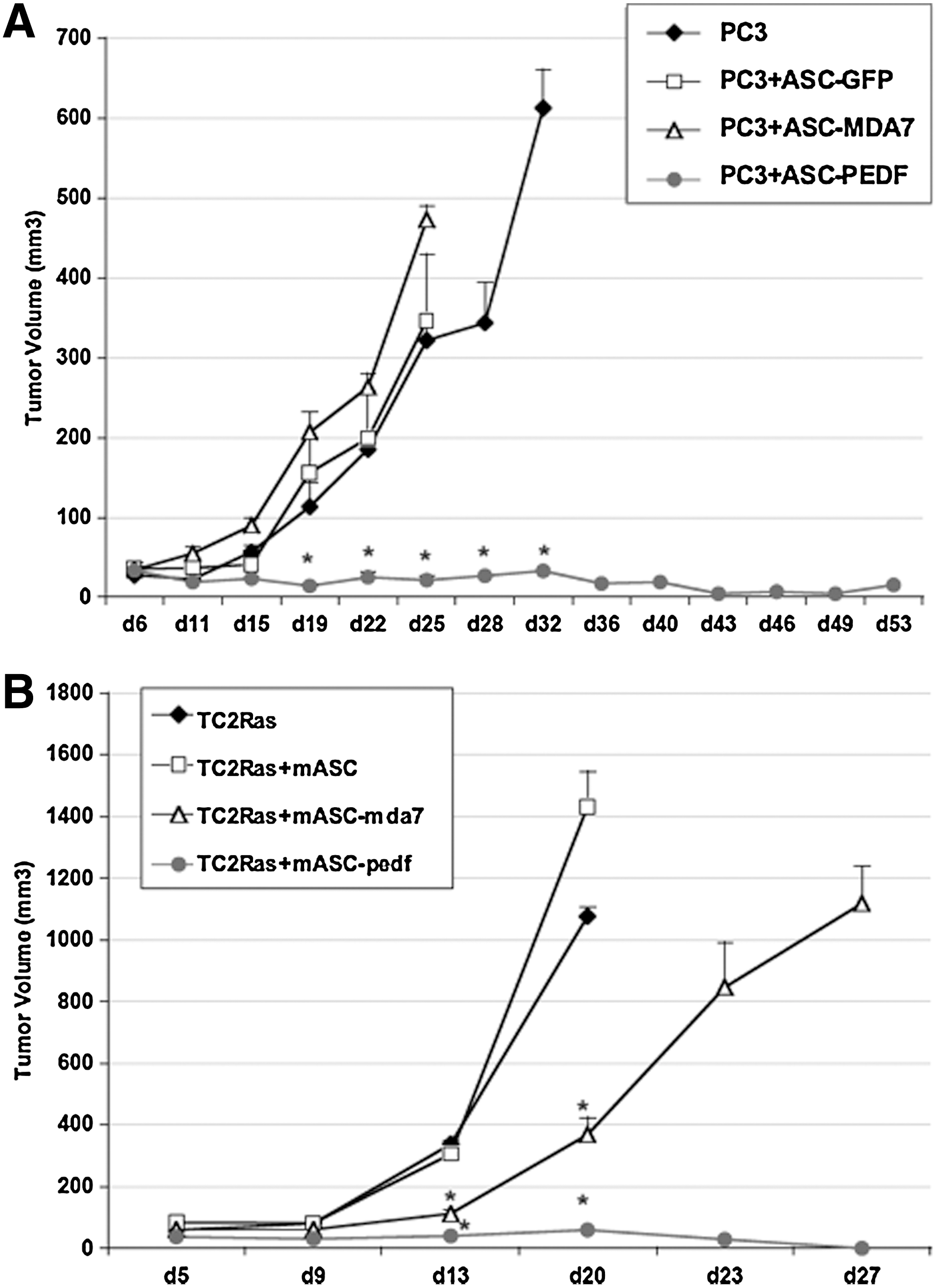
ASC therapies are effective against prostate cancer in vivo.
Discussion
There are no current effective therapies for prostate cancer. Gene therapy for prostate cancer has typically employed viral vectors, and these approaches have been hampered by the immunologic response mounted against the vectors, which renders them less effective. A novel approach to gene therapy of cancer is to employ a cellular carrier, MSCs, which can potentially be delivered intratumorally or systemically for therapy. The use of a cellular carrier can be enhanced when secreted gene therapy molecules are utilized, that is, cytokines.
A particular type of MSC than can be relatively easily obtained from adipose tissue and expanded to large quantities required for therapy is the adipose-derived MSC or ASC. In fact, reports of testing the potential of ASC preclinically for treating cancer are increasing and promising studies have utilized ASC to deliver either TRAIL to cervical, pancreatic, and colon tumors [7], CD for melanoma, breast, and prostate cancer [22], and herpes simplex virus thymidine kinase for glioblastoma treatment [23]. Given the recently reported potency of MDA7 and PEDF cytokines as proapoptotic and antiangiogenesis agents, we hypothesized that ASC could serve as a better gene therapy delivery agent than viral vectors commonly used preclinically and clinically. Adenovirus and others typically elicit robust immunogenicity in the host that can dramatically reduce the efficacy of gene therapy. We tested whether expression of either PEDF or MDA7 in ASC cells could provide a novel, effective, and improved tool with which to manage prostate cancer in vivo.
The results presented herein are promising in that they suggest ASC can be modified by stable (lentiviral transduction) or transient means (lipofection), delivering PEDF or MDA7 cytokines into the culture-conditioned media without adverse effects on ASC biology. The ASC retain viability, and the ability to differentiate into adipose and osteoblastic lineages and form equivalent numbers of colonies in vitro. The ASC expressing cytokines were able to effectively reduce tumor cell viability in coculture against a range of heterologous prostate and breast cancer cell lines. Also observed were induction of apoptosis in tumor cells by MDA7 and PEDF and reduction in endothelial tube formation, a measure of in vitro angiogenesis, by PEDF. Cytokine therapies induced a different pattern of gene expression in ASC-MDA7 and ASC-PEDF cells as compared with control. Both MDA7 and PEDF induced upregulation of SDF1 in ASC, whereas MDA7 alone upregulated TNF, and PEDF alone upregulated TRAIL and downregulated CXCR4 and TNF. Given the effect of cytokine expression on modulation of SDF1/CXCR4 genes, and reports that BM-MSC migrate toward tumor cells [24], we examined whether ASC could also migrate toward tumor cells and whether they might use the SDF1/CXCR4 signaling pathway for this purpose. For instance, when the CXCR4 receptor is blocked by specific antibodies, ASC migration toward cancer cells is reduced, suggesting that at least, in part, this signaling pathway is involved in their migration. The CXCR4/SDF-1(CXCL-12) axis has been considered the main axis of signaling for MSC migration toward tumors, whereby increased SDF-1 acts in an autocrine manner to increase survival, proliferation, and migration of MSCs in the tumor microenvironment [25]. However, one report suggests that in addition to SDF-1, MSCs exposed to CM might produce a number of additional chemokines, including CXCL-2 and CCL2, both of which have previously been demonstrated to protect cardiomyocytes from apoptosis during myocardial ischemia [26]. Therefore, the CXCL2 and CCL2 chemokine axes could also contribute to the migration of ASC toward tumor cells, as the CXCR4/SDF-1 might not be the only chemokine axis regulating migration of ASC. In particular, future studies will address this hypothesis regarding the migration of ASC toward CWR22Rv1 cells, for example, observed here to be refractory to CXCR4 blocking.
One major consideration for potentially translating ASC cytokine therapies or other cellular therapies is that they are nontumorigenic in nature once introduced into the recipient. We examined this potential in vitro using soft agar assays and in vivo using subcutaneous ASC implantation in immunocompetent and immunocompromised mouse models. Soft agar assays indicated that ASC of human or mouse origin are not tumorigenic, as they are unable to form clones in this assay as compared with positive and negative controls. Also, it appears that transduction with lentivirus at low MOI (MOI=1) or lipofection does not transform cells in vivo or in vitro, as the ASC-GFP or mouse ASC+plasmid do not grow into clones or tumors. Future studies utilizing AAV would be of interest before translation of such therapies, as this vector might provide further safety over lentiviral transduction. One interesting result was that human ASC (but not mouse ASC) formed a small swelling sub-cutaneously (s.c.) postimplantation, although the size remained unaltered throughout the experiment. These differences could be due to biological differences in ASC from different species, the fact that ASC are able to sustain minimal initial growth in an immunodeficient environment (nu/nu), or the fact that human ASC (but not mouse ASC) were implanted in Matrigel, a growth-promoting extracellular matrix material. Nevertheless, the mouse ASC are nontumorigenic in vivo, and human ASC form a small swelling that does not appear to be transformed over time and rather appears to develop locally into adipose tissue. These results support a recent report in which wild-type ASC or ASC modified to express CD (ASC-CD) were found to be nontumorigenic after testing of subcutaneous inoculation doses of up to 15×106 cells in immunocompromised mice [27]. Additionally, in the same study, ASC, ASC-CD, and MSC isolated from human lipoma were further treated with the chemical carcinogen 4-nitroquinoline-1-oxide in an attempt to induce cellular transformation. However, cells that survived this genotoxic stress did not undergo transformation but rather underwent replicative senescence displaying adipogenic terminal differentiation, rather than apoptosis. Regarding translational applications, some caution is advised in the dosage of ASC utilized for tumor therapy. It is known that BM-MSCs can be isolated and expanded to large numbers and can be used safely and are well tolerated in clinical trials for host-versus-graft disease [1,28], where importantly, no MSC-related tumors have been reported after transplantation of MSC into over 1,000 patients. However, caution likely has to be exercised at least for hematologic malignancies, where cotransplantation of BM-MSC with hematopoietic stem cells could yield a higher relapse rate [29]. In a related note, careful titration of the ASC dosage is necessary to avoid any tumor promotion ability. There is a trend in our in vivo data of ASC not enhancing the growth of prostate tumors at the doses utilized. Although this is in apparent discrepancy with published reports that MSC can enhance the growth of tumors and promote tumorigenesis, it is likely that this effect is rather dose dependent. For example, at high doses, BM-MSC can promote tumorigenesis, as shown by Karnoub et al., where BM-MSC concentrations exceeded those of coinjected breast tumor cells by 3-fold [30]. Under these conditions (66% MSC plus 33% tumor cells), the tumor growth was accelerated in comparison to breast cancer cells alone. Similarly, other reports used 2-fold more BM-MSC than colon tumor cells [31], and equal amounts of BM-MSC and MCF7 cells [32], and in both conditions the growth of tumors was enhanced. When ASC were injected in a 10-fold excess to 4T1 breast tumor cells, an enhancement of tumor growth was also observed [33]. In contrast, in our studies, when a 1 ASC: 5 PC3 cell ratio (20% therapeutic cells) dose was used, we did not observe significant enhancement in PC3 tumorigenesis. However, if a dose of 40% wild-type ASC (not carrying therapeutic gene therapy) is used, a tumor-promoting effect can be observed in PC3 tumors [22]. Interestingly, when ASC were given at the 40% dose containing cytotoxic CD gene therapy (CD-ASC), this dose was no longer tumorigenic. Also, lower therapeutic doses such as of 20% CD-ASC were as efficient a therapy as the 40% CD-ASC dose but were not tumor promoting.
In vivo, ASC appeared to be quite promising for delivering cytokine therapies to prevent establishment and/or growth of implanted tumor cells s.c., in both immunodeficient and immunocompetent mouse models. Aggressive models of prostate tumor growth were selected using either human prostate cancer cells (PC3) or mouse prostate cancer cells (TRAMP-C2 modified with Ras oncogene). The study is of substantial clinical relevance, as PC3 and TC2Ras cells are among the most aggressive models available and difficult to treat. These cell lines grow rapidly in vivo and recapitulate critical features of human prostate cancer, for example, migration and growth in bone and formation of osteolytic/osteoblastic lesions. However, ASC therapies were able to retard or completely prevent tumor establishment in vivo. Future studies will examine the potential of ASC to treat the prostate tumors and the surrounding bone lesions. The ASC-MDA7 therapy strategy significantly retarded the growth of TC2Ras in the context of the immunocompetent mouse, suggesting that MDA7 might modulate some level of immune system antitumor activity. This potential activity has been associated with MDA7 in some studies. For example, "bystander" effects are associated with MDA-7 that include immune stimulation (pro-Th1), some antiangiogenesis, and IL20-receptor-mediated cytotoxicity [34]. Interestingly, PEDF's effects appeared to be stronger, and ASC-PEDF were able to completely prevent tumor establishment of PC3 and TC2Ras cells, suggesting that PEDF does not need potential immune system antitumor activation within the tumor itself. Future studies will examine the potential of ASC cytokine therapies to recruit specific subsets of effector immune cells into prostate tumors and the surrounding bone lesions as a potential antitumor mechanism.
An adenoviral delivery system for PEDF (Ad-PEDF) strongly supports the use of this cytokine for prostate cancer therapy [19], whereby PC3 cells had a 3-fold increase in Annexin V apoptotic cells after treatment with Ad-PEDF as compared with an Ad-LacZ control. Also, in the same report, a ∼36% growth rate reduction was seen in Ad-PEDF treated PC3 tumors. Although lower numbers of PC3 cells were injected s.c. in our studies, with ASC-PEDF tumors never exceeded 25 mm3, representing an impressive ∼96% reduction in growth rate compared with untreated PC3 or ASC control tumors. Nevertheless, the Ad-PEDF system has been translated to Phase I macular degeneration trials with promising antiangiogenesis results [35]; therefore, this therapeutic strategy has high potential to also benefit patients with prostate and other cancer. Future studies can compare these strategies within the same study to examine the doses of Ad necessary to achieve equivalent therapeutic efficacy as ASC therapeutics.
Footnotes
Acknowledgments
We thank M. Dietrich for flow cytometry analysis for GFP expression, and J. Zhu and R. Watzel for assistance with plasmid vector preparation. This work was supported in part by funding from the Pennington Biomedical Research Foundation (J.M.G., G.Y.) and Louisiana State University School of Veterinary Medicine Seed CORP grant (M.L.F.).
Author Disclosure Statement
M.L.F., O.Z., and G.Y. have no conflicts to declare. J.M.G. has leadership roles at LaCell LLC to declare.