
Abstract
Although amniotic fluid cells can differentiate into several mesenchymal lineages and have been proposed as a valuable therapeutic cell source, their ability to undergo terminal neuronal differentiation remains a cause of controversy. The aim of this study was to investigate the neuronal differentiation ability of the c-Kit-positive population from GFP-transgenic rat amniotic fluid, amniotic fluid stem (AFS) cells, and to assess how they affected injury response in avian embryos. AFS cells were found to express several neural stem/progenitor cell markers. However, no overt neuronal differentiation was apparent after either treatment with small molecules known to stimulate neuronal differentiation, attempts to differentiate AFS cell-derived embryoid body-like structures, or grafting AFS cells into environments known to support neuronal differentiation (organotypic rat hippocampal cultures, embryonic chick nervous system). Nonetheless, AFS cells significantly reduced hemorrhage and increased survival when grafted at the site of an extensive thoracic crush injury in E2.5 chick embryos. Increased embryo survival was induced neither by desmopressin treatment, which also reduced hemorrhage, nor by grafting other mesenchymal or neural cells, indicating a specific effect of AFS cells. This was found to be mediated by soluble factors in a transwell coculture model. Altogether, this study shows that AFS cells reduce tissue damage and increase survival in injured embryos, providing a potentially valuable tool as therapeutic agents for tissue repair, particularly prenatal/perinatal repair of defects diagnosed during gestation, but this effect is mediated via paracrine mechanisms rather than the ability of AFS cells to fully differentiate into neuronal cells.
Introduction
T
There is much evidence that cells present in the amniotic fluid have the potential to give rise to several differentiated mesenchymal tissues, such as bone, cartilage, and adipocytes [9,11,12]. In contrast, their ability to undergo neural differentiation and produce functional neurones has been a matter of contention [4,7,9,13 –16]. In vitro, amniotic fluid cells appeared to differentiate into neurones expressing voltage-gated Na(+)channel with properties similar to those found in native neurones [17]. When transplanted into the rat brain, human amniotic fluid cells were able to migrate in the brain parenchyma and express immature neuronal markers, and AFS cells primed toward neuronal differentiation prior to transplantation seemed to integrate within host tissues and apparently differentiated into dopaminergic neurones [9,13]. Interestingly, amniotic fluid cells transplanted into animals following induction of an ischemic stroke not only were found to upregulate the neural markers Dcx and GFAP, but also appeared to ameliorate behavior in ischemic animals [13,18]. A more recent study, however, did not find any significant evidence that amniotic fluid cells could differentiate into mature dopaminergic neurones and suggested that these cells did not survive long term following transplantation [14].
Given the great interest recently emerged in the potential use of amniotic fluid cells in clinical contexts not only for mesenchymal tissue repair but also for neural repair [19], it is important to better understand their true potential and possible limitations for their use. The main aims of this study were to establish whether AFS cells have a significant differentiation potential along the neural lineage and whether they aid repair in a severe embryonic injury model. AFS cell differentiation ability was studied both in culture following treatments successfully used to induce neural differentiation in other cell types with neural potential and by grafting them in environments where neural development normally occurs, to ensure exposure to appropriate cues for differentiation over time. To this purpose we studied the behavior of AFS cells challenged with small molecules, grown as embryoid body-like (EB-like), or grafted onto organotypic rat hippocampal slices and in the chick embryo. This is a valuable model given its accessibility in vivo and previous reports that mammalian hematopoietic and embryonic stem cells could integrate in the chick developing nervous system [20 –23]. The chick was also used as an embryonic injury model to investigate whether AFS cells affected the response to injury. Here we show that while no overt neural differentiation could be observed in AFS cells under different culture conditions and following transplantation in vivo, grafted cells greatly increased embryo survival following severe injury and that this effect was specific to AFS cells and mediated via paracrine mechanisms.
Materials and Methods
Cell lines
All cells were grown at 37°C in a humidified incubator with 5% CO2. The tissue culture reagents, unless otherwise specified, were from Gibco. A c-Kit-positive population of AFS cells isolated from amniotic fluid of E16 rats expressing GFP was used. AFS cells were grown in bacteriological dishes (BD Bioscience) in complete Chang medium [α-MEM supplemented with 15% fetal bovine serum (FBS), 20% Chang B and 2% Chang C (both from Metachem Diagnostics), and 1% penicillin/streptomycin] [9] (De Coppi et al., in preparation). The C17.2 neural cell line (kind gift of C. Cepko) derived from postnatal mouse cerebellum by v-myc immortalization was maintained in DMEM containing 10% FBS, 5% horse serum, 2 mM glutamine, and 1% penicillin/streptomycin. Mouse NIH3T3 fibroblasts were grown in DMEM containing 10% FBS, 2 mM glutamine, and 1% penicillin/streptomycin. For grafting experiments, C17.2 and NIH3T3 cells were labeled with the green fluorescent dye, 5-chloromethylfluorescein diacetate (CMFDA), as previously described [24].
Formation of EB-like spheres was induced by plating AFS cells in complete Chang medium either onto PLL-LM (100 μg/mL poly-
Induction of differentiation
AFS cells were seeded at a plating density of 10,000 cells per 13-mm coverslip in complete Chang medium. The medium was changed to N2 medium (1% N2 supplement, 2 mM glutamine, and 1% penicillin/streptomycin in DMEM/F12) containing the compounds to be tested; 0.5 mM 3-isobutyl-1-methylxanthine (IBMX) and 1 mM dbcAMP (N6, 2'-O-dibutyryl-cAMP) were used either alone or in combination. In some experiments, after 48 h of chemical treatment, the medium was switched to Chang medium or DMEM containing 10% FBS. Cultures were monitored over 7 days and fixed for immunohistochemistry at days 1 and 7.
HD-derived EB-like spheres were plated onto LM-coated coverslips and maintained in either Chang medium or N2 medium (5 days) and N2 medium plus 10% FBS (1–10 days).
Organotypic cultures
Three-hundred-micrometer-thick entorhino-hippocampal slices were prepared from 5-day-old Wistar rats. Slices were grown individually onto a 0.45-μM white Millipore® FHLC membrane on a 0.4-μM Millipore®-CM culture plate insert according to Stoppini and coworkers [26]. Entorhino-hippocampal slices (n=9) were grown in serum-containing medium (MEM with Glutamax, 5% EBSS, 36 mM
Two-hundred-micrometer-thick E15 chick spinal cord cross sections were cultured onto a 0.4-μM Milipore®-CM culture plate insert in AFS cell medium (complete Chang medium) with or without AFS cells (100,000 cells/well) present in the lower chamber. Propidium iodide (PI, 5 μg/mL) was added to the medium to assess cell death at specific time points and some slices were processed for immunohistochemistry. PI staining intensity per organotypic slice area was measured at 24 h using ImageJ software available at
In vivo transplantation into chick embryos
Fertilized Brown Leghorn eggs (Needle Farm) were incubated at 37°C in a humidified forced flow incubator. All procedures were carried out under the Animals Scientific Procedures Act 1986. The total number of animals used for each experimental condition is indicated in the Results section and each experiment was carried out at least twice using controls from the same batch of eggs. Cells were grafted in the developing chick nervous system at different Hamburger and Hamilton stage (H&H) using a glass capillary with very gentle air pressure under a fluorescent LEICA MZ FL III stereomicroscope. Cells were grafted either in the anterior neuropore (H&H9-10) or in normal and injured spinal cords (E2–E2.5). Injury was induced by crushing the spinal cord and surrounding tissues cranially to the vitelline blood vessels (∼2 somites above).
In some experiments, injured embryos were treated with desmopressin, an inhibitor of fibrinolysis, which has been used to treat patients with factor XI deficiency and was recently used to induce hemostasis in an established chick model of spinal cord injury [27]. Injured E2.5 chick embryos were treated with 100 μg desmopressin/embryo at the time of injury. Embryos were observed daily to monitor gross anatomy changes, heart beating, vascularization, and GFP- or CMFDA-positive cells for up to 5 days following surgery and surviving embryos were processed for immunohistochemistry.
Significance of changes in embryo survival over 5 days was assessed by the Log-rank (Mantel-Cox) test and Gehan-Breslow-Wilcoxon test using Prism5 software; P<0.05 was taken to be significant.
RT-PCR
Total RNA was extracted from rat AFS cell monolayers, EB-like cultures, and E16 rat brains (positive controls) using TRIzol® reagent (Invitrogen) according to the manufacturer's instruction. Five hundred nanograms of the extracted RNA was used for reverse transcription using Superscript II (Invitrogen) with random hexamers. Primers (Appendix Table A1.) were designed using Primer3 software [28]. Cycler was programmed for either 31 or 40 cycles as follows: 1 cycle at 95°C for 4 min, 1 cycle at 95°C, 62°C, and 72°C for 30 s, 1 cycle at 95°C, 60°C, and 72°C for 30 s, 29 or 40 cycles at 95°C, 58°C, and 72°C for 30 s, and finally, 1 cycle at 72°C for 5 min. Following RT-PCR, reaction products were resolved using a 1.5% agarose gel.
Immunocytochemistry and immunohistochemistry
Immunostaining of cells, slices, and frozen sections fixed in 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) was carried out essentially as previously described [29,30]. Monolayer cells and EB-like spheres were washed with PBS and fixed in 4% PFA at room temperature for 20 min. They were permeabilized and blocked in 3% bovine serum albumin (BSA), 10% FBS, and 0.2% Triton X-100 in PBS. Chick embryos were fixed in 4% PFA overnight, cryoprotected by incubation in 30% sucrose containing 0.02% sodium azide in PBS at 4°C for approximately 24 h, and OCT embedded. Cryosections (14–16 μm) were permeabilized and blocked in 3% BSA, 10% FBS, and 1% Triton X-100 in PBS. Hippocampal slices were fixed in 4% PFA for 5 min at room temperature and postfixed in 20% cold methanol in PBS for 30 min. Slices were permeabilized in 0.5% Triton X-100 in PBS for at least 12 h at 4°C and blocked in 20% BSA in PBS either for 4 h at room temperature or overnight at 4°C.
The primary and secondary antibodies used are listed in Supplementary Table S2. Primary and secondary antibodies were diluted in blocking solution according to the type of culture to be stained. Incubation was either for 3 h at room temperature or overnight at 4°C for primary antibodies and for 1–3 h at room temperature for secondary antibodies. Stained specimens were visualized and digitally scanned using an Axiophot 2 (Zeiss) with Hamamatsu ORCA-ER digital camera, or an Axiovert 135 (Zeiss) with a ProgRes C14 digital camera, or by confocal laser scanning microscopy using an LSM 710 (Zeiss). Image collection and analysis were performed using Openlab, Volocity (Improvision), or ImageJ software. Complete “z” series optical sections were collected and projected onto a single plane using ImageJ software [31]. The percentage of cells positive for each antibody was evaluated by counting at least 400 cells in different representative fields.
Results
To assess the AFS cell neural potential, we first investigated the expression of neural stem cell markers and markers of neural differentiation by RT-PCR (Fig. 1A) and immunocytochemistry (Fig. 2 and Table 1). AFS cells expressed the neural stem cell markers Sox2, Pax6, and nestin and the neural crest marker Snai1, but not FoxD3 (Fig. 1A). Expression of Sox2, Pax6, and nestin was also detected at the protein level (Fig. 2A–C and Table 1). The transcript for βIII-tubulin, which is usually considered a marker of neuronal differentiation, was detected in AFS cells (Fig. 1A), but the protein was hardly detectable by immunocytochemistry. The neural marker A2B5, however, was clearly expressed by a subset of AFS cells (Fig. 2D–F and Table 1).

Analysis of gene expression in amniotic fluid stem (AFS) cells grown as either monolayers or embryoid body (EB)-like spheres by RT-PCR.

Expression of neural progenitor and neural differentiation markers in rat AFS cells grown as either monolayers
Approximate percentages of positive cells are given in brackets. Bold indicates strong staining and underline indicates weak staining.
Not only in mesoderm (eg, also in neural stem cells).
Embryonic stem cells; 2neural stem cells; 3neuroblast; 4early neurones; 5neurones; 6glial progenitors; 7astrocytes; 8oligodendrocyte; 9neural crest cells; 10mesenchymal stem cells.
As the expression profile of neural markers in AFS cells was suggestive of a neural differentiation potential, we performed a number of treatments using small molecules known to induce neural differentiation (not shown). The most significant change in cell morphology was observed following treatment with agents that increase cAMP levels such as 1 mM IBMX and 0.5 mM dbcAMP. Treated cultures were still nestin positive (not shown) and some of the cells (<5%) displayed a neuronal morphology and expressed βIII-tubulin (Fig. 2G, H), but this was lost when the drug was removed after 1-week treatment, suggesting that no bona fide neuronal induction might have occurred.
AFS cells plated onto PLL-LM, but not onto other substrates (not shown), rapidly underwent morphological changes and EB-like sphere appearance was observed within 5 days (Fig. 2I, J). The same type of structures also formed when AFS cells were grown in suspension as hanging drops. We tested whether these aggregates were indeed EBs containing all 3 germ layers and could undergo neural differentiation. Expression of markers of the 3 germ layers (endoderm: Sox17, Gata4; mesoderm: Brachyury, Sm22-a; neuroectoderm and derivatives: Pax6, βIII-tubulin) was assessed by RT-PCR. Sm22, Pax6, and βIII-tubulin could be easily detected in AFS-derived EB-like spheres, whereas the expression of Sox17, Gata4, and Brachyury was very low and could be detected only when a high cycle number (40 cycles) was used (Fig. 1B). Under these conditions, Sox17, Gata4, and Brachyury PCR products were not detected in AFS monolayers at either confluency or low density (Fig. 1B and Supplementary Fig. S1). At the protein level (Fig. 2J–P and Table 1), the EB-like spheres continued to express the stem cell markers vimentin and nestin and the neuroblast marker Dcx, but no significant βIII-tubulin (Fig. 2I, K, M, N). GFAP staining was also observed (Fig. 2L), but unlike that of other intermediate filaments (eg, nestin and vimentin) was rather granular, suggesting that though GFAP protein was synthesized, it did not become properly organized in a filamentous structure. Cytokeratin reactivity appeared to be more extensive in EB-like spheres grown either under normal or differentiating culture conditions (not shown) than in AFS monolayers, where patches of cytokeratin-positive cells were sometimes observed (Fig. 2O, P).
To rule out that failure to induce clear neural differentiation in AFS cells might have been due to nonoptimal in vitro conditions, an organotypic culture system was used. We grafted AFS cells onto hippocampal slices, as neurogenesis in the dentate gyrus continues postnatally and therefore provides a permissive neurogenic environment (Fig. 3). Cells seeded onto the dentate gyrus of rat hippocampal slices cultured either in serum-containing medium (n=9; Fig. 3) or in serum-free media (n=22; not shown), but not onto the enthorinal cortex (not shown), were able to migrate and proliferate (Appendix Table A2) and underwent morphological changes. However, neither 2 weeks (not shown) nor 4 weeks after grafting, costaining of GFP and βIII-tubulin (Fig. 3A, B) or GFAP (not shown) was observed.
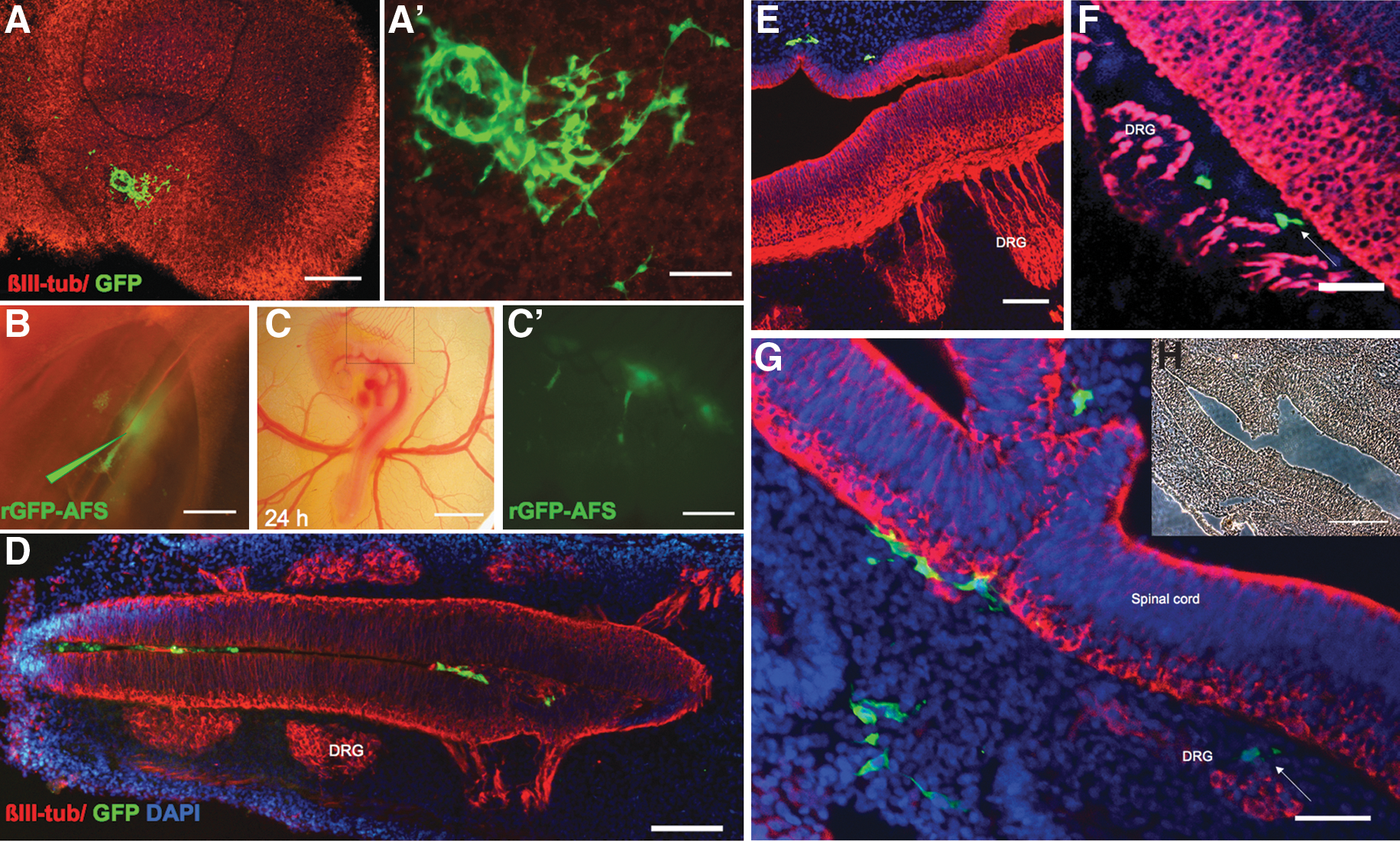
AFS cells grafted either onto hippocampal slices
It is conceivable that the postnatal organotypic hippocampal slices/culture conditions used might not have provided the factors required for neural differentiation of AFS cells. Therefore, we grafted these cells in different locations within the chick central nervous system (CNS) in vivo at different developmental stages when neurogenesis is occurring (Fig. 3). No obvious damage appeared to be caused by grafting AFS cells and the vascular network was not affected (Fig. 3B, C). Cells injected in the anterior neuropore at H&H 9–10 (n=6; not shown), in E2 (H&H 14–15; n=10; Fig. 3E, F), and in E2.5 (H&H 15–16; n=14; Fig. 3D) spinal cords were monitored for up to 5 days, initially in vivo and at the later stages in tissue sections. AFS cells were observed in the E2.5 spinal cord at 1 day after grafting (Fig. 3C) and were still detectable after 4 days (E6), although they were mainly found in proximity of the dorsal root ganglia rather than in the spinal cord (Fig. 3D, E). These cells did not express βIII-tubulin.
Finally, we assessed the effect of AFS cells and their behavior in an embryonic injury model. AFS cells were grafted into E2.5 chick embryo spinal cords at the time of a thoracic crush injury. As at early developmental stages chick embryos can repair small injuries, we performed a lesion that resulted in severing of the spinal cord and nearby tissues that did not repair itself. The response to injury in control and grafted embryos was monitored over 5 days (Figs. 3F and 4).

Effect of AFS cell grafts on chick embryos injured at E2.5.
Although no significant hemorrhage was usually observed shortly after injury in control injured embryos (n=27), by 24 h diffuse heavy bleeding was apparent. In contrast, in AFS cell-grafted injured embryos (n=18), hemorrhage was more localized to the injury site (Fig. 4A, B). By 48 h, injured chick embryos had slower heart rate than noninjured embryos and by day 3 high mortality was observed in this group. In contrast, in the cell graft group, 60% of the embryos appeared to be developing normally, although in some cases morphological defects were observed at the microscopic level at the site of injury, they were always less severe than those observed in control embryos at earlier stages after injury (Fig. 3F and inset in Fig. 4A). Altogether, AFS cell-grafted embryo survival was significantly higher than in injured controls (P<0.02), particularly at the earlier time points; 43% of the grafted embryos appeared normal at day 4 (Fig. 5).

Effect of grafting AFC cells (AFSCs) or different cell types and of desmopressin treatment on injured chick embryos. Survival of embryos injured at E2.5 was monitored over 5 days following different treatments at the time of injury and the cumulative survival probability with 95% confidence intervals was assessed. Survival was significantly increased (*) in embryos grafted with AFS cells when compared to injury alone (log-rank Mantel-Cox test, P=0.009; Gehan-Breslow-Wilcoxon test, P=0.0136). In contrast, no significant difference in survival was observed in embryos grafted with 3T3 or C17.2 cells or treated with the antihemorrhage drug desmopressin.
To assess the specificity of the survival effect induced by the AFS cells, we repeated the injury experiment grafting either neural progenitor cells, C17.2 (n=6), or fibroblasts (n=12). Both cell types showed no significant effect on chick survival; by day 3 after surgery, less than 25% of the embryos grafted with C17.2 cells or fibroblast were alive, versus 58% of AFS cell-grafted embryos (Fig. 5).
As the extent of hemorrhage greatly differed in control and AFS cell-grafted injured embryos, we investigated whether a reduction in hemorrhage would be sufficient to reduce mortality. To this purpose we treated injured embryos (n=8) with the hemostatic drug desmopressin, which we had previously shown to reduce hemorrhage and spinal cord injury at later developmental stages [27]. Although reduced hemorrhage was observed in desmopressin-treated embryos, survival in these embryos was lower than under all the other conditions tested, as almost (n=6) 80% of the animals had died by day 2 after surgery (Fig. 5).
We then tested whether this AFS cell rescue effect was due to neural differentiation by staining for Dcx, GFAP (not shown), and Tuj1 (Fig. 3F); the grafted cells were negative for these neural markers, although most of them continued to express nestin (not shown). As the rescue effect did not appear to be due to AFS cell differentiation, we further investigated whether AFS cells could increase cell survival and whether this was mediated via paracrine mechanisms using organotypic cultures (Fig. 6). Chick spinal cord slices were cocultured in a transwell system either with or without AFS cells (n≥15/group). Control slices grown in medium alone displayed extensive cell death at 2 days in culture as indicated by PI staining of live cultures (Fig. 6A). In contrast, hardly any PI-positive cell was observed when slices were cocultured with AFS cells (Fig. 6B). Consistent with this observation, both NeuN and βIII-tubulin staining showed a healthier neuronal population in spinal cord slices maintained in the presence of AFS cells (Fig. 6C–F); beading, which is indicative of axonal degeneration, was hardly observed in spinal cords cocultured with AFS cells.
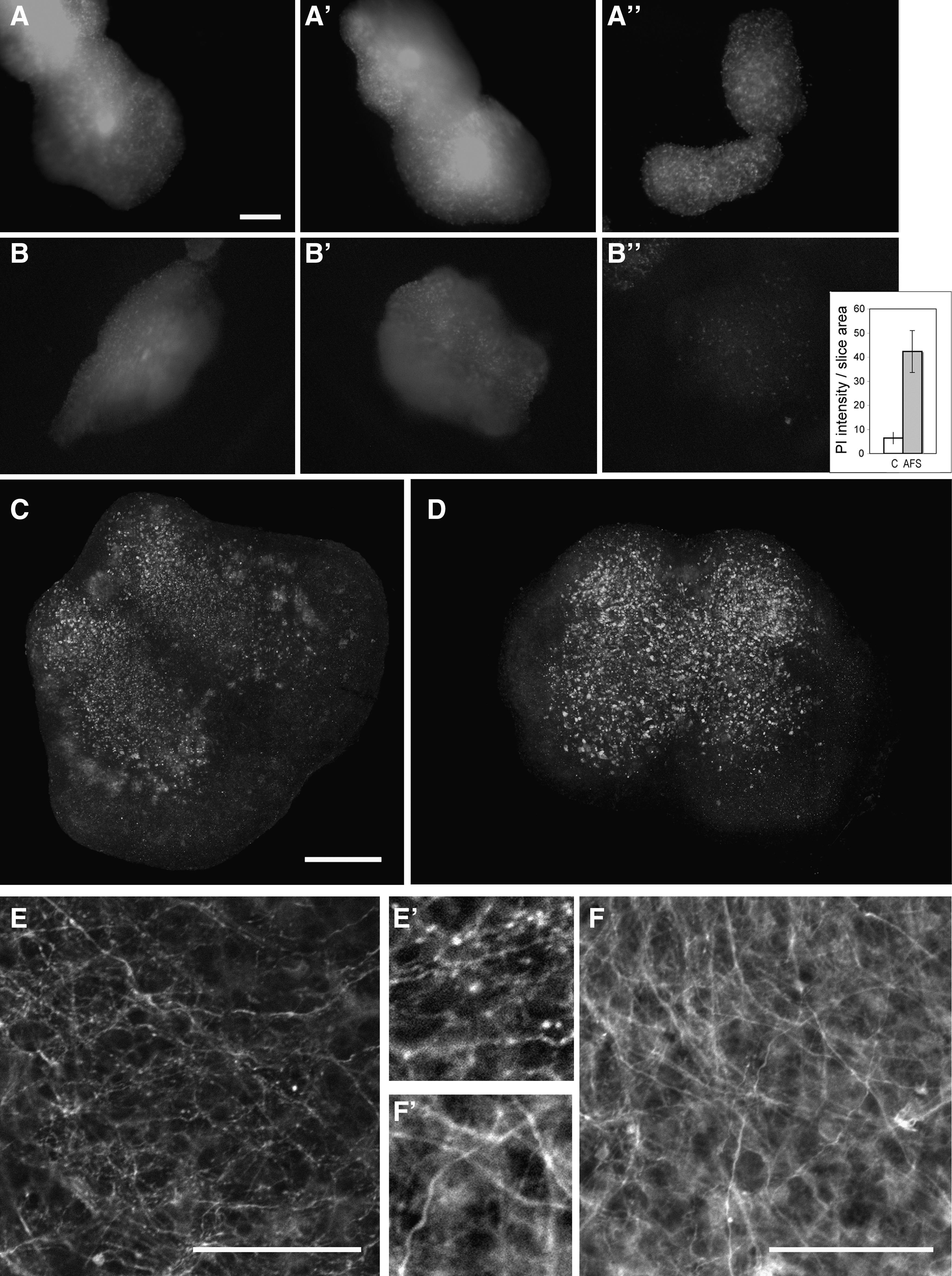
AFS cells increase survival of E15 chick spinal cord organotypic slices. Spinal cords were either cultured alone
Discussion
In this study, we have shown that AFS cells are of potential value for aiding repair, although this ability does not correlate with a significant potential to terminally differentiate along neural lineages.
AFS cells have limited neural differentiation potential
Although a number of published studies have suggested a neural potential for AFS cells from different sources, the evidence that they have full neural differentiation potential has not been extensively tested and has been mainly based on neuronal markers and morphological changes [9,14,16,32]. Electrophysiological analysis has indicated presence of some channel/neuronal responses, but the density of these channels is much lower than in mature neurones [17].
Most of the markers, and neural markers in particular, that we have detected in AFS cell monolayers are expressed by the majority of the cells, although to different extents, with A2B5 being the exception. Altogether, this suggests an AFS cell population relatively homogeneous, at least in the context of the markers investigated. This is not surprising given that our AFS cells had been initially selected on the basis of their c-kit expression, whereas other authors have studied unsorted amniotic fluid cells [4,15]. Notwithstanding AFS cells express several neural markers, consistent with work published while this manuscript was in preparation [4], none of the in vitro and in vivo approaches we used revealed a terminal differentiation potential along the neural lineage. Of the many treatments tested, only IBMX and dbcAMP in combination induced AFS cells to acquire a neuronal-like morphology and express βIII-tubulin protein. These 2 agents, which increase intracellular cAMP availability, have been shown to induce neuronal differentiation in various stem cell types [33,34]. However, the fact that the morphological changes and increase in βIII-tubulin were seen within 24 h after treatment, and that neuron-like morphology could be reversed by removing the treatment, suggest that the changes observed reflect more induction of a stress response than of a differentiation process. This is a crucial point to be considered for the interpretation of results from neural differentiation studies.
Another possible explanation of the phenotypic changes observed following treatment of AFS cells with small molecules could be that they induce a labile phenotype that needs to be “stabilized” by other factors, as recently suggested for differentiation of Schwann cell from mesenchymal stem cells [35]. However, the fact that AFS cells grafted in permissive environments did not undergo neuronal differentiation is not consistent with this hypothesis. Interestingly, when grafted onto hippocampal slices, AFS cells survived only when placed onto the dentate gyrus, where neurogenesis occurs during development and in the adult brain, but not on the enthorinal cortex (not shown), indicating a response of AFS cells to specific local cues. Nonetheless, although the grafted cells changed morphology and extended processes, they did not differentiate into neurones.
Therefore, the neurogenic niche appears to provide factors that support survival of rat GFP-AFS cells, but it does not seem to provide differentiation factors. We do not know at this stage which type of interactions is established between AFS cells and hippocampal cells, and whether there is any effect of the former on the latter.
The developing chick embryo provides a neurogenic environment easily accessible in vivo and has been previously used to graft embryonic and somatic stem cells to study their neural potential [20 –23]. As both rodent and human cells were shown to differentiate into neurones when grafted in chick CNS, it is most unlikely that lack of terminal differentiation of AFS cells into neural cell types is due to species differences. Although grafted AFS cells expressed neither βIII-tubulin nor GFAP, some of the grafted cells became nestin negative. As these cells were found in the mesenchyme rather than in the spinal cord, loss of nestin may be indicative of differentiation along mesenchymal lineages, as the AFS cells we used could undergo skeletogenic differentiation (not shown). This is consistent with their extensive mesenchymal differentiation capability [9,36,37].
While our work was in progress, formation of EBs from amniotic fluid cells was reported [38]. That study did not provide any indication of the levels of expression of the 3 germ layer markers found in the EBs and did not assess the EB differentiation potential. As Valli et al., we could detect some markers of the 3 germ layers in EB-like spheres generated under either of the culture conditions used, but in most cases only when a very high cycle number was used in the RT-PCR studies, although expression of some of these markers was never observed in the AFS cell monolayers. However, there was no evidence that EB-like spheres could undergo terminal neural differentiation. Altogether, our work suggests that if any true reprogramming of rat AFS cells occurs spontaneously in a culture condition-dependent manner, this is a rare event and not sufficient to obtain overt differentiation along a neural lineage. Inconsistency in the results reported in the literature may reflect the presence of very small subsets of pluripotent cells, or their absence, in different AFS cell preparations. In contrast to AFS cell neural potential, differentiation along mesenchymal lineages is much more robust, and hence, it might be more valuable for certain clinical applications, for example, for intrauterine repair when damaged tissue/organs are identified prenatally and autologous cells from the amniotic fluid could be used.
AFS cells display a specific repair potential
Considering the size of injury induced in our model and the several tissues affected, the beneficial effect of grafted AFS cells is particularly striking. Interestingly, reduction of hemorrhage with desmopressin alone is not sufficient to reduce mortality, suggesting that the effect of AFS cells goes well beyond this initial response to injury and preservation of the vasculature.
It is clear that AFS cell integration/differentiation is not increased in the injury environment and cannot underlie improved recovery. Further, the effect of AFS cells on survival of operated chick embryos is an early effect, and it should be noted that later mortality seems to correlate with disappearance of injected cells at 5 days. As other cell types do not support survival, it is unlikely that the ability of AFS cells to decrease injury response is simply due to a “cement” effect that fills the gap created by the injury. The observed effect might be due to their ability to either increase survival within the injured tissues or boost endogenous repair mechanisms, or both. The extensive difference in tissue damage observed in live embryos with or without grafted AFS cells within the first 24 h after surgery suggests that the AFS cell graft plays an important trophic role. This is further supported by the great reduction in cell death and parallel increased maintenance of healthy neurones in spinal cords cocultured with AFS cells. This effect is clearly due to factors secreted by the AFS cells, as there is no direct contact between these cells and the spinal cord slices. The paracrine mechanism modulating the AFS cell-dependent survival effect in severe embryonic injury identified here is consistent with reports suggesting that amniotic fluid cells, particularly when transduced with growth factors, can facilitate peripheral nerve regeneration and wound healing [39 –43]. The identity of the factors involved in the increased survival observed both in vivo and in vitro has yet to be established. Several growth factors known to be expressed by AFS cells (eg, VEGF, Tgfβ) are also expressed by fibroblasts, which do not reduce cell death in our in vivo model. It is most likely that the AFS cell-induced effect reported here is due to specific combinations and levels of growth factors. A comparative analysis of secreted factors in AFS cells and cells that do not support survival following injury will have to be carried out to identify such “cocktail.”
Conclusions
Although we cannot rule out that differences in the literature are due to selective properties of different AFS cell lines, the overall difficulty encountered by many researchers in inducing AFS cell neural differentiation suggests that it is at the least premature to consider using these cells for cell therapies aimed at repopulating specific neural populations in the CNS. AFS cells, however, could be a very useful cell source for aiding recovery in injured tissues, given their ability to reduce tissue damage via paracrine mechanisms. They could provide a particularly useful tool for in-uterus repair of defects diagnosed during gestation, as autologous cells could be easily obtained. It will therefore be important to investigate the molecular basis underlying the effects reported here as this could lead to the development of novel therapeutic approaches for neural and nonneural tissue damage.
Footnotes
Acknowledgments
The authors thank the Office of the Commission on Higher Education of the Royal Thai Government, Thailand (Higher Educational Strategic Scholarship for Frontier Research to W.P.), Great Ormond Street Hospital Children's Charity and BBSRC for financial support and Bertrand Vernay for help with confocal imaging.
Author Disclosure Statement
The authors have no conflicting financial interests.
Appendix
| Primary antibody | Species | Specificity | Dilution | Source |
|---|---|---|---|---|
| Oct3/4 (c-10) | Mouse | IgG2b | 1:100 | Santa Cruz Biotechnology, Inc. |
| Nanog | Rabbit | Ig | 1:100 | Abcam |
| Sox2 | Rabbit | IgG | 1:1,000 | Millipore |
| c-Kit | Mouse | IgG | 1:100 | Santa Cruz Biotechnology, Inc. |
| c-Myc | Mouse | IgG1 | 1:100 | Developmental Study Hybrydoma Bank |
| SSEA1 | Mouse | IgM | 1:100 | Developmental Study Hybrydoma Bank |
| SSEA3 | Mouse | IgM | 1:100 | Developmental Study Hybrydoma Bank |
| SSEA4 | Mouse | IgG3 | 1:100 | Developmental Study Hybrydoma Bank |
| Nestin (Rat-401) | Mouse | IgG | 1:200 | Developmental Study Hybrydoma Bank |
| Vimentin | Mouse | IgG | 1:100 | DAKO |
| GFAP | Rabbit | IgG | 1:1,000 | Millipore |
| Pax6 | Mouse | IgG1 | 1:100 | DSHB |
| A2B5 | Mouse | IgM | 1:100 | R&D Systems |
| Tuj1 | Mouse | IgG | 1:1,000 | Promega |
| MAP2 (clone HM-2) | Mouse | IgG1 | 1:100 | Sigma |
| CRMP2 | Rabbit | Ig | 1:200 | Covalab |
| HuC/D | Mouse | IgG | 1:100 | Molecular Probe, Invitrogen |
| Dcx | Rabbit | IgG | 1:200 | Cell Signalling |
| O4 | mouse | IgM | 1:100 | R&D Systems |
| MBP | Rat | IgG | 1:100 | Millipore |
| HNK-1 | Mouse | IgG | 1:100 | Sigma |
| NGFR P75 | Rabbit | IgG | 1:500 | Sigma |
| VEGF (VG-1) | Mouse | IgG1 | 1:100 | Santa Cruz Biotechnology, Inc. |
| flk-1 | Rabbit | IgG | 1:400 | Fisher Scientific |
| TGF β1 | Rabbit | IgG | 1:100 | Santa Cruz Biotechnology, Inc. |
| TGF βIII | Rabbit | IgG | 1:100 | Santa Cruz Biotechnology, Inc. |
| vWF | Rabbit | Ig | 1:500 | Abcam |
| PH3 (ser10) | Rabbit | IgG | 1:200 | Millipore |
| Cit * H3 | Rabbit | Ig | 1:250 | Abcam |
| Iba-1 (C-20) | Goat | IgG | 1:100 | Santa Cruz Biotechnology, Inc. |
| Snail | Rabbit | IgG | 1:100 | Santa Cruz Biotechnology, Inc. |
| Runx2 | Rabbit | IgG | 1:100 | Santa Cruz Biotechnology, Inc. |
| GFP | Rabbit | IgG | 1:500 | Molecular Probe, Invitrogen |
| GFP | Mouse | IgG | 1:100 | Cell Signaling |
| CD11 | Mouse | IgG | 1:100 | Developmental Study Hybrydoma Bank |
| CD45 | Mouse | IgG1 | 1:100 | Developmental Study Hybrydoma Bank |
| Clusterin β (N-18) | Rabbit | IgG | 1:100 | Santa Cruz Biotechnology, Inc. |
| Patched | Rabbit | IgG | 1:100 | Santa Cruz Biotechnology, Inc. |
| Pax7 | Mouse | IgG1 | 1:100 | Developmental Study Hybrydoma Bank |
| Myosin | Mouse | IgG2a | 1:100 | Developmental Study Hybrydoma Bank |
| Myogenin | Mouse | IgG1 | 1:100 | Developmental Study Hybrydoma Bank |
| PEB2αA | Rabbit | IgG | 1:100 | Santa Cruz Biotechnology, Inc. |
| FGFR2 (BEK) | Rabbit | IgG | 1:100 | Santa Cruz Biotechnology, Inc. |
| Secondary antibody | Species | Type | Dilution | Source |
|---|---|---|---|---|
| Anti-mouse IgG1a Alexa Fluor® 568 | Goat | IgG1a | 1:400 | Molecular probe, Invitrogen |
| Anti-mouse IgM Alexa Fluor® 594 | Goat | IgM | 1:400 | Molecular probe, Invitrogen |
| Anti-rabbit IgG Alexa Fluor® 594 | Goat | IgG | 1:400 | Molecular probe, Invitrogen |
| Anti-mouse IgG Alexa Fluor® 488 | Goat | IgG | 1:400 | Molecular probe, Invitrogen |
| Anti-goat IgG Alexa Fluor® 594 | Rabbit | IgG | 1:400 | Molecular probe, Invitrogen |
Ig, immunoglobulin.
, citrullinated histone 3.