
Abstract
Embryonic stem cells (ESCs) are promising donor sources in cell therapies for various diseases. Although low levels of reactive oxygen species (ROS) are necessary for the maintenance of stem cells, increased ROS levels initiate differentiation and cell damage. We and others have previously demonstrated that heme oxygenase (HO)-1, a stress response protein with antioxidative and anti-inflammatory properties, plays critical protective functions in cardiovascular and other diseases. However, the functions of HO-1 in ESCs remain to be elucidated. Our goal was to investigate the roles of HO-1 in ESC survival and differentiation. Due to the lack of HO-1-deficient ESCs, we used Oct3/4, Sox2, c-Myc, and Klf4 retroviruses to reprogram mouse embryonic fibroblasts into induced pluripotent stem (iPS) cells of different HO-1 genotypes. These iPS-HO-1 cells exhibited characteristics of mouse ESCs (mESCs) and formed teratomas that were composed of cell types of all 3 germ layers after injected into severe combined immunodeficiency mice. In response to oxidant stress, iPS-HO-1–/– cells accumulated higher levels of intracellular ROS compared with D3 mESCs or iPS-HO-1+/+ cells and were more prone to oxidant-induced cell death. Spontaneous differentiation experiments revealed that Oct4 levels were significantly lower in iPS-HO-1–/– cells after leukemia inhibitory factor withdrawal and removal of feeders. Further, during the course of spontaneous differentiation, iPS-HO-1–/– cells had enhanced Erk1/2 phosphorylation, which has been linked to ESC differentiation. By the loss-of-function approach using iPS-HO-1–/– cells, our results demonstrate that a lack of HO-1 renders iPS cells more prone to oxidative stress-induced cell death and differentiation.
Introduction
P
Heme oxygenase-1 (HO-1), a stress response protein, is upregulated in response to various stimuli, including but not limited to oxidative and inflammatory stress [8]. HO-1 degrades the pro-oxidant heme and generates biologically active molecules such as carbon monoxide (antiproliferative and anti-inflammatory), biliverdin and bilirubin (potent antioxidants), and ferrous iron, which can induce ferritin expression for iron sequestration [8 –10]. With these unique properties, HO-1 can protect cells and tissues from pathological and oxidative stress-induced injury [8]. We and others have demonstrated a critical protective function of HO-1 in cardiovascular and other diseases [11 –17]. Trigona et al. reported that HO-1 is expressed in human ESCs [18]. In a mixed lymphocyte reaction where irradiated plate-bound human ESCs were incubated with responder peripheral blood mononuclear cells (PBMCs), the pharmacological inhibition of HO-1 activity in human ESCs increased PBMC proliferation against human ESCs, suggesting that HO-1 may have a role in modulating the recipient immune response to ESCs [18]. Given the importance of redox status in the maintenance of ESCs [2,6] and that the functions of HO-1 in ESCs have not been completely elucidated, the aim of this study was to investigate the role of HO-1 in ESC survival and differentiation.
The breakthrough that pluripotent ESC-like cells can be generated from somatic cells by introduction of defined transcription factors underscores the possibility of patient-specific cell therapy [19 –21]. Furthermore, the induced pluripotent stem (iPS) cell technology holds great potential for developing disease models. In this study, we reprogrammed mouse embryonic fibroblasts (MEFs) into iPS cells of different HO-1 genotypes. Our results showed that iPS cells which are deficient in HO-1 were more susceptible to oxidant-induced cell death than D3 ESCs or wild-type iPS cells. HO-1-deficient iPS cells tend to undergo spontaneous differentiation and, consequently, Oct4 levels were significantly lower than wild-type iPS cells or D3 ESCs. Our results demonstrated that HO-1 is critical for iPS cell survival and differentiation.
Materials and Methods
Cell culture
MEFs were isolated from embryonic day 13.5 embryos after removal of head and visceral tissues and cultured in Dulbecco's modified Eagle medium (DMEM) containing 10% fetal bovine serum (FBS; Thermo Scientific HyClone) as described [19]. Plat-E cells (kindly provided by Dr. Toshio Kitamura, Institute of Medical Science, University of Tokyo) were maintained in DMEM containing 10% FBS, 1 μg/mL puromycin, and 10 μg/mL blastocidin S as described [22]. D3 mouse ESCs (mESCs) [11] and mouse iPS-HO-1 cells were cultured on mitomycin C (Sigma-Aldrich)-treated MEF feeders in ESC medium [DMEM supplemented with 15% FBS, 3.7 g/L sodium bicarbonate, 100 units/mL penicillin, 100 μg/mL streptomycin, 1.7 mM L-glutamine, 0.1 mM β-mercaptoethanol, 0.1 mM nonessential amino acids, and 1,000 units/mL leukemia inhibitory factor (LIF; Millipore)]. All culture reagents were from Invitrogen unless otherwise stated.
HO-1 mice previously generated [11,12] were backcrossed to a C57BL/6 genetic background and maintained in a mixed 129sv/C57BL/6 background. MEFs of different HO-1 genotypes (wild-type HO-1+/+ , heterozygous HO-1+/– , and knockout HO-1–/– ) were isolated from day 13.5 embryos obtained from pregnant female HO-1+/– mice mated with male HO-1+/– mice. MEFs from individual embryo were initially cultured separately. Genotyping was performed on DNA samples isolated from tail of each embryo by polymerase chain reaction (PCR) as described [23]. After confirmation of genotypes, primary MEFs of the same genotype were then pooled.
Retroviral infection and generation of iPS-HO-1 cells
The pMXs-based retroviral vectors pMXs-c-Myc, pMXs-Oct3/4, pMXs-Sox2, and pMXs-Klf4 were obtained from Addgene and separately transfected using FuGENE 6 (Roche) into Plat-E packaging cells essentially as described [19]. Forty-eight hours after transfection, the virus-containing supernatant was used for retroviral infection. Mouse iPS cells of different HO-1 genotypes were generated according to the protocol described by Takahashi et al. [24]. Briefly, 5×105 MEFs of HO-1+/+ , HO-1+/– , and HO-1–/– were plated onto feeder cells in 100 mm dishes. After overnight incubation, the culture medium was replaced by an equal volume of retroviral medium supplemented with 4 μg/mL polybrene (Sigma). After 24 h of retroviral transduction, fresh MEF medium was replaced. After further incubation for another 48 h, the medium was changed to ESC medium. Embryonic stem (ES)-like colonies started to appear ∼7 days after the initial retroviral infection. Different genotypes of iPS-HO-1 colonies were picked 10–14 days after the retroviral infection and subsequently expanded.
Characterization of iPS-HO-1 cells
To confirm the genotypes of iPS-HO-1 cells, we performed western blot analysis to detect HO-1 expression. To measure HO activity, cell extracts of D3 and various iPS-HO-1 cells were prepared in a homogenization buffer (30 mM Tris, pH 7.5, 0.25 M sucrose, and 0.15 M NaCl) containing Complete™ protease inhibitor (Roche), microsomal fraction isolated, and HO enzymatic activity measured by bilirubin generation as described [25]. HO enzyme activity was expressed as nmoles of bilirubin formed per mg of protein per hour. To determine the growth rates of iPS-HO-1 cells, we measured the doubling time of D3 mESCs and various iPS-HO-1 clones of different genotypes. Cells were plated at a density of 1×104 cells per well in triplicate on 12-well plates, and cell numbers were counted every 24 h for 4 days. The doubling time was calculated with an online software (
Teratoma formation and histological analysis
D3 mESCs or iPS-HO-1 cells (1–2×106 cells) were injected subcutaneously into the middorsal intrascapular region of 6–8 week-old severe combined immunodeficiency (SCID) mice (Biolasco). Mice were followed for tumor formation. Tumors were dissected ∼4–6 weeks after the injection, fixed in formalin (Sigma), processed, and embedded in paraffin. Sections were stained with hematoxylin and eosin. All experimental procedures were approved by the Institutional Animal Care and Use Committee of the National Health Research Institutes, Taiwan.
Measurement of intracellular ROS content
D3 and iPS-HO-1 cells were plated on matrigel (BD Biosciences)-coated 96-well plates (3×104 cells/well) in triplicate and allowed to attach overnight. Cells were treated with 500 μM H2O2 for 1 h and then incubated with 20 μM 2′,7′-dichlorodihydrofluorescein diacetate (DCFDA; Invitrogen) for 30 min at 37°C. The intracellular ROS accumulation was measured by DCF fluorescence using Plate CHAMELEON™ microplate reader (Hidex), with excitation and emission wavelengths of 485 and 535 nm, respectively, as described [26,27].
Hydrogen peroxide-induced cytotoxicity
D3 mESCs and iPS-HO-1 cells (3×104 cells/well) were plated on matrigel-coated 96-well plates in phenol red-free media (Invitrogen) and allowed to attach overnight. Cells were treated with different concentrations of freshly diluted H2O2 (Sigma) for 24 h. Cell viability was determined using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (Sigma) assay as described [28].
Analysis of apoptotic cells by terminal deoxynucleotidyl transferase dUTP nick end labeling and flow cytometry
D3 ESCs and iPS-HO-1 cells were grown on feeders and treated with 1 mM H2O2 for 6 h. DNA breaks in apoptotic cells were detected with a TUNEL assay kit (Promega), and nuclei were counterstained with DAPI. Green staining in the nuclei indicated positive terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining. For flow cytometry, cells were treated with 1 mM H2O2 for 6 h, harvested, and incubated in PI staining solution for sorting. Flow cytometry analysis was performed using an FASCalibur flow cytometry system (BD Biosciences). Forward light scatter characteristics were used to exclude the cell debris from the analysis. The sub-G1 population was calculated as an estimate of the apoptotic cell population. The percentages of the cells within the G1, S, and G2/M phases of the cell cycle were determined by analysis with the program CellQust.
Spontaneous differentiation
Spontaneous differentiation experiments were performed by feeder removal and LIF withdrawal. Approximately 4×105 D3 mESCs or iPS-HO-1 cells were plated on 60 mm CellBIND® (Corning Incorporated) culture dishes without feeder in ESC medium without LIF. Total protein was then isolated at different time points after feeder removal and LIF withdrawal. ESC marker gene Oct4 expression and Erk1/2 phosphorylation were examined by western blot analysis. To measure mRNA levels of Oct4, total RNA was isolated from D3 and iPS-HO-1 cells using the RNeasy Mini RNA isolation kit (Qiagen), and 1 μg RNA was reverse transcribed to cDNA using the SuperScript III first-strand synthesis system (Invitrogen). Real time quantitative PCR was performed with the 7500 real-time PCR system (ABI) using KAPA™ SYBR® FAST qPCR kit (Kapa Biosystems) with specific Oct4 primer set (5′-TTGGGCTAGAGAAGGATGTGGTT-3′ and 5′-GGAAAAGGGACTGAGTAGAGTGTGG-3′) that had been used to identify Oct4 expression in pluripotent ESCs [29]. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was also amplified for normalization using the primer set (5′-CCTCCTGCACCACCAACT-3′ and 5′-GACCTTGCCCACAGCCTT-3′). Quantitation was performed by the comparative CT method, and Oct4 expression was normalized to GAPDH. To examine SSEA-1 expression, D3 and iPS-HO-1 cells were harvested at different time points, fixed, and incubated with SSEA-1 antibody (MAB4301, Millipore) for flow cytometry using an FACSCalibur (BD Biosciences). Data analysis was carried out using Cell Quest (BD Biosciences). To detect the expression of alkaline phosphatase of D3 and iPS-HO-1 cells at different time points during the course of spontaneous differentiation, we performed alkaline phosphatase activity staining assays with the alkaline phosphatase detection kit (Millipore).
Inhibition of HO activity and suppression of HO-1 expression
To inhibit HO activity, D3 mESCs were resuspended in medium (without LIF) containing vehicle dimethyl sulfoxide (DMSO) or 5 μM zinc protoporphyrin IX (ZnPP; an HO activity inhibitor, Frontier Scientific) and plated on CellBIND dishes for spontaneous differentiation experiments. Medium containing vehicle or ZnPP was changed after 2 days. Protein extracts were isolated at different time points for western blot analysis.
To suppress HO-1 mRNA levels in D3 ESCs, we performed HO-1 knockdown experiments with small interfering RNA (siRNA) against mouse HO-1. The ON-TARGET plus SMART pool HO-1 siRNA (5′-ACACAUCCAAGCCGAGAAU-3′, 5′- CCUCAC AGAUGGCGUCACU-3′, 5′- AAGAGGAGAUAGAGCG CAA-3′, 5′- AGAACUUUCAGAAGGGUCA-3′) and negative control siRNA were obtained from Dharmacon. D3 cells were transfected with negative control siRNA or HO-1 siRNA with Effectene transfection reagents (Qiagen) according to the manufacturer's instructions with slight modifications. Since transfection per se causes certain cell death to D3 ESCs, we used more cells for transfection experiments such that the cell density after plating following transfection would be similar to that of the routine plating for spontaneous differentiation. Briefly, 4 μg siRNA was mixed with buffer EC to a volume of 300 μL, to which 32 μL of enhancer was then added. After incubation at room temperature for 5 min, 20 μL Effectene was added to the siRNA/enhancer mixture. This transfection mixture was incubated at room temperature for 5–10 min. D3 ESCs (1.8×106) in 10 mL medium (without LIF) was subsequently added to the transfection mixture and plated on one 100 mm or two 60 mm CellBIND culture plates. The medium was changed next morning and again at day 2. Total protein was isolated at different time points for western blot analysis.
Western blot analysis
Total proteins from D3 mESCs or iPS-HO-1 cells were prepared by lysing cells in extraction buffer [25 mM Tris, pH 7.4, 150 mM NaCl, 0.5% Na deoxycholate, 2% Nonidet P-40, and 0.2% sodium dodecyl sulfate (SDS)] containing Complete™ protease inhibitor (Roche) and Halt Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific). Aliquots containing 20 μg of protein were subjected to SDS-polyacrylamide gel electrophoresis (PAGE) using 4%–15% Mini-PROTEAN® TGX™ precast gels (BIO-RAD) and then transferred to Immobilon-FL filters (Millipore), followed by immunoblotting. Blots were incubated with antibodies against HO-1 or HO-2 (Enzo Life Sciences) to detect HO-1 and HO-2 expression, respectively. To detect pluripotency marker Oct4 expression, blots were incubated with an Oct4 antibody (Oct3/4 (C-10): sc-5279; Santa Cruz Biotechnology), which does not cross react with Oct3/4 isoform B and has been commonly used to asses pluripotency of stem cells [19,30]. Membranes were processed with an enhanced chemiluminescence reagent (Millipore) and exposed to film. Blots were subsequently probed with α-tubulin (Sigma) or actin (Millipore) antibodies to verify equivalent loading. To assess Erk1/2 activation, membranes were incubated with rabbit anti-phospho-Erk1/2 (Cell Signaling Technology) and mouse anti-Erk1/2 (Cell Signaling Technology). After washing, membranes were incubated with IRDye 800CW goat anti-rabbit IgG secondary antibody (LI-COR Biosciences) and Alexa Fluor 680 goat anti-mouse IgG secondary antibody (Invitrogen) to detect Erk1/2 phosphorylation and total Erk1/2, respectively. The blots were scanned using Odyssey Infrared imaging system (LI-COR Biosciences).
Rescue experiments
To restore expression of HO-1 in iPS-HO-1–/– cells, we first generated an HO-1 expression plasmid pcDNA3.1-HO-1 by subcloning the 1-kb human HO-1 cDNA open reading frame [31] into the expression vector pcDNA3.1(−) (Invigtrogen). Since transfection per se causes certain cell death to iPS-HO-1–/– cells, we used more cells for transfection experiments such that the cell density after plating following transfection would be similar to that of the routine plating for spontaneous differentiation. iPS-HO-1–/– cells were transfected with vector control pcDNA3.1(−) or pcDNA3.1-HO-1 with Effectene transfection reagents (Qiagen) according to the manufacturer's instructions. Briefly, 2 μg plasmid DNA was mixed with buffer EC to a volume of 300 μL before 16 μL of enhancer was added. After 5 min incubation, 20 μL Effectene was added to the DNA-enhancer mixture. After incubation at room temperature for 5–10 min, iPS-HO-1–/– cells (1.8×106) in 10 mL medium (without LIF) was added to the transfection mixture and plated on one 100 mm or two 60 mm CellBIND culture plates. The medium was refreshed next morning and at day 2. Total protein was isolated at different days for western blot analysis.
To determine whether reaction products of HO-1 or antioxidant can overcome the effects due to HO-1 deficiency, iPS-HO-1–/– cells were treated with bilirubin, CO, or antioxidant N-acetyl-cysteine (NAC). Approximately 4×105 iPS-HO-1–/– cells were resuspended in medium (without LIF) containing vehicle DMSO or 2 μM bilirubin (Tokyo Chemical Industry) and plated on 60 mm CellBIND dishes for spontaneous differentiation experiments. Medium containing vehicle or bilirubin was changed after 2 days. The CO releasing molecule tricarbonyldichlororuthenium (II) dimer (CORM2, Sigma) has been widely used as a CO donor. After plating of iPS-HO-1–/– cells on CellBIND culture dishes in medium without LIF overnight, medium was refreshed daily with that containing 100 μM CORM2 or the control compound ruthenicum(III) chloride (RuCl3, Sigma). To determine the effect of NAC, cells were allowed to attach overnight, and the medium containing 1 μM NAC or vehicle H2O was refreshed daily. Protein extracts were isolated at day 4 for western blot analysis.
Statistical analysis
Data are presented as mean±standard error of the mean. Statistical analysis was performed by Student's t-test, and the statistical significance was accepted at P<0.05.
Results
HO-1 expression in mESCs
HO-1 protects many cell types and tissues against oxidative stress [8,10]. However, the role of HO-1 in ESCs has not been fully investigated. As a first step, we examined whether HO-1 was expressed in D3 mESCs that are often used for gene targeting experiments [11,32,33]. Western blot analysis revealed that D3 mESCs expressed a significant level of HO-1 compared with that of spleen, which has the highest level of endogenous expression among all organs, suggesting a potential role of HO-1 in ESCs (Fig. 1A).
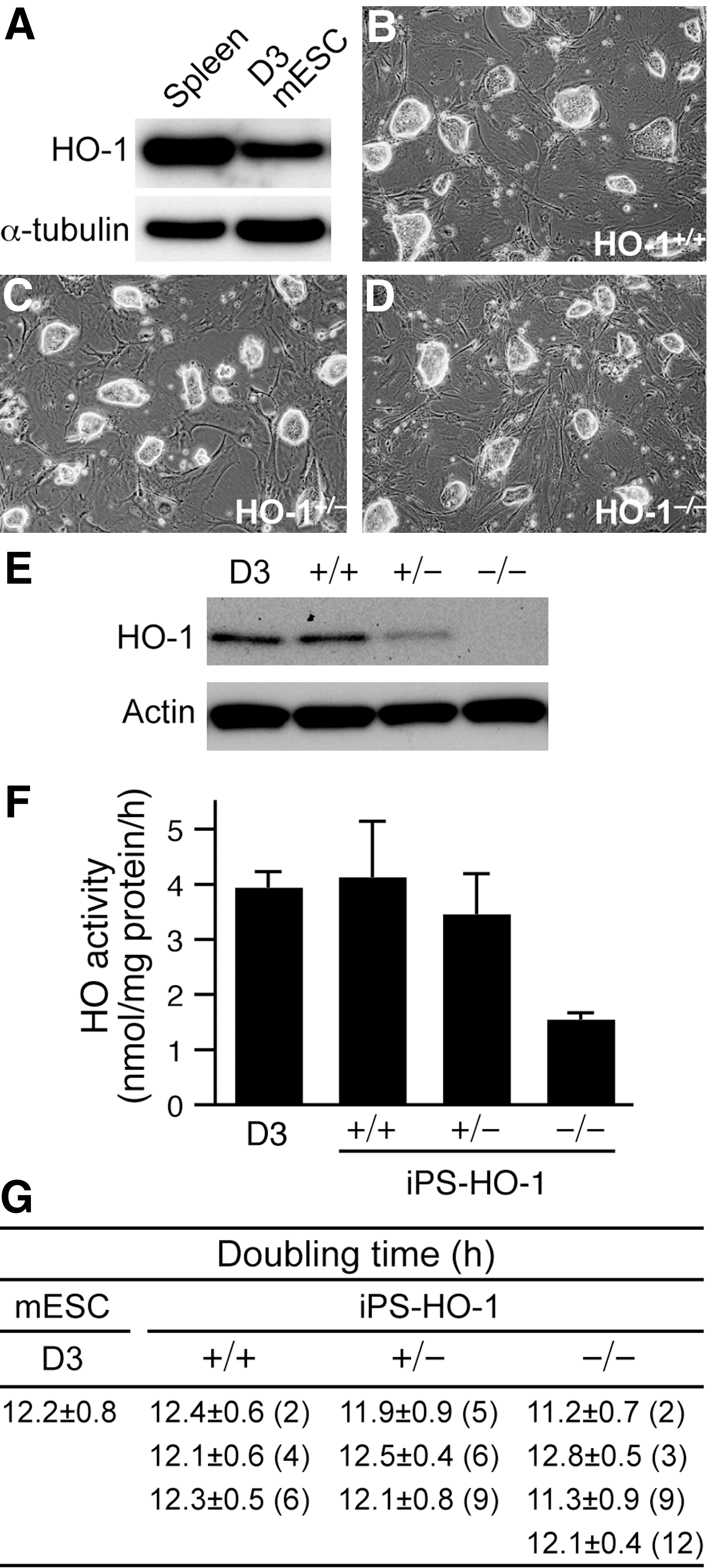
Generation and characterization of iPS-HO-1 cells.
Generation and characterization of iPS-HO-1 cells
We next wanted to use a genetic loss-of-function approach to investigate the role of HO-1 in ESCs; however, due to the lack of HO-1-deficient ESCs, we took advantage of the iPS technology [19]. By transduction of Oct4, Sox2, c-Myc, and Klf4 transcription factors into genetically modified fibroblasts, Takahashi et al. successfully isolated iPS cells by G418 selection [19]. Subsequently, Meissner et al. demonstrated that reprogrammed iPS cells could be isolated from genetically unmodified somatic cells solely based on morphological criteria [34]. Thus, we first isolated MEFs of different HO-1 genotypes and used retroviruses encoding the 4 transcription factors Oct4, Sox2, c-Myc, and Klf4 to reprogram wild-type (+/+), heterozygous (+/–), and knockout (–/–) MEFs into iPS-HO-1+/+ , iPS-HO-1+/– , and iPS-HO-1–/– cells, respectively, by morphology selection (Fig. 1B–D). The mESCs are characterized by large nuclei with scant cytoplasm and formation of colonies with shiny edges when grown on feeders. Consistent with these characteristics, the reprogrammed iPS-HO-1 cells also showed typical colony formation with shiny edges on feeders (Fig. 1B–D). The genotypes of these iPS cells were further confirmed with western blot analysis (Fig. 1E). The expression levels of HO-1 in iPS-HO-1+/+ were comparable to those of D3 mESCs and ∼2-fold of iPS-HO-1+/– (Fig. 1E). Importantly, no HO-1 expression was detectable in iPS-HO-1–/– cells (Fig. 1E). Next, we measured HO enzymatic activity in these cells (Fig. 1F). D3 and iPS-HO-1+/+ cells had comparable activity (3.9±0.3 and 4.1±1.0 nmol/mg protein/h, respectively), while iPS-HO-1+/– cells had slightly lower activity of 3.5±0.7 nmol/mg protein/h. In contrast, iPS-HO-1–/– cells exhibited much reduced activity (1.5±0.1 nmol/mg protein/h), which may represent the activity from HO-2. We then measured the doubling times of D3 and different iPS-HO-1 cells (Fig. 1G). As expected, D3 ESCs had a rather short doubling time of 12.2±0.8 h (Fig. 1G). In comparison, multiple clones of iPS-HO-1 cells with different HO-1 genotypes also had doubling times similar to those of D3 (Fig. 1G), further indicating the successful reprogramming of iPS-HO-1 cells.
Pluripotency and differentiation potential of iPS-HO-1 cells
The undifferentiated state of the mESCs is characterized by strong alkaline phosphatase activity and expression of ESC marker genes such as Oct4 and SSEA-1 [35]. Consistent with the morphology assessment, the iPS-HO-1 cells of all 3 genotypes possessed strong alkaline phosphatase activity (Fig. 2A–C, pink staining). Further, immunostaining revealed that iPS-HO-1 cells were strongly positive for both Oct4 (Fig. 2D–F, green) and SSEA-1 (Fig. 2G–I, red); the iPS colonies were revealed by DAPI staining (Fig. 2J–L, blue). These data indicated that the iPS-HO-1 cells indeed exhibited ESC characteristics. To further test in vivo the pluripotency of iPS-HO-1 cells, we used a teratoma formation model with immunodeficient SCID mice. Injection of D3 (as a positive control), iPS-HO-1+/+ , iPS-HO-1+/– , and iPS-HO-1–/– cells into SCID mice all resulted in teratoma formation. Histological analysis revealed that these teratomas were composed of different cell types from ectoderm, such as epidermis and neural tissue (Fig. 3A–C), mesoderm, such as cartilage, adipose, and muscle (Fig. 3D–F), and endoderm, such as GI-epithelium (Fig. 3G–I), demonstrating the pluripotency of the iPS-HO-1 cells we generated.
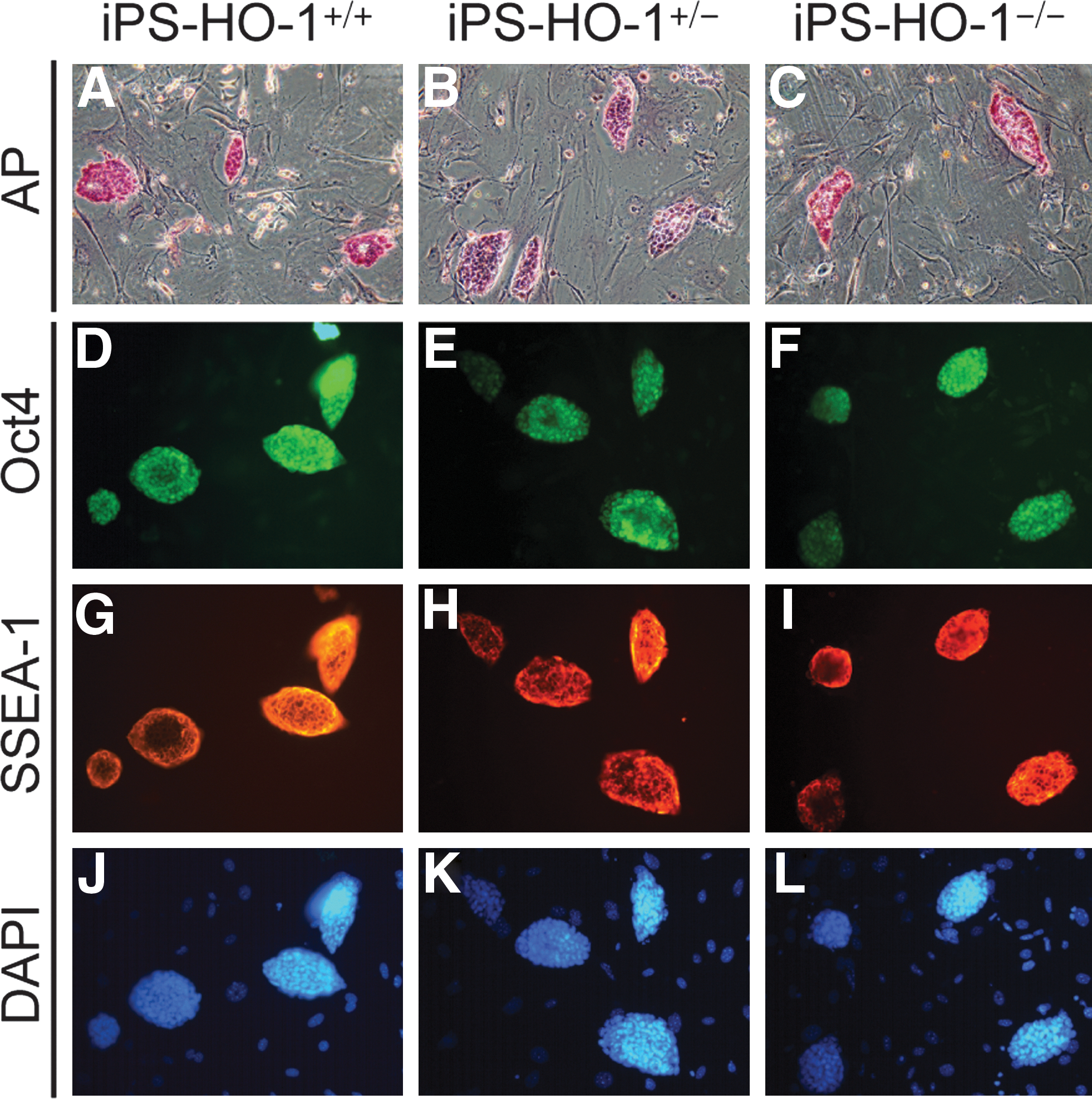
Expression of pluripotency markers in iPS-HO-1+/+
, iPS-HO-1+/–
, and iPS-HO-1–/–
cells.
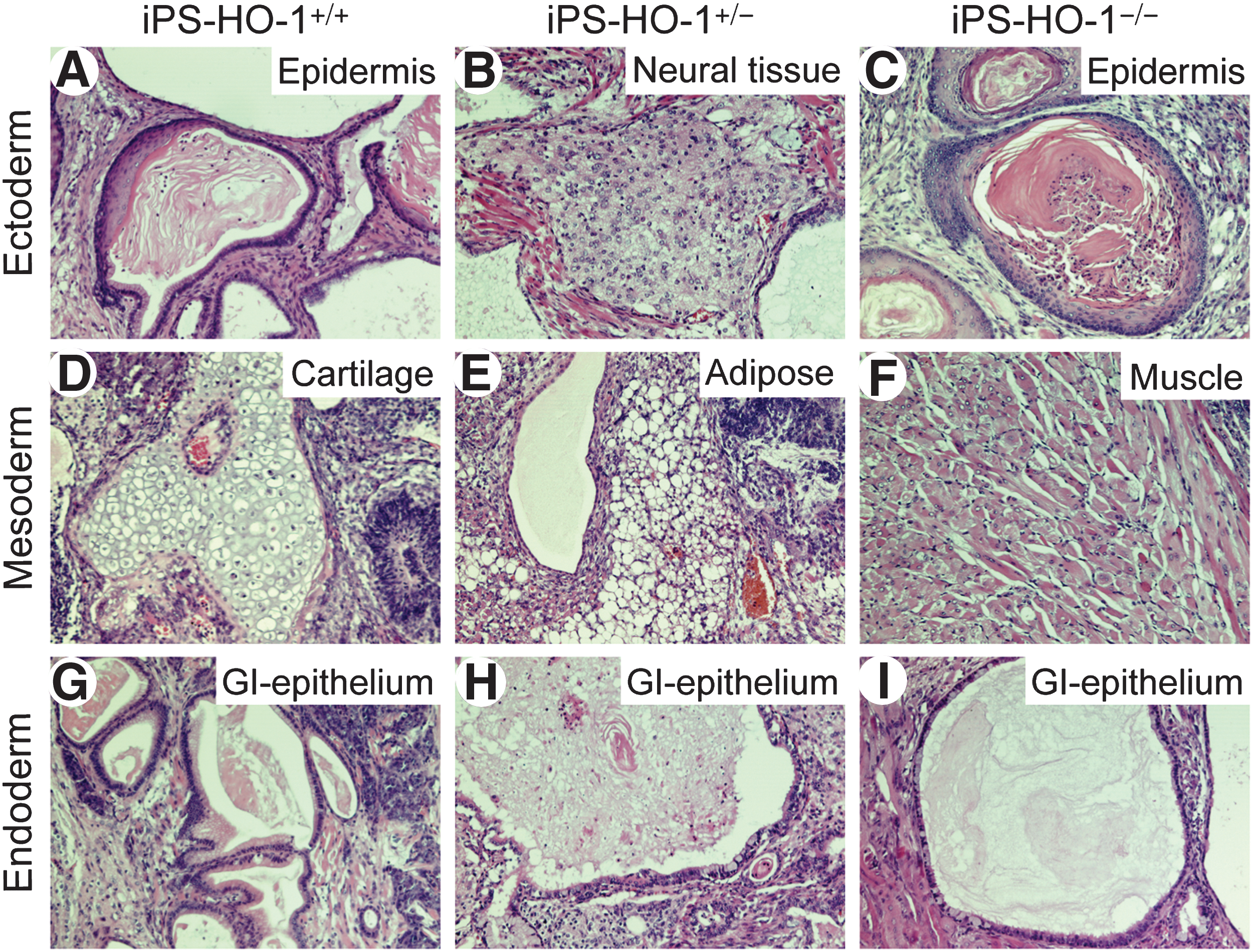
Teratoma formation of iPS-HO-1
+/+, iPS-HO-1+/–
, and iPS-HO-1–/–
cells. Different genotypes of iPS-HO-1 cells (1–2×106 cells) were injected subcutaneously into the middorsal intrascapular region of SCID mice. Teratomas were dissected from the mice ∼4–6 weeks after the injection, fixed, processed, and embedded in paraffin. Sections were stained with hematoxylin and eosin. Cell types of ectoderm were present in
Elevated intracellular ROS accumulation of iPS-HO-1–/– cells after oxidant treatment
In light of the antioxidative property of HO-1, we hypothesized that in response to oxidant stress, a lack of HO-1 may render ESCs more prone to accumulating intracellular ROS. To test this hypothesis, we treated D3 ESCs and iPS-HO-1 cells with hydrogen peroxide (H2O2) for 1 h and then evaluated their ROS levels by DCFDA. The intracellular ROS content of iPS-HO-1–/– cells was significantly higher than that of D3 ESCs (Fig. 4), while ROS content of iPS-HO-1 +/+ and iPS-HO-1+/– cells was comparable to that of D3 ESCs (Fig. 4). These results indicate that an HO-1 deficiency indeed leads to elevated ROS levels after oxidant stress.

Increased intracellular ROS accumulation in iPS-HO-1–/– cells. D3 ESCs and iPS-HO-1 cells were plated on 96-well plates in triplicate and allowed to attach overnight. Cells were then exposed to H2O2 for 1 h, and ROS accumulation was measured by 2′,7′-dichlorodihydrofluorescein diacetate ester flurorescence. Intracelluar ROS accumulation is expressed relative to D3 mESCs. Values are mean±SE of 4 experiments. *P<0.05 versus D3 mESCs. ROS, reactive oxygen species; DCF, 2′, 7′-dichlorofluorescin fluorescence.
iPS-HO-1–/– cells are more susceptible to oxidative stress
Given that excessive ROS results in oxidative damage and cell death [36] and that iPS-HO-1–/– cells had enhanced ROS levels in response to oxidant stress (Fig. 4), we next wanted to determine whether iPS-HO-1–/– cells are more susceptible to oxidant-induced cell death by treating the cells with H2O2. Viability assays after 24 h treatment revealed that both D3 and iPS-HO-1+/+ cells tolerated H2O2 concentrations up to 400 μM, with only a slight decrease in viability (Fig. 5). Although iPS-HO-1+/– cells had a small decrease in viability at 400 μM, it did not reach statistical significance (Fig. 5). In comparison, the viability of iPS-HO-1–/– cells decreased to 60% and 50% at 200 and 400 μM, respectively (Fig. 5). At a higher concentration of 600 μM, only ∼25% of iPS-HO-1–/– cells were viable, whereas ∼50%–60% of D3, iPS-HO-1+/+ , and iPS-HO-1+/– cells survived (Fig. 5). These data suggested that iPS-HO-1–/– cells were more susceptible to oxidant H2O2-induced cell death in comparison with D3 ESCs, iPS-HO-1+/+ , or iPS-HO-1+/– cells. To investigate the mechanisms leading to increased cell death of iPS-HO-1–/– cells, we performed TUNEL assays to detect the presence of DNA fragmentation and apoptosis after 6 h of H2O2 treatment. Given that iPS-HO-1+/– cells responded similarly as iPS-HO-1+/+ cells to oxidant stress in ROS accumulation and viability, we did not include iPS-HO-1+/– cells for the next apoptosis experiments. Some TUNEL-positive cells were detectable in D3 or iPS-HO-1+/+ cells (Fig. 6A, green). In iPS-HO-1–/– cells, however, a significant number of TUNEL-positive cells were detected (Fig. 6A, green). To assess apoptotic cells in a quantitative manner, we used flow cytometry to sort cells of the cell cycle based on DNA contents after staining with PI. The results showed that before H2O2 treatment, ∼3% of cells were apoptotic from all genotypes (Fig. 6B, C), indicating that under normal conditions, an absence of HO-1 did not increase cellular apoptosis. As expected, H2O2 treatment induced an increase in apoptosis of D3 and iPS-HO-1+/+ cells to ∼9% (Fig. 6B, C). In contrast, apoptosis of iPS-HO-1–/– cells was further increased to 21%±4.7% (Fig. 6B, C), indicating that increased apoptosis contributed to exaggerated oxidant-induced cell death in iPS-HO-1–/– Cells.
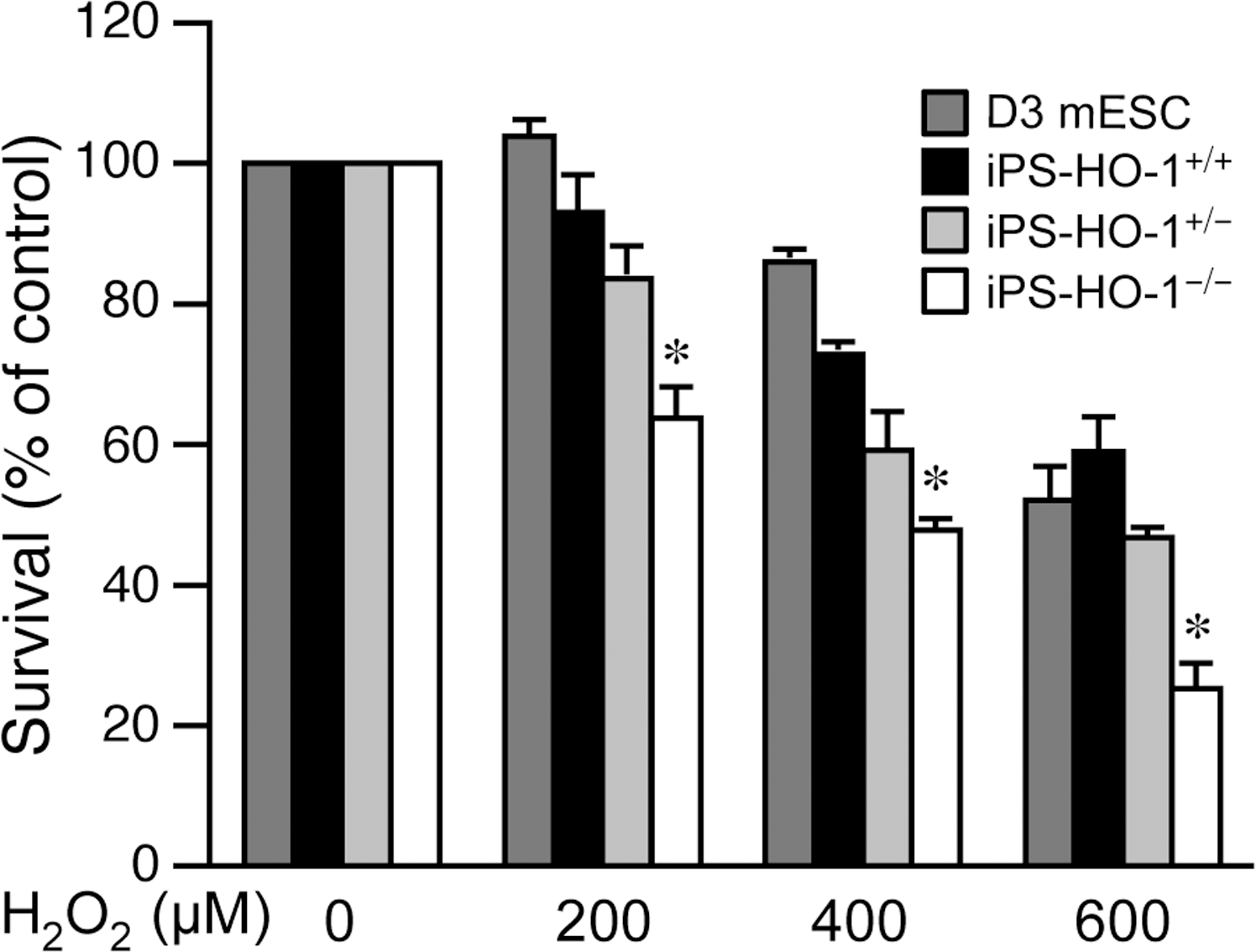
Reduced viability of iPS-HO-1–/– cells in response to oxidant stimulation. D3 mESCs (dark gray bars), iPS-HO-1+/+ (black bars), iPS-HO-1+/– (light gray bars), and iPS-HO-1–/– (white bars) cells were treated with increasing concentrations of H2O2 for 24 h, and cell viability was measured using MTT assays. Cell survival is expressed relative to that without H2O2 treatment. Values are mean±SE of 4 to 5 experiments. *P<0.05 versus D3 mESCs exposed to the same concentration of H2O2. MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.

Increased apoptosis of iPS-HO-1–/–
cells after H2O2 treatment. D3 mESCs, iPS-HO-1+/+
, and iPS-HO-1–/–
cells were treated with H2O2 for 6 h, and TUNEL assays were performed to detect apoptotic cells.
Lack of HO-1 accelerates ESC differentiation
Given that ROS could facilitate differentiation of ESCs and progenitor cells [5,37], we hypothesized that a lack of HO-1 may render ESCs more prone to differentiation. To test this hypothesis, we performed spontaneous differentiation experiments by LIF withdrawal and removal of feeders and assessed differentiation status by expression levels of the ESC marker gene Oct4. Before LIF withdrawal, Oct4 protein levels in iPS-HO-1–/– cells were comparable to those of D3 and iPS-HO-1+/+ cells (Fig. 7A). Two days after LIF withdrawal, Oct4 levels decreased slightly (∼10%) to those of control levels in D3, iPS-HO-1+/+ , and iPS-HO-1–/– cells (Fig. 7A). Interestingly, 4 days after LIF withdrawal, Oct4 levels in iPS-HO-1 −/− cells were significantly lower than those in D3 or iPS-HO-1+/+ cells (Fig. 7A). To further assess Oct4 expression at mRNA levels, we performed real-time PCR experiments with specific primers for Oct4. As with protein levels, 2 days after LIF withdrawal, Oct4 mRNA levels decreased slightly in D3, iPS-HO-1+/+ , iPS-HO-1+/– , and iPS-HO-1–/– cells when compared with those of d0 (Fig. 7B). Consistent with the findings from protein expression, 4 days after LIF withdrawal, Oct4 mRNA levels in iPS-HO-1–/– cells were significantly decreased in comparison with D3, iPS-HO-1+/+ , or iPS-HO-1+/– cells (Fig. 7B).

Lack of HO-1 promotes ESC differentiation during spontaneous differentiation. D3 mESCs, iPS-HO-1+/+
, and iPS-HO-1–/–
cells were plated on culture dishes without feeder and cultured in ESC medium without LIF. Total proteins were isolated before plating (d0) and at 2 (d2) and 4 days (d4) after plating.
Flow cytometry analysis with SSEA-1 antibody revealed that before plating for spontaneous differentiation experiments, more than 95% of cells of all genotypes were SSEA-1 positive (Fig. 7C, d0). SSEA-1 positive cells were slightly decreased in all cells 2 days after LIF withdrawal (Fig. 7C). At day 4, ∼80% of cells were SSEA-1 positive in D3, iPS-HO-1+/+ , or iPS-HO-1+/– cells, while only ∼50% of iPS-HO-1–/– cells were SSEA-1 positive (Fig. 7C). Moreover, strong alkaline phosphatase activity was detected in all genotypes of cells at d0 before plating for differentiation experiments as indicated by intense pink staining of ES colonies (Fig. 8, d0). One day after plating, D3, iPS-HO-1+/+ , iPS-HO-1+/– , or iPS-HO-1–/– cells retained strong alkaline phosphatase activity (Fig. 8, d1). As the cells continued to proliferate and committed toward differentiation, the intensity of pink staining began to decrease at day 2 and 3, and almost diminished at day 4 in D3, iPS-HO-1+/+ , and iPS-HO-1+/– cells (Fig. 8). In comparison, iPS-HO-1–/– cells had an accelerated decline in pink staining at day 3 (Fig. 8). Taken together, the results from Oct4 protein and mRNA expression, SSEA-1 flow cytometry, and alkaline phosphatase activity strongly suggested a role of HO-1 in protecting ESCs against differentiation.

HO-1 deficiency accelerates decline of alkaline phosphatase activity during spontaneous differentiation. Alkaline phosphatase activity was determined during the course of spontaneous differentiation in D3, iPS-HO-1+/+
, iPS-HO-1+/–
, and iPS-HO-1–/–
cells by an activity staining kit. The intensity of pink color indicates the extent of alkaline phosphatase activity. The activity staining of D3, iPS-HO-1+/+
, iPS-HO-1+/–
, and iPS-HO-1–/–
cells before plating at d0, d1, d2, d3, and d4 are shown. Data are representative of 3 independent experiments. Color images available online at
To further confirm our findings, we took a pharmacological approach. Although ZnPP may have nonspecific effects, ZnPP has been commonly used to inhibit HO activity. Two days of ZnPP treatment did not significantly alter Oct4 levels of D3 ESCs when compared with that of day 0 or vehicle controls (Fig. 9A). In comparison to day 0 controls, although there was a decrease of Oct4 levels at day 4 in vehicle-treated cells (45%±5%), ZnPP further reduced Oct4 levels to 29%±5%. These data indicated that consistent with the results from iPS-HO-1–/– cells, pharmacological inhibition of HO activity also resulted in accelerated differentiation of ESCs. Since ZnPP can induce HO-1 expression [38], we examined HO-1 protein levels after ZnPP treatment. Interestingly, HO-1 was substantially induced at day 2 in vehicle controls, and ZnPP treatment further increased the induction (Fig. 9A). In comparison, at day 4, HO-1 expression (either with or without ZnPP treatment) returned to the levels similar to that at day 0 (Fig. 9A). These results suggested that withdrawal of LIF and feeder might cause stress to ESCs, and HO-1 induction may serve as an endogenous adaptive mechanism to protect against stress. Furthermore, although ZnPP did increase HO-1 expression (at d2), its inhibition of HO activity resulted in accelerated differentiation of ESCs.
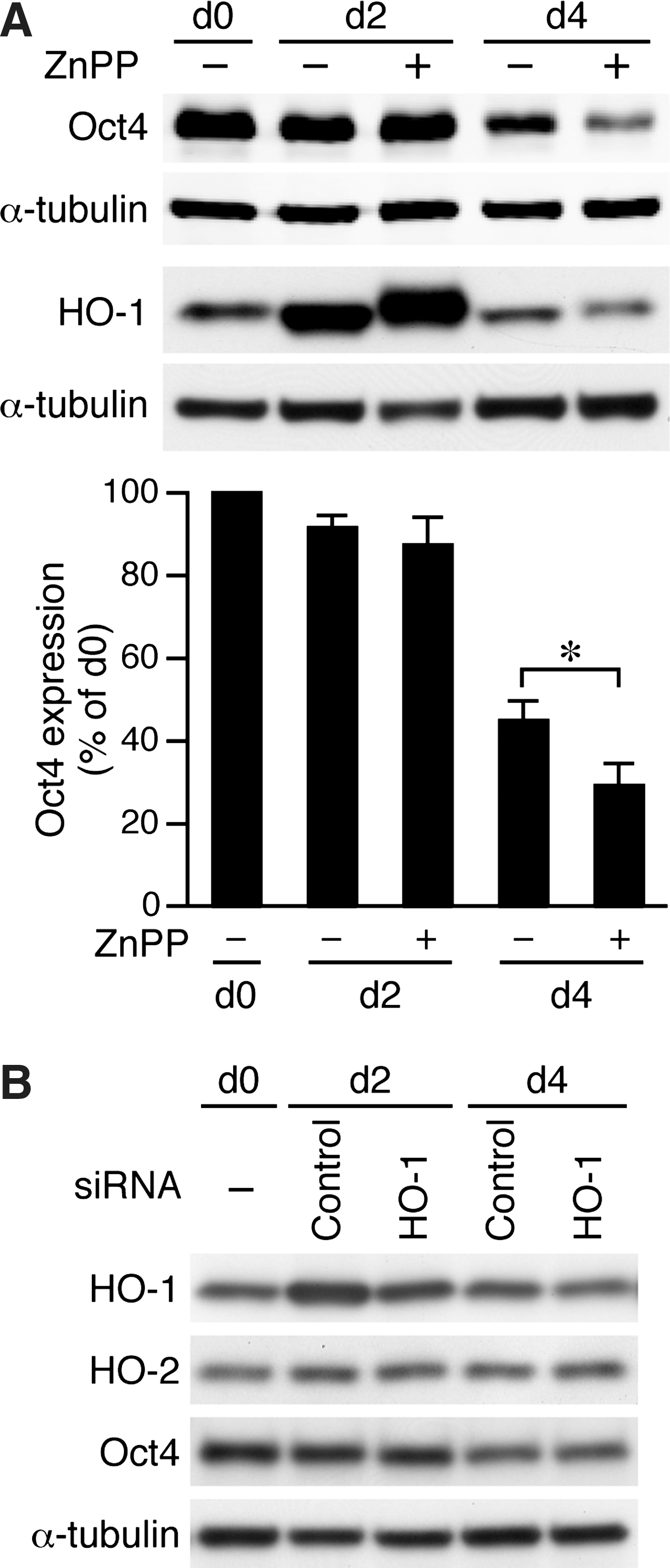
Inhibition of HO activity reduces Oct4 expression during spontaneous differentiation.
Next, in an attempt to differentiate the effects of HO-1 and HO-2 in a nongenetic approach, we transfected D3 ESCs with HO-1 or contorl siRNA before plating for differentiation experiments. Given that there was a substantial induction of HO-1 at day 2 (Fig. 9A) and that transfection with higher amount of siRNA resulted in significant cell death, we were only able to suppress ∼35% of HO-1 expression with HO-1 siRNA at day 2 and marginally at day 4 when compared with control siRNA (Fig. 9B). The specificity of HO-1 knockdown was further supported by that HO-2 levels were not affected (Fig. 9B). At this condition, we did not observe a reduction in Oct4 protein levels by HO-1 siRNA at either day 2 or 4 when compared with controls (Fig. 9B). This might be due to the fact that cells continued to proliferate after plating (see also Fig. 8) and, thus, diluting the knockdown effect of those cells transfected with HO-1 siRNA. Additionally, these results suggested that the remaining HO-1 level after siRNA knockdown was still sufficient to maintain Oct4 levels and further emphasized the importance of genetic loss-of-function experiments.
Rescue of Oct4 expression in iPS-HO-1–/– cells during differentiation
To determine whether restoring HO-1 expression in iPS-HO-1–/– cells can reverse the decrease of Oct4 expression during differentiation, we transfected iPS-HO-1–/– cells with an HO-1 expression plasmid or the control vector and performed differentiation experiments. Western blot analysis revealed that compared with vector, transfection with HO-1 expression plasmid restored HO-1 expression at day 2 and a consistent ∼30% increase of Oct4 (Fig. 10A), further supporting a critical role of HO-1 in maintaining Oct4 levels. In comparison, HO-1 expression was not detectable at day 4 and Oct4 level was similar to that of the vector control (Fig. 10A). We reasoned that as with siRNA transfection, cells continued to proliferate after plating and thus diluting the effect of those cells expressing HO-1 at day 4. Alternatively, due to the nature of transient transfection, those cells transfected with HO-1 plasmid might have lost the expression by day 4.
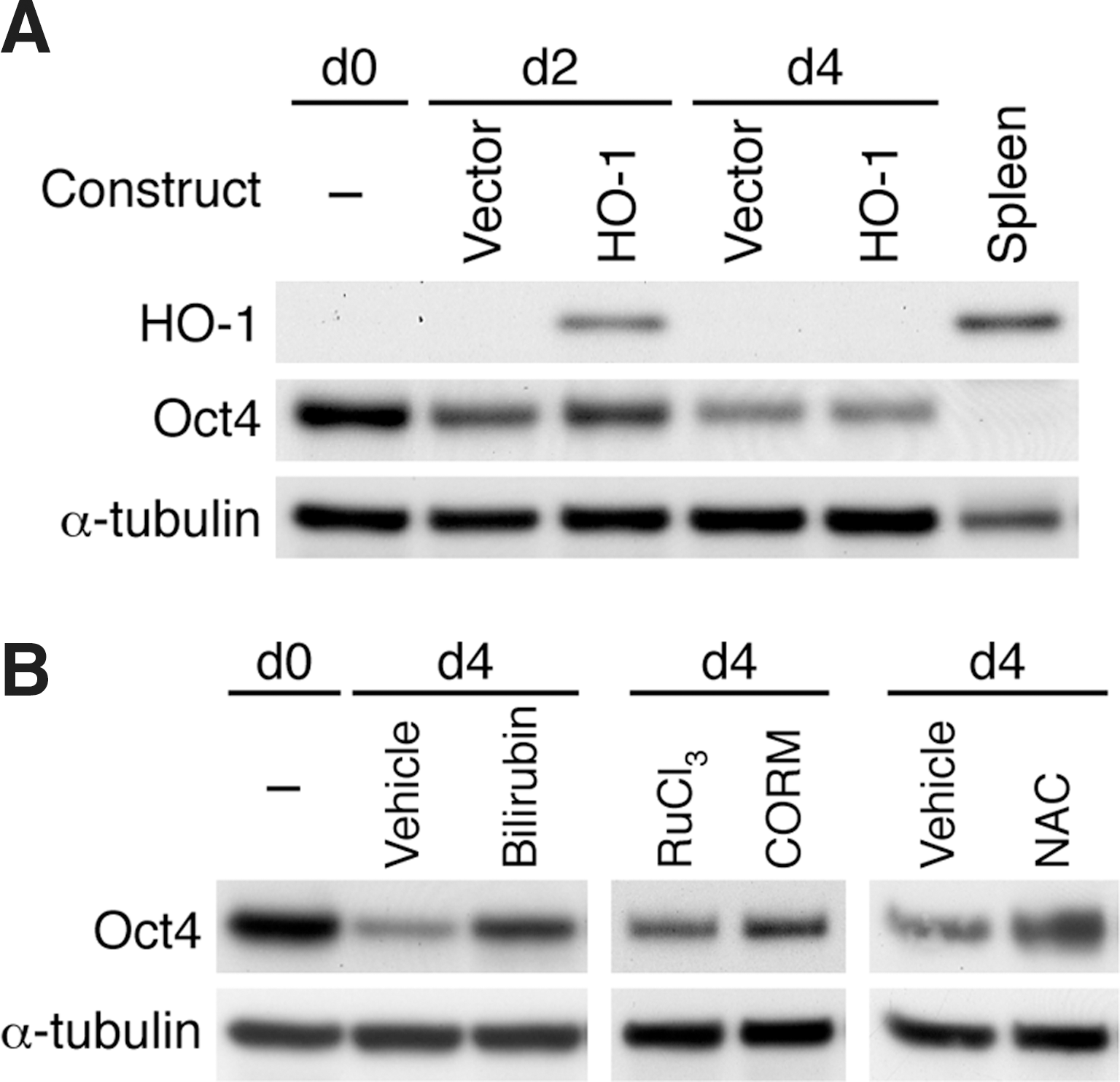
Rescue of Oct4 expression in iPS-HO-1–/–
cells during differentiation.
We next set out to investigate whether the reaction products of HO-1 can rescue Oct4 expression. To this end, we first treated iPS-HO-1–/– cells with bilirubin and examined Oct4 expression at day 4. Compared with vehicle DMSO, 2 μM of bilirubin increased Oct4 expression (Fig. 10B), while higher concentrations of bilirubin caused cytotoxicity (data not shown). Treatment of CO releasing molecule CORM2 also increased Oct4 levels when compared with control compound RuCl3 (Fig. 10B). These data indicated that the reaction products of HO-1 enzymatic activity bilirubin and CO both contributed to maintaining Oct4 expression during differentiation. Since HO-1 has an antioxidant property, we then tested whether antioxidant NAC can prevent the loss of Oct4. Interestingly, treatment of iPS-HO-1–/– cells with 1 μM NAC was sufficient to reverse the decline of Oct4 (Fig. 10B).
Enhanced Erk1/2 activation after LIF withdrawal in the absence of HO-1
Erk1/2 signaling has been linked to ESC differentiation [39 –41]. To investigate the mechanisms by which HO-1 modulates differentiation, we, thus, examined Erk1/2 activation during the course of spontaneous differentiation. Significant levels of Erk1/2 were phosphorylated in D3, iPS-HO-1+/+ , and iPS-HO-1–/– cells before differentiation (Fig. 11A). One day after LIF withdrawal and feeder removal, there was a substantial drop in Erk1/2 activation in all 3 lines of cells. After initial drop at day 1, phospho-Erk1/2 levels increased at day 2 and 3 before a drop again at day 4 (Fig. 11). The observations that an initial drop and a subsequent increase of Erk1/2 activation are consistent with previous findings during neural differentiation of mESCs [40]. Notably, the reactivation of Erk1/2 at day 2 and 3 in iPS-HO-1–/– cells was more enhanced than that in D3 or iPS-HO-1+/+ cells (Fig. 11A). Erk1/2 phosphorylation levels increased to 60%–65% that of d0 in iPS-HO-1–/– cells compared with ∼35% in D3 or iPS-HO-1 ++ cells (Fig. 11B). Intriguingly, Oct4 levels were substantially lower in iPS-HO-1–/– than D3 or iPS-HO-1+/+ cells at day 4 (Fig. 11A), indicating the preceding higher Erk1/2 activation might be a requisite for accelerated Oct4 reduction in iPS-HO-1–/– cells. Interestingly, phospho-Erk1/2 levels were reduced at day 4 to ∼15% of d0 (Fig. 11B), suggesting that at this stage most cells might have committed to differentiation and, thus, no longer require Erk1/2 activation. Taken together, our results suggested that an absence of HO-1 renders iPS cells more prone to lose pluripotency, which might be due to enhanced Erk1/2 signaling.
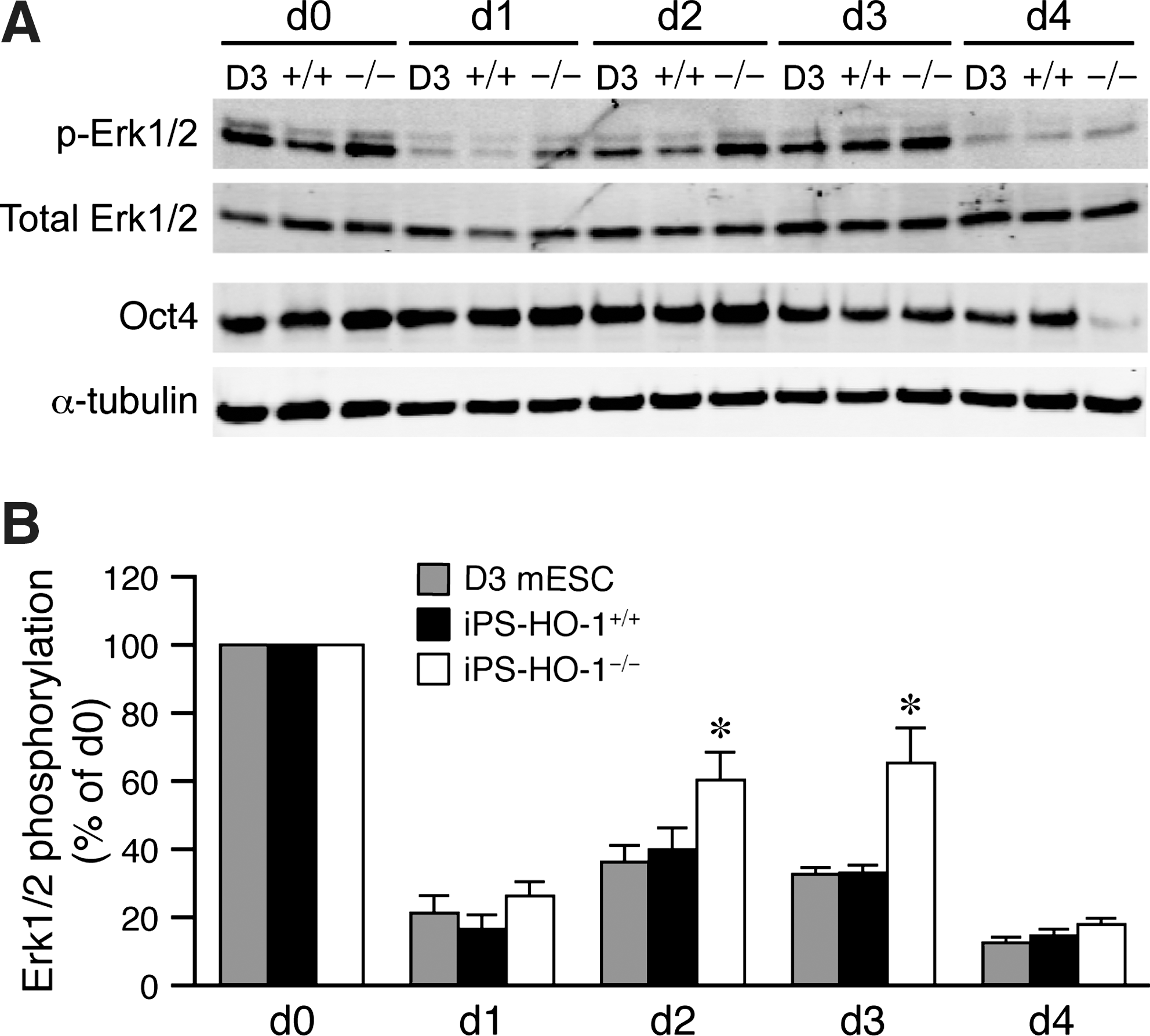
Enhanced Erk1/2 activation after LIF and feeder withdrawal in the absence of HO-1. D3 mESCs, iPS-HO-1+/+
, and iPS-HO-1–/–
cells were plated on culture dishes without feeder in ESC medium without LIF. Total proteins were isolated before plating (d0) and every 24 h after plating for 4 days.
Discussion
In the current study, we showed that HO-1 played an important role in protecting iPS cells from oxidant stress-induced cell death and differentiation. We took advantage of iPS technology and generated iPS-HO-1–/– cells that exhibited characteristic ESC morphology, marker gene expression, and in vivo pluripotency. Importantly, in a loss-of-function approach using iPS-HO-1–/– cells, we demonstrated that an absence of HO-1 rendered iPS cells more susceptible to oxidant stress-induced apoptosis and cell death. In addition, in the course of spontaneous differentiation iPS-HO-1–/– cells were more prone to lose pluripotency than D3 ESCs or iPS-HO-1+/+ cells, which might be due to enhanced Erk1/2 activation.
With the advent of iPS technology that enables the conversion of somatic cells back to pluripotent ESC-like cells, it opens up new avenues for patient-specific regenerative medicine and cellular models for studying disease mechanisms. Several iPS cell lines with genetic mutations have been generated [42,43]; of these, Rett syndrome iPS cells-derived neurons were used to investigate the cellular and molecular mechanisms underlying the abnormalities of the disease [43]. By gain- and loss-of-function experiments, we and others have demonstrated that HO-1 plays an essential role in maintaining cellular homeostasis and protecting cardiovascular and other tissues against pathological stress [11 –17]. Importantly, the critical role of HO-1 in diseases was highlighted in 2 cases of human HO-1 deficiency. Both patients had many clinical symptoms and died at young ages, 6- and 15-year-old, respectively [44 –46]. In light of the crucial role of HO-1 in many cell types and the minimal knowledge regarding the role of HO-1 in ESCs, we used iPS technology to investigate the function of HO-1 in ESCs.
In response to oxidant stress, iPS-HO-1–/– cells accumulated higher levels of intracellular ROS than D3 ESCs or iPS-HO-1+/+ cells (Fig. 4) and were more susceptible to oxidant-induced cell death (Fig. 5), which was in part mediated by increased apoptosis (Fig. 6). These protective properties are also seen in several cell types, including cardiomyocytes, vascular smooth muscle cells, and endothelial cells [11,12,47]. These somatic cells express little HO-1 under normal conditions; however, in response to stress, HO-1 is induced to exert its protective function. In contrast, HO-1 is significantly expressed in mouse (Fig. 1A) and human ESCs [18]. The high endogenous expressions of HO-1 further emphasize the critical importance of HO-1 in ESCs. Another major finding in our study is that an absence of HO-1, either genetically or pharmacologically, accelerated ESC differentiation on LIF and feeder withdrawal (Fig. 7). This finding is consistent with the idea that ROS could facilitate differentiation of ESCs and progenitor cells [5,37] and that iPS-HO-1–/– cells have a tendency to accumulate ROS under stress conditions (Fig. 4). On the other hand, billirubin, the reaction product of HO-1 activity with antioxidative properties, can rescue the accelerated loss of Oct4 in iPS-HO-1–/– cells (Fig. 10B). The antioxidant protection exerted by HO-1 in iPS cells was further supported by the findings that antioxidant NAC can reverse the loss of Oct4 in iPS-HO-1–/– cells (Fig. 10B). Another HO-1 reaction product CO can also overcome the accelerated loss of Oct4 (Fig. 10B). CO has anti-inflammatory and anti-proliferative properties; however, the mechanisms by which CO rescues Oct4 expression require further investigation. Although the molecular mechanisms underlying the accelerated differentiation of iPS-HO-1–/– cells are not yet clear; intriguingly, compared with D3 ESCs or iPS-HO-1+/+ cells, Erk1/2 phosphorylation was enhanced in iPS-HO-1–/– cells preceding a significant reduction of Oct4 after LIF and feeder withdrawal (Fig. 8). Supporting our findings, Erk1/2 activation has been linked to ESC differentiation [39 –41].
Interestingly, despite the importance of HO-1 in protecting ESCs against stress and differentiation, it is not required during reprogramming processes, because we were able to reprogram HO-1–/– fibroblasts into iPS-HO-1–/– cells (Fig. 1D–F). Further, HO-1 is not necessary for the maintenance of pluripotency under normal conditions either in vitro or in vivo (Figs. 2–3). The fact that homozygous HO-1-deficient mice and humans can be born and viable (although with a low survival rate in mice and rare cases in humans) further supports this notion [11,44,46]. Taken together, it is conceivable that although HO-1 is not required for pluripotency maintenance and reprogramming under basal conditions, it is essential for the adaptation of ESCs to pathophysiological stress.
In conclusion, we demonstrated that an absence of HO-1 renders iPS cells more sensitive to oxidative stress-induced apoptosis and cell death. Furthermore, in response to stress, HO-1-deficiencient iPS cells were more prone to lose pluripotency resulting in an accelerated differentiation, which might be due to enhanced Erk1/2 activation. More importantly, our findings may provide a new strategy in maintaining ESC survival and renewal by protecting ESCs against pathophysiological stress.
Footnotes
Acknowledgments
This work was supported in part by National Health Research Institutes (Taiwan) grant CS-100-PP-04 (to S.-F.Y.), National Science Council (Taiwan) grants NSC 98-3111-B-400-005 and NSC 99-3111-B-400-004 (to S.-F.Y.), NSC 99-3111-B-400-002 (to B.L.Y.), and NSC 99-3111-B-400-005 (to K.K.W.). This research was conducted in part under the Graduate Program of Biotechnology in Medicine sponsored by the National Tsing Hua University and National Health Research Institutes.
Author Disclosure Statement
No competing financial interests exist.