
Abstract
The transplantation of stem cells offers potential therapies for many neurodegenerative disorders that currently have limited or no treatment options. However, relatively little is known about how the host environment affects the development and integration of these cells. In this study we have engrafted immortalized human midbrain neural progenitor cells (NPCs) onto rat hippocampal brain slice cultures to examine the influence of a neural environment on differentiation. Patch clamp recordings revealed that the transplanted progenitor cells could express neuronal-type voltage-gated currents and rapidly receive synaptic input from the hippocampal brain slice. The distribution of progenitor cells across the hippocampal slices was strongly influenced by the neural architecture, with most cells located in the fissural regions and sending processes parallel to the laminar structure, while in contrast, cells located in the dentate gyrus showed no organized pattern. Almost no cells were found in the stratum radiatum or pyramidal cell layers. Together, these results demonstrate the potential for the architecture of the host environment to regulate the integration of transplanted cells, and highlight the utility of coculture systems for studying the mechanisms underlying the migration, integration, and differentiation of human NPCs in structured neural environments.
Introduction
T
For this purpose cell cultures are limited by their inability to recreate the complexity and signaling properties of this environment. Cell–cell interactions, diffusible factors, and the extracellular matrix (ECM) all regulate multiple signaling pathways during development [1,2], and variation in these signals between cell culture and in vivo conditions may alter the development of NS/PCs; indeed, the microenvironment is a key regulator of development in adult stem cell niches [3]. Neural activity is also in a position to influence NS/PC differentiation, as neurotransmitters can do so in cell cultures and during adult neurogenesis [4,5]. Furthermore, excitation is an important factor in neuronal survival [6,7], and synaptic integration into the host brain maybe required if NS/PC-derived neurons are to survive in large numbers.
Organotypic slice cultures provide a model neural environment while maintaining several of the advantages of working in vitro: controllable environment, ease of manipulation, high throughput, and accessibility for time lapse studies. The coculture system does not replicate in vivo transplantation per se, as cultures must be made using embryonic or postnatal tissue, at which times the nervous system is still developing, but many of the basic features, such as neural networks, glia, and a structured environment, are present. It has previously been demonstrated that when seeded onto organotypic hippocampal slice cultures, mouse embryonic stem cells can differentiate into functional neurons that receive synaptic input [8] and into glia [9,10]. Expression of neuronal markers has also been induced in mouse bone marrow stromal cells [11] and human cord blood–derived stem cells [12]; however, neural progenitor cells (NPCs) derived from adult rats predominantly differentiated into astrocytes when cocultured with hippocampal slices [13]. It is clear that the responsiveness to environmental cues varies between cell types, and as yet it is not clear how such environmental conditions might influence human cells. Here, we have engrafted cells of a human fetal NPC line (hNPCs) with rat hippocampal brain slices. We observed that the hNPCs could differentiate into neurons and could receive synaptic input from the host. Furthermore we found that the distribution of the hNPCs throughout the slices was dependent on the hippocampal architecture. We conclude that coculture systems are a valuable tool for studying the influence of a structured neural environment on the development and integration of human NS/PCs.
Methods
We used the immortalized hNPC line ReNcell VM (Millipore). This cell line possesses the potential to differentiate into functional neuronal cells, expressing markers like βIII-tubulin and tyrosine hydroxylase in standard culture systems [14,15], as well as in 3-dimensional scaffolds [16]. The cell line is characterized by a fast proliferation and a rapid onset of differentiation upon the withdrawal of growth factors [17]. The cell line was used as an in vitro model system in a variety of different studies dealing with neuronal differentiation [14 –17] of hNPCs, where it is clear that the immortalization prevents any application in human studies.
Generation of a stable green fluorescent protein-expressing cell line
ReNcell VM cells were transfected with pCAGIP-GFP [18] using Nucleofector technology (Amaxa) according to manufactuers' recommendations. Eighteen hours after transfection, cells were passaged and selection was started using 2.5 μg/mL Puromycin in medium containing growth factors for 2 weeks. A green fluorescent protein (GFP)-positive subpopulation was subsequently cultured in the standard proliferation medium as previously described [15]. Flow cytometry analyses revealed in proliferating cells 94.4%±3.6% and in cells differentiated for 3 days 90.5%±3.8% GFP-positive cells.
Slice culture and seeding of hNPCs
Slice cultures were prepared from P6 Wistar rats (Charles River, Germany) and cultured using the interface method [19]. The hippocampi were sectioned into 400 μm slices and cultured with a bicarbonate buffered medium principally comprised of 50% Dulbecco's modified Eagle medium, 25% Hank's balanced salt solution, and 25% horse serum, with penicillin and streptomycin (Sigma-Aldrich) and additional nutrients. The medium was exchanged on the first day after transplantation and every 2–3 days thereafter. To prevent the slices drying out during medium changes the slices were covered with a drop of slice medium, with the excess removed before return to the incubator. The seeding of hNPCs was performed one day after slice preparation where this time point was set to day 0 of days in vitro (DIV). ReNcell VM cells transfected with GFP were suspended in slice culture medium at 100,000 cells/μL. Approximately 0.1 μL of suspended cells was placed onto each hippocampal slice. All experiments were performed in accordance with the German Animal Welfare Act.
Immunocytochemistry
Slices were fixed at room temperature for 30 min in 4% paraformaldehyde (PFA), washed with phosphate-buffered saline (PBS), and stored in 0.02% NaN3 at 4°C. Immunocytochemistry was performed for βIII-tubulin (1:500, mouse monoclonal; Santa Cruz Biotechnology), GFAP (1:1,000, rabbit polyclonal; Dako), Ki67 (1:200, rabbit polyclonal; Santa Cruz Biotechnology), nestin (1:50, mouse monoclonal; R&D Systems), MAP2ab (1:1,000, mouse monoclonal; Millipore), NeuN (1:100, mouse monoclonal; Millipore), and Olig2 (1:200, goat polyclonal; Santa Cruz Biotechnology). Blocking was carried out using 0.3% triton X-100 and either 10% normal goat serum (for βIII-tubulin, glial fibrillary acidic protein (GFAP), Ki67, and nestin) or 10% bovine serum albumin (BSA) (Olig2) for 24 h at 4°C. Primary antibodies were then incubated overnight at 4°C with 2% blocking agent and 0.3% triton X-100, followed by six 30-min washing steps in PBS. Alexa Fluor 568 (1:1,000, goat anti-mouse or rabbit, or rabbit anti-goat) was used as the secondary antibody, and incubated overnight at 4°C with 2% blocking agent and 0.3% triton X-100. Slices were then washed in PBS and stained for 4′,6-diamidino-2-phenylindole (DAPI; 20 min, 250 ng/mL), before being removed from the insert membrane and mounted on cover slips.
For immunostaining of cover slips and insert membranes, the specimen was fixed at room temperature for 15 min in 4% PFA, then blocked with 5% normal goat serum and 0.3% triton X-100. Primary and secondary antibodies were each incubated for 30 min with 1% normal goat serum. Fluorescence pictures were taken with a Biozero 8000 microscope system (Keyence). Each antibody staining was made in at least 3 independent experiments. A set of pictures was taken with a confocal microscope (FluoView FV1000; Olympus) to visualize the colocalization of GFP and DAPI.
Flow cytometry
The cells were cultured in 6-well plates up to 80% confluency and subsequently differentiated for 3 days. For fluorescence-activated cell sorting (FACS) analysis, cells were trypsinized and centrifuged at 100 g at room temperature for 5 min, washed with PBS without Ca2+ and Mg2+, and fixed with 1% PFA in PBS for 15 min. Cells were then resuspended in washing buffer (PBS+0.5% BSA+0.02% Na-azide) and stored at 4°C in the dark. For the staining, cells were centrifuged and resuspended in saponin buffer (PBS+0.5% saponin+0.5% BSA+0.02% Na-azide) containing βIII-tubulin (1:100) in which they were incubated for 2 h at room temperature. Subsequently cells were washed and incubated with the secondary antibody (Alexa Fluor 647 goat-anti-mouse, 1:1,000; Molecular Probes) for 1 h in saponin buffer. Cells were washed twice with saponin buffer and resuspended in wash buffer for analysis. Measurements were made using FACSCalibur (Becton Dickinson) in combination with Cell Quest Pro software.
Electrophysiology
Whole cell patch clamp recordings of engrafted hNPCs were performed at 37°C using an EPC10/2 amplifier (HEKA). Data were filtered at 2.9 kHz and sampled at 10 kHz. Series resistances were compensated by 60%. The intracellular solution contained in mM: 140 KCl, 1 CaCl2, 1 MgCl2, 10 HEPES, 11 EGTA, 2 Mg-ATP adjusted to pH 7.2 with KOH, ≈305 mOsm. The extracellular solution contained: 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 26 NaHCO3, 1.25 NaH2PO4, 20 Glucose, ≈305 mOsm. Patch pipettes were pulled from borosilicate glass tubing (1.5 mm OD, 0.5-mm wall thickness; Hugo Sachs), and had a resistance of between 4 and 8 MΩ. GFP-positive cells were identified visually and recordings were confirmed by the presence of GFP in the pipette (Fig. 6A). Voltage step families were applied in 10 mV increments from −60 to +50 mV, with holding potentials of −60 and −80 mV. A P/N protocol was used for an online leak subtraction of leak currents. Recordings of synaptic currents were made from a holding potential of −80 mV and filtered at 3 kHz.
Statistical analysis
Analysis of the data was carried out with GraphPad Prism 5 (GraphPad Software, Inc.). Mini Analysis 6 (Synaptosoft) was used to analyze recordings of postsynaptic currents (PSCs). Data are given as mean±standard error of the mean. Unless otherwise stated unpaired t-tests were used to test for significance, with P<0.05 considered to be statistically significant.
Results
Differentiation of hNPCs in standard cell culture
Human fetal midbrain NPCs were transfected with pCAGIP-GFP to produce a GFP-expressing subpopulation. During proliferation the cells expressed nestin (Fig. 1A, B) and GFAP (Fig. 1C, D), as well as the proliferation marker Ki67 (Fig. 1E, F). Upon the induction of differentiation, by withdrawal of the growth factors, βIII-tubulin–positive cells (βIII-tub+) were observed (Fig. 2A, B). Flow cytometry analysis revealed no difference in the amount of βIII-tub+ cells between GFP-expressing cells and nontransfected cells (data not shown). During differentiation the cells developed more mature morphologies, characterized by compact cell bodies and extensive processes; however, most continued to express GFAP (Fig. 2C, D) and nestin (Fig. 2E, F). This suggests that most of the cells remained in an intermediate state, though some astroglial differentiation may also occur. No cells positive for Olig2 were detected. These results are consistent with the proliferation and differentiation of nontransfected cells [17,22].
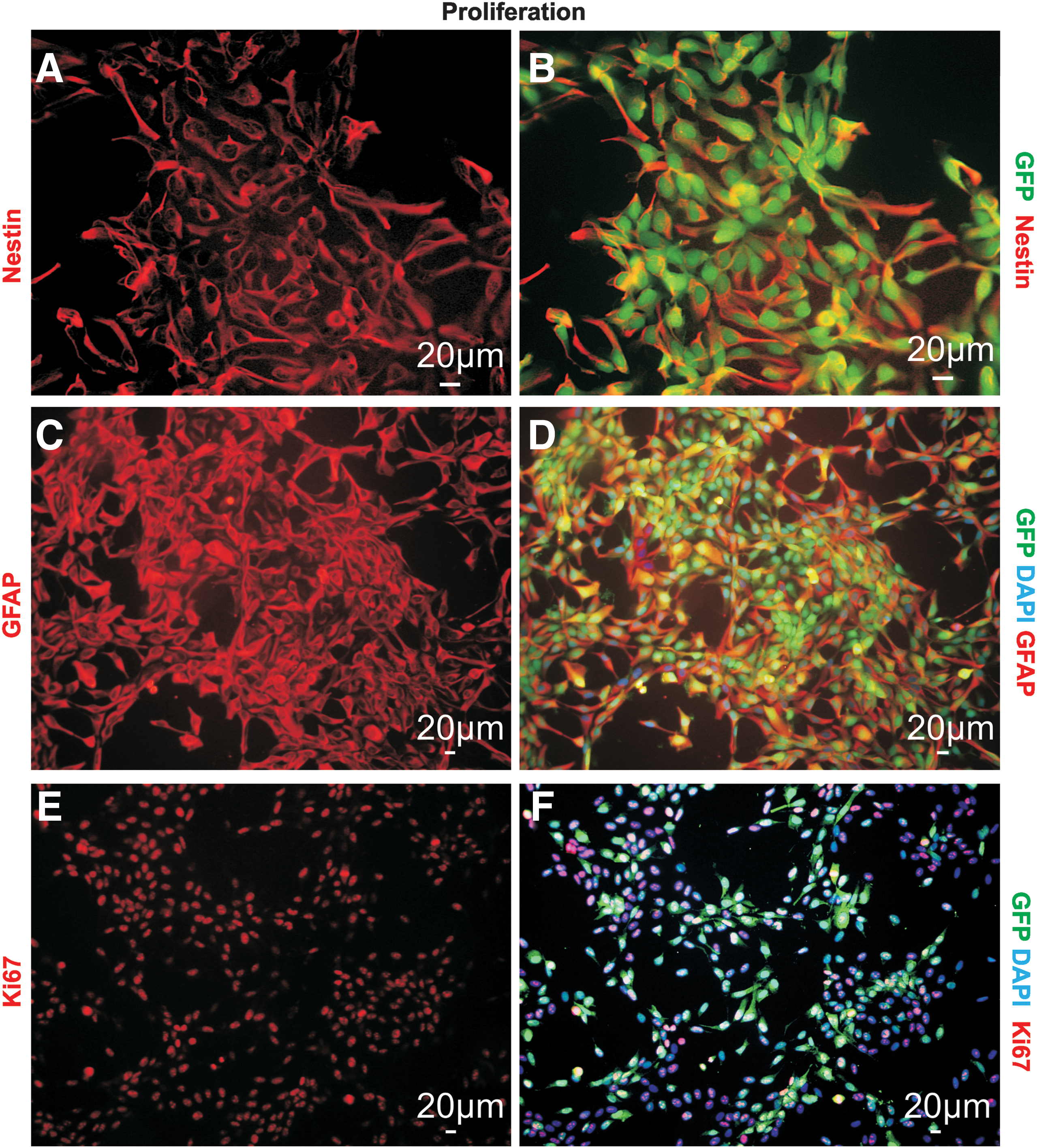
Proliferation of hNPCs in standard cell culture. GFP-expressing hNPCs were generated to allow identification of the cells in the coculture system. During proliferation, nestin,

Differentiation of hNPCs in standard cell culture. Immunostainings from hNPCs differentiated for three days. Upon differentiation βIII-tub+ cells could be detected
Coculture of hNPCs with hippocampal brain slices
We seeded the GFP-expressing hNPCs onto hippocampal brain slices from P6 rats and analyzed their development between 3 and 24 DIV (day of engraftment=DIV0). Up to 24 h after the seeding, the cells displayed no preferential location on the slice (Fig. 3A). During the cultivation, the hNPCs penetrated the slice (Fig. 3Bi/ii) and formed a distinctive distribution along the regions around the hippocampal fissure, namely the stratum moleculare and stratum lacunosum-moleculare (Fig. 3C, D). Cells were also frequently found in the dentate gyrus, though with lower density. Occasionally cell bodies were present in the stratum radiatum, but cells were rarely found in the pyramidal cell layer or the stratum oriens. The hNPCs developed differentiated morphologies, typically uni- or bipolar, and extended processes throughout the slice. A clear pattern of process outgrowth emerged in the fissal region, with many running parallel to the laminar structure (Fig. 3E). Other cells in the fissal region extended processes outward into the stratum radiatum or dentate hilus. The cells located in the dentate gyrus showed no consistent pattern in their projections (Fig. 3F). The few cells seen in the stratum radiatum almost always sent their processes perpendicular to the laminar structure of the hippocampus. These observations suggest that the hNPCs are differentially responding to guidance cues present in the slice, and that characteristics of the hippocampal architecture, known to regulate the growth of axons during development, can also regulate the integration of hNPCs.
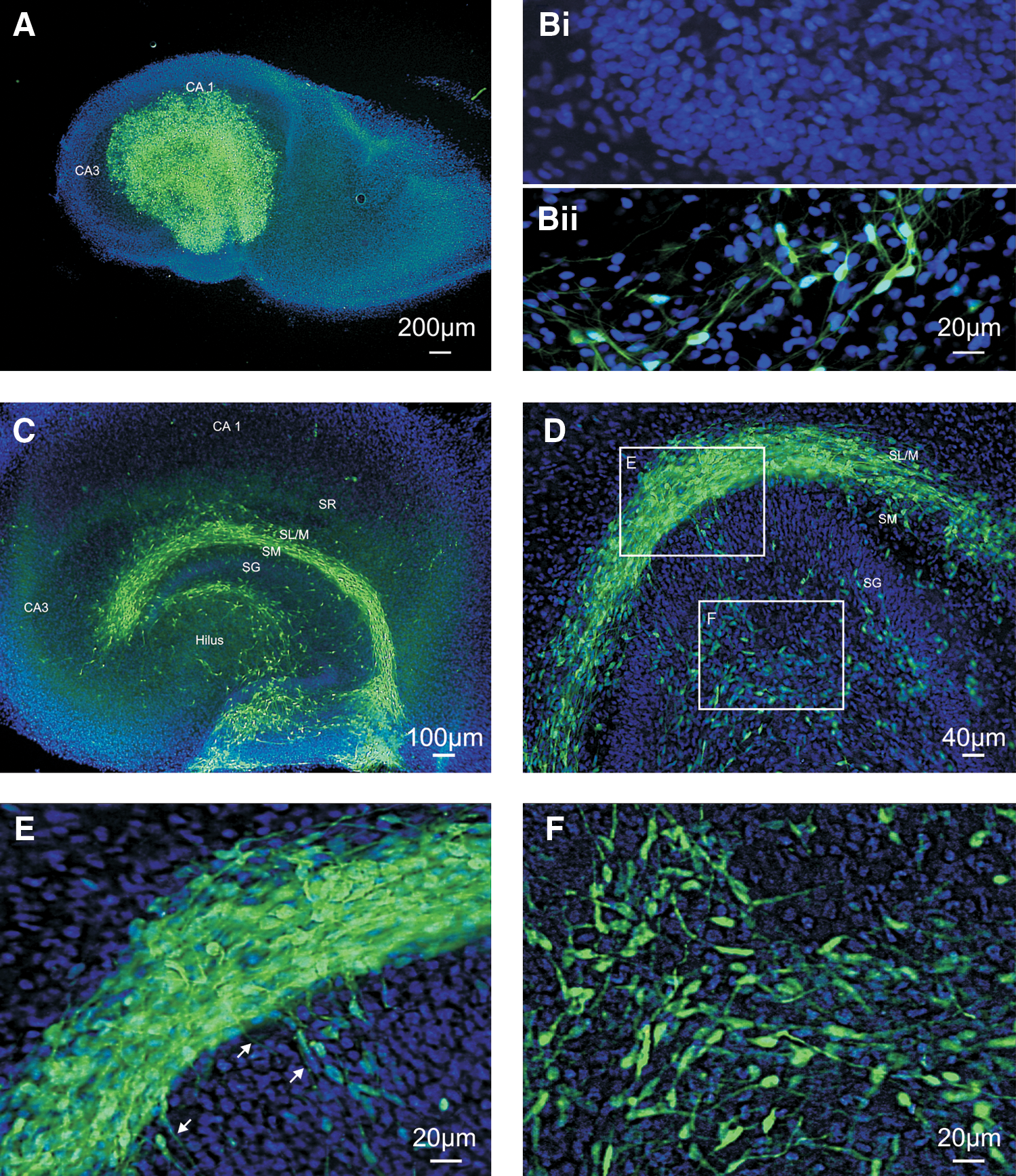
Seeding of hNPCs on rat hippocampal slices. GFP-expressing cells were seeded on to the rat hippocampal slices and cultured for up to 24 days.
Immunocytochemical characterization of hNPC differentiation in slice cultures
We examined the fate of engrafted hNPCs by immunostaining for βIII-tubulin, GFAP, and Olig2 as markers for neurons, astrocytes, and oligodendrocytes, respectively. As with differentiation in cell cultures, a small proportion expressed βIII-tubulin (Fig. 4A, B). Similarly the detection of βIII-tub+ cells was rarer and more variable at later time points, with only one βIII-tub+ cell detected between 14 and 24 DIV, consistent with previous findings that in this cell line the number of neurons declines after several days in cell culture [15,16]. βIII-tub expression was not quantified since the density of cells prevented accurate determination of their total number. To determine whether the loss of βIII-tub reflected a maturation of the neuronal cells, stainings for MAP2ab and NeuN were made at 14 and 24 DIV. No cells positive for either marker were detected (data not shown). Very few of the hNPCs expressed GFAP at any of the time points tested (Fig. 4C), and no cells positive for Olig2 were found (data not shown). Most of the cells continued to express the progenitor marker nestin (Fig. 4E, F), indicating that they had not fully differentiated, though no cells continued to proliferate, shown by the absence of any Ki67-positive cells (Fig. 4D). Therefore most of the cells remained in an intermediate state and did not differentiate into any of the 3 major neural cell types.

Differentiation of hNPCs on hippocampal slices.
In contrast to the hNPCs seeded onto the slices, those growing on the culture insert membranes, away from the slices, had immature morphologies and some continued to proliferate as demonstrated by the expression of Ki67 (Fig. 5A). Those cells adjacent to the slice did not express Ki67 and had the fine processes and compact cell bodies of differentiated cells (Fig. 5B). The majority of hNPCs on the membrane also expressed nestin (Fig. 5C) and, unlike those on the slice, GFAP (Fig. 5D). As both hNPCs on the slice and membrane are exposed to the medium, and hNPCs close to the slice show differentiated morphologies, these results suggest that the slice environment is promoting the switch to differentiation.
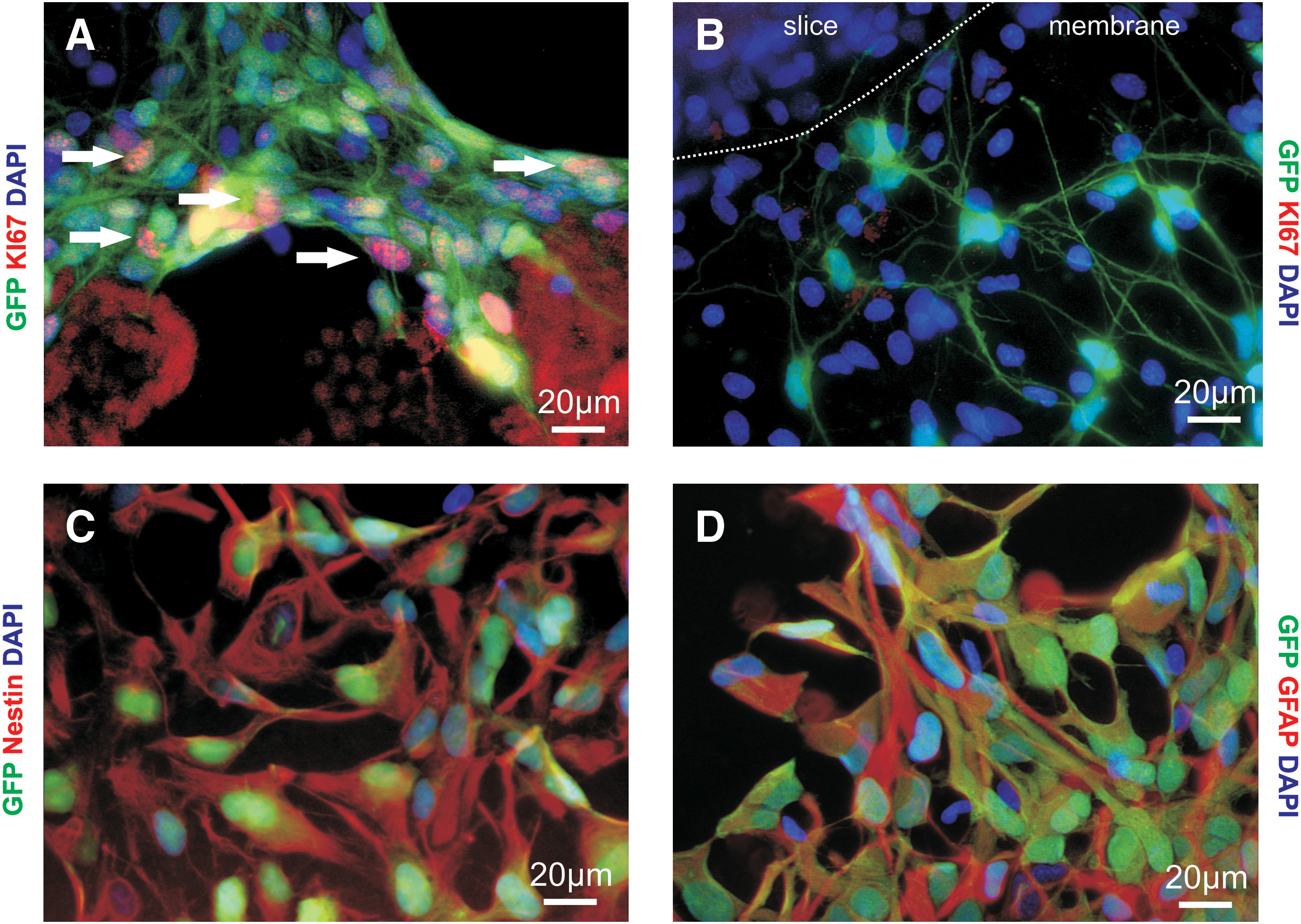
Development of hNPCs on culture insert membranes.
Electrophysiological properties of cocultured hNPCs
To examine the functional development of the cocultured hNPCs we made whole cell patch clamp recordings from GFP-labeled cells between 3 and 12 DIV (Fig. 6A). Voltage-dependent currents were elicited by a series of voltage steps from −60 to +50 mV. The K+ (Kv) and Na+ (Nav) currents expected of neuronal cells could be found (Fig. 6C, B), and upon current injection 6/10 of these cells produced a single action potential (Fig. 6D). Nav currents were detected in cells between 3 and 10 DIV. There was no evidence of increased current expression or maturation over time. Another population of cells expressed Kv currents alone (Fig. 6E, F). In many cases the current traces were dominated by an inactivating leak current (Fig. 6G, H), which has also been observed in standard cell cultures. We categorized the cells based on these currents. The Nav and Kv currents were found in 9% of cells (12/132, Fig. 7A). Kv currents alone were found in 39% (51/132), and 36% (48/132) expressed the inactivating leak current. In 14% (19/132) of hNPCs no voltage-gated currents were expressed. In comparison, the hNPCs with immature morphologies growing on the insert membrane did not express Nav or Kv currents, consistent with their undifferentiated state. Several did, however, exhibit the inactivating leak current (8/14).

Electrophysiological properties of hNPCs.
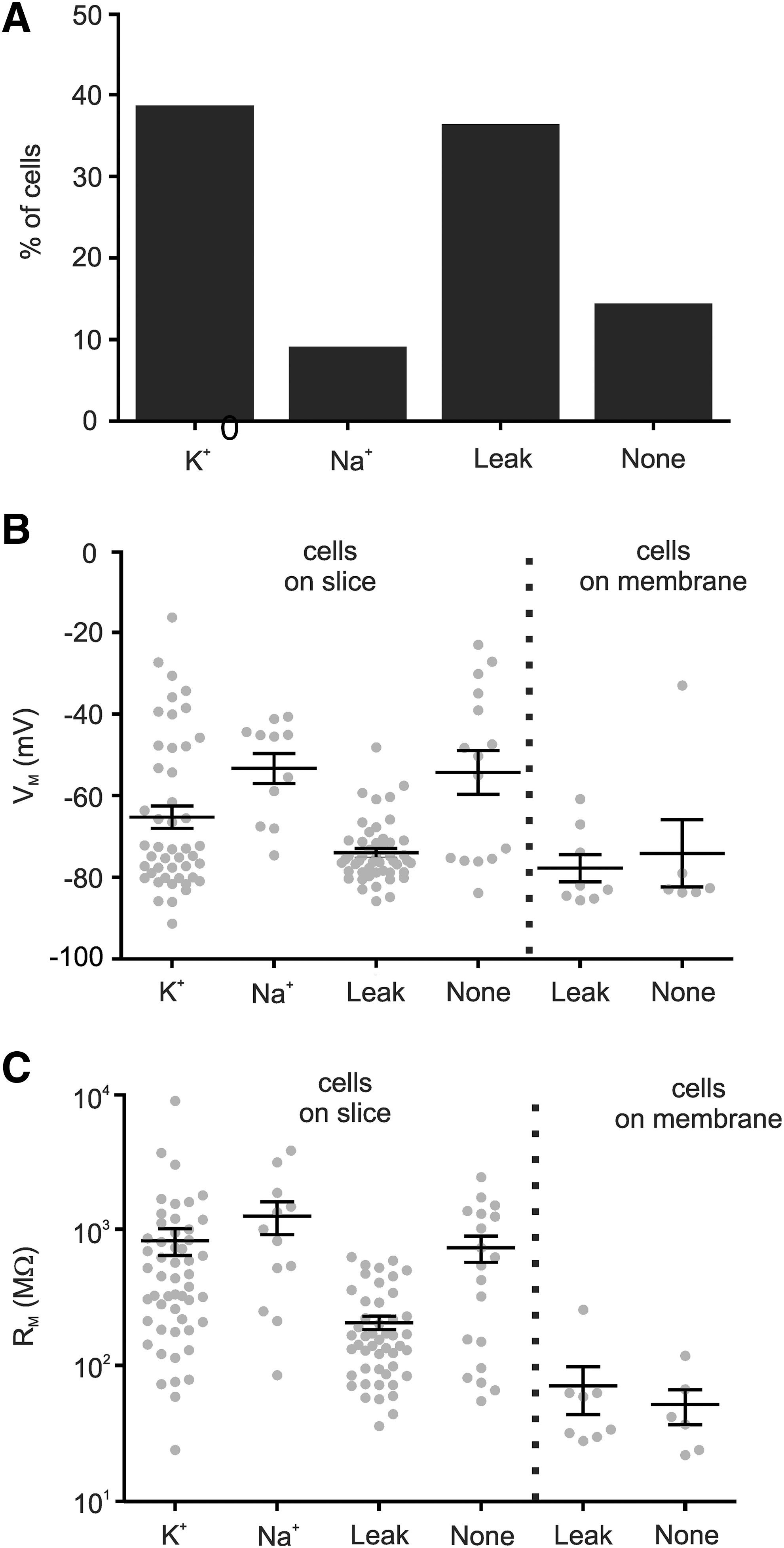
Distribution of different current types of hNPCs.
Comparison of the membrane potentials between groups (Fig. 7B) showed that neuronal cells tended to be depolarized relative to cells expressing Kv or inactivating leak currents (Kv −65.3±2.8 mV, n=46 vs. Nav −53.5±3.7 mV, n=11 and leak −74.0±1.1 mV, n=48, P<0.05, vs. none −54.3±5.4 mV, n=15, P>0.05), consistent with reduced dependence on the potassium equilibrium potential. The mean membrane potential of Kv cells was significantly higher than the inactivating leak group; however, there was greater variability within the Kv group and many Kv cells also showed evidence of inactivating leak currents. Therefore, there is probably extensive overlap between these groups. The cells on the membrane also showed hyperpolarized membrane potentials (membrane leak: −77.8±3.5 mV, n=8, membrane none: −74.1±8.3 mV, n=6). No trend was seen over time within any groups (one-way analysis of variance, P>0.05).
The membrane resistance of cells expressing inactivating leak currents, both on the slice (207±23 MΩ, n=48) and membrane (52±15 MΩ, n=8), was lower compared with other cell groups (Kv 826±184 MΩ, n=52, Nav 1,252±338 MΩ, n=12, None 734±159 MΩ, n=19; Fig. 7C). The cells on the membrane without currents similarly had a low membrane resistance (71±27 MΩ, n=6).
hNPCs receive synaptic input
Synaptic connections are a fundamental part of neuronal functioning and therefore it is important for any NPC-derived neuron to be capable of forming them. We observed spontaneous PSCs (Fig. 8A) at very low frequencies in several cells (8/45). These PSCs could be detected as early as 4 DIV, similar to the time course of neuronal differentiation. To examine the extent to which hNPCs received synaptic input we stimulated the slices with 5 μM kainate or 35 mM KCl. Both kainate (Fig. 8A) and KCl (data not shown) could induce synaptic activity. Stimulation of the cell resulted in an increase of the PSC frequency from 0.14 to 0.73 Hz (Fig. 8B). A slightly higher amplitude was observed for the PSCs measured in the presence of kainate (control: 23.0±0.9 pA, 42 events, kainate: 34.1±1.2 pA, 98 events). Analysis of the decay kinetics revealed a fast rise time in both groups (control 1.3 ms; kainate 0.6 ms), indicating that the currents originate from synaptic input rather than from an activation of postsynaptic glutamate receptors. The decay kinetics of the PSCs recorded in the control were best fitted by a monoexponential function, whereas the PSCs in the presence of kainate were best fitted by a biexponential function (control τ: 2.6 ms; kainate τ 1: 11 ms; τ 2: 37 ms; Fig. 8C). The differences between the time constants may indicate different types of synaptic input, with the fast decay of the monoexponential events suggesting excitatory input and the slow, biexponential decay suggesting inhibitory input. [20,21]. The difference in PSCs under control and kainate probably represents a change in network dynamics, where under broad stimulation inhibitory neurons dominate the network activity. The low number of cells exhibiting PSCs and the low frequency of this activity prevented further characterization of their kinetic and pharmacological properties.
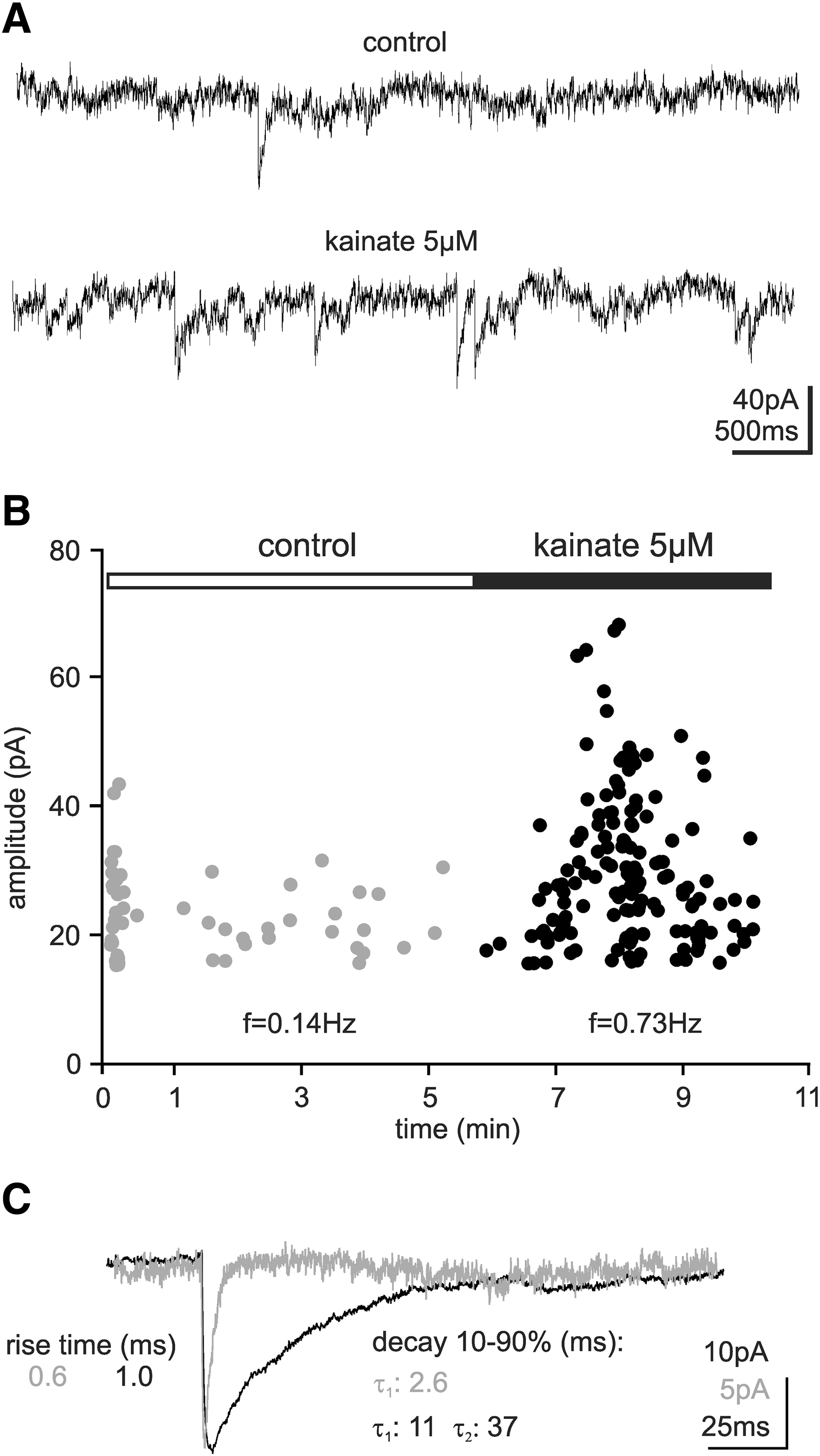
Synaptic connections between host tissue and hNPCs. Patch clamp recordings of PSCs indicating synaptic input to the hNPCs.
Discussion
We have shown here that after seeding onto an organotypic neural environment the distribution of hNPCs is highly dependent on the structure of that environment. We also found that the hNPCs could differentiate into functional neurons capable of producing action potentials and receiving synaptic input from the host tissue. These observations demonstrate that the composition of the host environment can regulate how hNPCs integrate, and thereby their potential to provide therapeutic effects. Furthermore, they demonstrate the validity of using organotypic slice cultures to study the functional integration and migration of human NS/PCs.
Neuronal differentiation and synaptic integration
Many studies have shown that fetal, embryonic, and induced pluripotent stem cells can all differentiate into neurons when transplanted in vivo [23 –27]. It is clear, therefore, that neurogenesis from grafted NS/PCs is possible in the adult brain; however, optimizing this process and regulating integration into the host in a way that will produce functional benefits remain a distant prospect. We have shown here that hNPCs can differentiate into functional neurons in hippocampal slice cultures and, significantly, that they could receive synaptic input and produce immature action potentials. We also noted that several of the hNPCs with PSC activity did not express Nav currents, indicating that either some neurons had not developed functionally or that nonneuronal progenitors were capable of responding to synaptic activity, as progenitor cells can express neurotransmitter receptors [28]. Given the low number of neurons on any given slice, their limited ability to generate action potentials, and the absence of any synaptic activity in cell cultures, it is unlikely that any of the PSCs observed were the result of connections between hNPCs.
It is notable that the hippocampal neurons recognized the human midbrain-derived hNPCs as appropriate synaptic partners, since neural connectivity is highly specific and tightly regulated during development [29,30]. The specificity of synaptic connections is achieved through several mechanisms, such as the expression of adhesion molecules like N-cadherin [31,32], gradients of morphogens such as Wnts [33], and through local signals provided by glial cells [34 –36], and it appears that the hNPCs respond to these cues, which may then influence their integration and subsequent differentiation. Though these mechanisms are only partly understood, guiding the expression and availability of the signals involved, for example via artificial matrices [37], may form a method of managing the integration of transplanted hNPCs. Early synaptic integration of transplanted NS/PCs may be important as during development many neurons undergo programmed cell death, regulated in part by the level of synaptic input [38]. We have shown previously in this cell line that excitation is neuroprotective [15], as has also been reported for other stem cells [39], and in in vivo models of Parkinson's disease the number of stem cell–derived dopaminergic neurons has been seen to decline over tine [40,41]. This suggests that promoting synaptic integration of transplanted neurons may promote their survival and subsequent therapeutic benefits.
As yet there have been no studies on the role of activity in the survival of stem cells transplanted in vivo; one reason for a lack of such studies may be the technical difficulty of investigating the influence of activity on survival in vivo. The demonstration of synapse formation in ours and earlier studies [8] shows that organotypic slice cultures are a promising system for studying the mechanisms underlying synaptic integration of NS/PCs. Compared with in vivo models they are highly accessible and easily manipulated, while compared with primary coculture systems they maintain the tissue structure, and subsequently the organization of extracellular signaling molecules.
Environmental regulation of process growth and migration
We observed a distinct distribution of the hNPCs within the hippocampal slices, which could be caused by the host environment restricting migration and process growth, or preferential survival of cells within specific regions. Preferential adhesion may also contribute, though data from 3 to 9 h postseeding indicate that this is not the principle cause.
Preferential migration along fiber pathways has been described for mouse embryonic stem cell–derived astrocytes [9]; however, this is the first description we are aware of demonstrating such specific interaction between NS/PCs and the host architecture.
To provide optimal benefit, therapeutic application of NS/PCs requires a balanced migration of NS/PCs across the target region, but too extensive migration increases the risk of aberrant integration and adverse side effects. Identifying the mechanisms underlying NS/PC migration in neural environments may make it possible to regulate this process, and for studying such organotypic slice cultures provide a valuable model system.
Several features of the hippocampal chemoarchitecture could be involved in the localization of the hNPCs to the fissural regions. Reelin, an ECM molecule secreted by Cajal-Retzius cells, is prominent in these regions, and appears to act as a stop signal for migration through stabilization of the cytoskeleton [42,43]. However, any interaction would necessarily be more complex than simply preventing changes in cell morphology, as the hNPCs manage to extend processes throughout this region. Another ECM molecule, hyaluronan, acts as an anchor for many signaling molecules and has also been associated with adhesion and lamination in the stratum moleculare and stratum lacunosum-moleculare [44,45], and therefore could result in greater hNPC adhesion in these regions.
The extension of processes by hNPCs in parallel to the hippocampal fissure follows the pathway of entorhinal afferents entering the hippocampus, and it may be that the hNPCs responded to signals guiding this pathway. However, other hNPCs sent projections up through the layers, oriented parallel to the projection neurons, suggesting that within the hNPCs there are populations that differentially respond to guidance cues present in the hippocampus.
Hippocampal environment alters the identity of the progenitor pool
We found that many of the cocultured hNPCs remained in an intermediate progenitor state and did not adopt a mature phenotype, even after 24 DIV. Significantly, with the absence of GFAP this intermediate state appears different to that of the nestin-expressing cells in cell cultures. The finding does not appear to result from differences in the medium composition, as hNPCs growing on the insert membrane continued to express both markers. Interestingly, a similar suppression of GFAP expression was reported for human cord blood stem cells [12], whereas studies using mouse stem cells observed astrocytic differentiation in hippocampal cultures [9,10] and in striatal cultures [46], hinting at species-specific responses to the environment. The cause of this change and the extent to which the cells differ as a result, particularly in their signaling properties, require further investigation.
The failure of the hNPCs to differentiate into astrocytes or oligodendrocytes while showing some degree of neuronal differentiation hints that the hNPCs remain neurogenic, as neurogenesis and gliogenesis are temporally segregated in the developing brain [47,48]. Therefore, given the appropriate cues, a much greater proportion of neurons might be obtained from this and similar immortalized hNPC lines. Despite the aim of inducing neuronal differentiation, many of the functional benefits seen in transplantation studies are probably due to protective actions of NS/PCs that have not differentiated into neurons. In a recent study using both striatal organotypic slice cultures and in vivo SCA1 mutant mice models, gap junction connections between nestin-positive mouse NSCs and host cells were identified as a mechanism by which transplanted cells provide protection [46]. Whether a similar mechanism is present with hNPCs requires further study. Because gap junction coupling requires contact between transplanted and host cells the extent of any therapeutic benefit will depend on the distribution of cells throughout the afflicted region, which, as we have shown here, maybe strongly influenced by the structure of the environment.
The benefits provided by immortalized progenitors could be counteracted should they continue to proliferate and increase the risk of tumorigenesis. However, we observed that the slice environment appeared to suppress proliferation, given the mature morphology and absence of proliferation markers in hNPCs growing close to or on the slices. This could be caused by short-range diffusible factors or to contact mediated signals present on the debris that surrounds the slices; for example, the ECM component hyaluronan has been shown to inhibit proliferation of progenitor cells through the activation of toll-like receptors [49,50].
Functional properties of hNPCs
In analyzing the functional development of the hNPCs we observed cells with currents expected of differentiating neurons and NPCs; however, alongside these we also observed an inactivating leak current that has not been described previously. This current was correlated with a hyperpolarized membrane potential and a low membrane resistance. One possible cause is heterotypic gap junction coupling [51], which may also explain the low membrane resistance. A volume-regulated chloride current that also appears superficially similar has been described in glial cells [52], and volume regulation processes are involved in differentiation. But as equiosmolar solutions were used for the patch clamp recordings it is unlikely that this current is activated in response to changes in volume; however, it is possible that the current is activated by hNPCs to induce changes in volume. Further studies are required to determine the identity of the channel and its biological significance.
In sum, our findings demonstrate diverse influences of the host environment on hNPCs, which, taken together, will affect the potential of transplanted cells to provide therapeutic benefits. Furthermore they highlight differences in the differentiation of many cells after transplantation compared with in cell cultures, emphasizing the need for caution in extrapolating effects on in vitro differentiation to in vivo situations.
Footnotes
Acknowledgments
The authors thank Dr. James Whiteford for the kind gift of pCAGIP-GFP plasmid. We thank Ellen Ewald, Norman Krüger, and Steffen Albrecht for their excellent technical support.
Author Disclosure Statement
No competing financial interests exist.