
Abstract
Argonaute2 (Ago2) is a well-known factor that has intrinsic endonuclease activity and is a part of the fundamental gene regulatory machinery. Recently, we showed that nuclear Ago2 regulates voltage-gated potassium (Kv) channels and that Ago2/Kv1.3 has crucial functions in the self-renewal and cell de-aging processes in adipose tissue-derived stromal cells (ATSCs). In the nucleus, Ago2 bound to the promoter regions of calcium-activated potassium channel 3, potassium channel subfamily K member 1 (KCNK1), and voltage-gated potassium channel 2, and the expression of these genes was significantly upregulated at the level of transcription. We detected an active K+ channel that plays a critical role in Ago2-mediated ATSC self-renewal through the control of membrane potential during cell self-renewal and differentiation. Among the several regulatory subunits of voltage-dependent K+ (Kv) channels, Kv1.3 and Kv1.5 have been shown to impact tissue differentiation and cell growth in cultured ATSCs following their direct binding to the regulatory region of the Kv channel gene. In ATSCs, interference with Ago2 or K+ channel gene expression or treatment with tetraethylammonium significantly downregulated stemness gene expression, induced cell cycle arrest, and inhibited the ability of cells to transdifferentiate into neurons or β-cells via Oct4 knockdown. Blockage of the K+ channel significantly induced protein kinase C (PKC) α, β, and δ phosphorylation and negatively affected Ago2 and Oct4 expression. This K+ channel blockage also resulted in the upregulation of p53 and p21 expression and the inactivation of mitogen-activated protein kinase (MEK), extracellular signal-regulated kinase 1/2 (ERK 1/2), AKT, β-catenin, and STAT3. Our results suggest that the nuclear Ago2 regulation of the K+ channel or stemness-related gene expression plays a critical role in adult stem cell self-renewal and differentiation.
Introduction
S
Potassium (K+) channels are both ubiquitous and diverse and are involved in many physiological functions, such as solute transport, volume control, enzyme activity regulation, secretion, excitation-contraction coupling, and intercellular communication. Voltage-dependent K+ (Kv) channels control the action potential and electrical excitability of nerves and muscles [9]. Additionally, Kv channels are involved in myogenesis and myoblast proliferation [10 –12]. Kv channel antagonists arrest the cell cycle at G1 or G1/S transition [12,13]. Thus, Kv channels control membrane potential and cell volume during cell self-renewal and differentiation. Among the several regulatory subunits of Kv channels, Kv1.3 and Kv1.5 are the members that function in tissue differentiation and cell growth. The Kv1.3 channel is expressed in a range of organs and tissues [12 –16] including the hippocampus, the olfactory bulb, epithelia, adipose tissue and muscle [11,16,17]. Kv1.5 has been isolated from the human ventricle, and the immune system [11,12,17,18]. The level of Kv1.3 or Kv1.5 expression was shown to affect cell self-renewal, differentiation, or activation in several tissue types. Additionally, Kv channels were differentially expressed during development [19,20]. Dividing cells usually pass through the mitotic cell cycle, comprised of G1, S, G2, and M phases. Embryonic stem cells (ESCs) have unique capacities for self-renewal and pluripotency that are revealed following the G1 phase of ESCs, when cells would either proceed to enter the S phase for self-renewal or to the G0 stage for differentiation [13]. During ESC self-renewal, a Kv channel blocker decreased cell self-renewal in a concentration-dependent manner, revealing the involvement of Kv channels in ESC self-renewal. Moreover, Kv channels may also be involved in cell survival and pluripotency [21]. Kv channel blockage in ESCs induces the expression of the stemness genes Oct4, Sox2, and Nanog [21]. Mammalian neuron-derived neural progenitor cells express voltage-gated ionic currents. Current intensity depends on a cell's differentiation or proliferation state. When embryonic hippocampus progenitor cells are cultured under conditions that foster proliferation, low level Na+ currents are observed [22]. Previous reports have concluded that E15 SVZ neural progenitor cells express Kv2.1 and Kv4.3 and that these channels have important roles in embryonic neural progenitor cell differentiation in in vitro culture or at later developmental stages [23 –25].
Transcription factors that control stem cell plasticity and self-renewal have been identified [26,27], and the core regulatory circuitry by which these factors exert their effects on protein-coding genes has been described [5,28 –34]. Mature miRNAs are specified as post-transcriptional gene repressors that arise from primary transcripts before processing [3]. Studies comparing ESCs and differentiated cells with regard to transcription factor occupancy and mRNA expression have yielded information regarding the transcriptional regulation of miRNA genes during the various processes that occur in stem cells, such as differentiation, proliferation, and dedifferentiation [28 –33].
Our recent study showed that the miRNA processing protein Ago2 actively induces ATSC proliferation. Here, we show that Ago2-overexpressing ATSCs exhibit primitive stem cell behaviors, including an increased capacity for self-renewal and differentiation compared with control ATSCs, due to the direct binding of Ago2 to the regulatory regions of functional genes. Ago2 also induces the demethylation of stemness genes and the overexpression of specific functional genes. We also demonstrate that nuclear localized Ago2 controls Kv channel expression via direct binding to regulatory regions of Kv channel genes in ATSCs. Additionally, we show that Ago2 directly positively regulates cell self-renewal and differentiation in other tissues or cells via Oct4 expression and the activation of specific signal mediators. Finally, we focus on the novel electrophysiological functions of nuclear Ago2 with Kv and Oct4 in ATSCs.
Materials and Methods
Culture of ATSCs
Donor-derived raw adipose tissues were processed in accordance with established methodologies to determine the stem vascular functions. Human raw fat tissue obtained from the patient abdomen (female; n=5, male; n=5; following patient's approval document) was processed according to established methodologies. To isolate the stem cells, the samples were digested with 0.075% collagenase IV (Sigma) and centrifuged at 1,200g for 10 min to create a high-density cell pellet. The pellet was then suspended in red blood cell (RBC) lysis buffer (BioWhittaker) and incubated for 10 min at room temperature to lyse contaminating RBCs. The stem cell pellet was then collected and incubated overnight at 37°C and 5% CO2 in 10% fetal bovine serum containing α-minimum essential medium (GIBCO BRL). For Ago2 overexpression or knockdown experiment, we used P8-P10 of cultured ATSCs. This work was approved by the Seoul National University Institutional Review Board (IRB No. 0603/001–002), and the ethics committee specifically approved this procedure.
Nonradioisotopic telomerase assay
Telomerase activity was assessed using a modified telomeric repeat amplification protocol assay in accordance with the manufacturer's instructions (BD Science). Protein extracts were prepared from the ATSCs, and 0.5 μg of protein extract prepared from each cell line was incubated in the presence of a synthetic oligonucleotide (telomerase-specific primer, 5′-AATCCGTCGAGCAGAGTT-3′) that could be the substrate for the addition of telomeric repeats by telomerase. If telomerase activity was detected in the extracts, the oligonucleotide was elongated and could function as a template in subsequent polymerase chain reactions (PCRs). PCR was conducted in the presence of nucleotides, and the formation of the amplification products was assessed via monitoring of telomerase repeat amplification. PCR products were separated on 12.5% nondenaturing acrylamide gels and stained using SYBR-Gold dye (Molecular Probes). Quantification of telomerase for comparisons with telomerase activity in the Ago2-overexpressing ATSCs and the ATSCs controls was conducted via the PCR enzyme-linked immunosorbent assay procedure suggested by the manufacturer (BD Science).
Chromatin immunoprecipitation analysis
The monoclonal antibodies, anti-Ago2, c-Myc, and Sox2 were obtained from Santa Cruz Biotechnology and rabbit IgG (PP64B) antibody were obtained from Upstate. Cells were harvested and chemically crosslinked with 1% formaldehyde (Sigma) for 20 h at 4°C. Fixation was quenched by 2.5 M glycine for 5 min at room temperature. Cells were pelleted at 4°C (500 g), washed with ice-cold phosphate-buffered saline and with lysis buffer including 0.5% IGEPAL and 1 mM fresh phenylmethonesulfonylfluoride (PMSF), and pelleted. Pellets were resuspended in pre-immunoprecipitation dilution buffer containing 4% GEPAL, 1 mM PMSF, and 60 mL PMSF. Cells were sonicated using a Branson Sonifier 450D at 50% amplitude, followed by 1 min rests in ice water. Sonicated fragments ranged in size from 200 to 1,000 bp. Postsonication, samples were centrifuged at 14,000 rpm for 10 min at 4°C and flash frozen in liquid nitrogen. Sonicated cell extracts equivalent to 26,106 cells were used for immunoprecipitations. Samples were precleared with protein G Dynabeads (Dynal) in 1,000 mL dilution buffer [0.01% sodium dodecyl sulfate (SDS), 1.1% Triton X-100, 1.2 mM ethylenediaminetetraacetic acid (EDTA), 16.7 mM Tris-HCl (pH 8.1), 167 mM NaCl, Upstate protease inhibitor cocktail II]. Cell extracts were incubated with 1 mg antibody overnight at 4°C. Chromatin antibody complexes were isolated with protein G Dynabeads, and washed once with low salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, 150 mM NaCl), once with high salt buffer (same as low salt with 500 mM NaCl) and twice with tris-EDTA buffer. Protein/DNA complexes were eluted from the beads in 10 mL 20% SDS, 20 mL 1 M NaHCO3, and 170 mL H2O at 65°C with occasional vortexing. Crosslinking was reversed by addition of 8 mL 5 M NaCl and incubation overnight at 65°C. Extracts were then treated with RNase A and proteinase K, and DNA was purified using an Upstate EZ ChIP kit. 5 mg purified DNA (Ago2-chip, c-Myc-chip, Sox2-chip, and Input) was amplified using a GenomePlexH Whole Genome Amplification Kit (Sigma).
Bisulfite modification and sequencing of genomic DNA
Genomic DNA was purified via phenol/chloroform/isoamylalcohol extraction, followed by 1 chloroform extraction, after which the DNA was ethanol-precipitated. The DNA was dissolved in distilled water. Bisulfite conversion was conducted using the EZ DNA Methylation–Gold kit (Zymo Research), as described by the manufacturer. Briefly, unmethylated cytosines in DNA were converted into uracil via the heat-denaturation of DNA and with a specially designed CT conversion reagent. DNA was then desulfonated and subsequently cleaned and eluted. The bisulfite-modified DNA was then immediately utilized for PCR or stored at or below −20°C. The converted DNA was amplified via PCR or designed with MethPrimer (
Real-time reverse transcription-PCR (primer sets JNK)
Total cellular RNA was extracted with Trizol (Life Technologies) and reverse transcribed into cDNA using an oligo-dT primer amplified by 35 cycles (94°C for 1 min; 55°C for 1 min; and 72°C for 1 min) of PCR using 20 pM of specific primers. The PCRs were conducted using an ABI 7700 Prism Sequence Detection System and SYBER green detection kit (Applied Biosystems). The primer sequences were designed with Primer Express software (PE-Applied Biosystems) using gene sequences obtained from the GeneBank database. For PCR product labeling, we used a Cyber green detection kit purchased from Applied Biosystems.
Western blot
For the confirmation of differentially expressed proteins in cultured ATSCs, cells were lysed in 500 μL of lysis buffer [20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM ethylene glycol tetraacetic acid (EGTA), 1 mM glycerophosphate, 1 mM Na3VO4, and 1 mM PMSF]. The lysates were clarified via 10 min of centrifugation at 15,000g and the total protein content was determined via a Bio-Rad protein quantification kit. For western blotting, equal amounts (40 μg) of protein extracts in a lysis buffer were subjected to 10% SDS-polyacrylamide gel electrophoresis analysis and transferred to a nitrocellulose membrane. Then anti-PI3K, anti-p-mitogen-activated protein kinase (MEK), anti-p-extracellular signal regulated kinase (ERK), anti-p-STAT3, anti-p-Akt, anti-p-stress-activated protein kinase/jun amino-terminal kinase (SAPK/JNK), anti-JAK2 (1:1,000, Cell Signaling), anti-p21, anti-p53, anti-c-Myc (1:200, Santa Cruz Biotechnology), anti-MAP2ab (1:500, Sigma), anti-NF160 (1:40, Sigma), anti-Nestin (1:500, BD Biosciences), anti-glial fibrillary acidic protein (GFAP) (1:3,000, Dako cytomation), antiacetyl Histone 3 (1:3,000), antiacetyl Histone 4, anti-Tuj (1:400, Sigma), and anti-β-Actin (1:500, Sigma) antibodies were incubated with membranes. After western blotting, relative band intensities were determined and compared using Quality-one 1-D analysis software (Bio-Rad).
Transfection with small interfering RNA
For knockdown of K+ channel genes, Oct4, Sox2, Nanog, and Ago2, we utilized synthesized siRNA duplexes. For transient transfection, cells at about 60% confluency were transfected with 10 μM siRNA using Lipofectamine as per manufacturer's instructions (Invitrogen), and cells were harvested after 24 h for RNA isolation. Silencer Negative Control siRNA (catalog number 4611; Ambion, Inc.) was utilized as a control for nonspecific gene silencing. The transfection of siRNAs was conducted using DharmaFECT siRNA transfection reagents, in accordance with the manufacturer's instructions (Dharmacon RNA Technologies). Two complementary hairpin siRNA template oligonucleotides harboring the 21 nt target sequences present in each of the human genes targeted were employed for each transient transfection using 50 nM siRNA. The quantity of siRNA transfected was optimized as recommended by the manufacturer. The siRNAs against each gene target (Silencer® predesigned siRNAs; Ambion) and scrambled siRNAs with the same nucleotide content were assessed. When compared with unrelated control siRNAs and scrambled siRNAs, the siRNAs against specific genes resulted in an 80%–90% reduction in the gene target's mRNA levels, as determined via real-time PCR. The siRNA that provided the most efficient inhibition (90%–95%) was utilized in the experiments shown here. We transfected siRNAs against each target gene into ATSCs and counted dye-exclusive viable cells for 6 days to evaluate any negative regulation of cell growth.
Electrophysiological recordings
Electrophysiological recording was performed in whole-cell configuration of the patch clamp technique with EPC 10 USB amplifier (HEKA Electronik). Cells were attached to glass cover slips and transferred to a 0.5 mL recording chamber, which were continuously superfused with normal Tyrode solution by gravity at a rate of ∼10 mL/min. Borosilicate glass capillaries (1.5 mm O.D., 1.17 mm I.D.; Harvard apparatus Ltd.) were fashioned into micropipettes using a PC-10 puller (Narishige Company), and the resistance of the pipettes was 4–7 MΩ when filled with pipette solution. The tip potentials were compensated before the pipette touched the cell. After a gigaseal was obtained by negative suction, the cell membrane was ruptured by gentle suction to establish whole-cell configuration. Series resistance was checked and compensated by 50%–80%. Data were filtered at 3 kHz, digitized at 10 kHz, and analyzed using Pulse program version 8.67 (HEKA Electronik) and Origin 6.1 software (MicroCal). Outward currents were recorded with the following normal Tyrode solution (143 mM NaCl, 5.4 mM KCl, 0.5 mM MgCl2, 1.8 mM CaCl2, 0.5 mM NaHPO4, 10 mM glucose, and 5 mM (4-(2 hydroethyl)-1-piperazineethanesulfonic acid (HEPES); the overall pH was adjusted to 7.4 with NaOH). The pipette solution contained 150 mM KCl, 1.0 mM MgCl2, 10 mM HEPES, 5 mM EGTA, 2 mM Mg-ATP, and the overall pH was adjusted to 7.2 with NaOH. Four-aminopyridine (4-AP) and tetraethylammonium (TEA) were purchased from Sigma-Aldrich. All experiments were performed at room temperature (22°C±1°C).
Statistical analysis
Significance was tested by t-test or analysis of variance (ANOVA) using GraphPad InStat 3.0 software. For repeated measures ANOVA and in vitro studies where Kaplan–Meier curves and log-rank analysis were performed, MedCalc software was used.
Results
Nuclear Ago2 induces ATSC self-renewal and stemness through the control of global gene regulation
We investigated the nuclear translocation of Ago2 from the cytosol and Ago2 binding activity in the regulatory regions of specific genes by chromatin immunoprecipitation (ChIP)-on-chip analysis. We also determined whether the binding of Ago2 to regulatory regions induced the activation or repression of target genes at the transcriptional level. Immunocytochemical staining for Ago2, bromodeoxyuridine (BrdU), Sox2, Nanog, and Oct4, in addition to BrdU/Ago2 co-staining, revealed that the expression levels of Ago2 and Sox2 were markedly higher in the nuclei of Ago2-overexpressing ATSCs (Ago2/ATSCs). In contrast, the control cells showed limited nuclear Ago2 localization. When we induced Ago2 knockdown in the Ago2/ATSCs, the amount of BrdU-positive mitotic cells was significantly decreased (Fig. 1). Ago2-overexpressing ATSCs possessed improved self-renewal activity, which was increased up to 150% (Fig. 1B). RNA interference-induced knockdown of Ago2 expression significantly decreased the nuclear localization of Ago2 in ATSCs (Fig. 1C). Ago2-expressing ATSCs displayed a markedly higher expression of the cell cycle controlling factors Oct4, Sox2, Nanog, and Klf4, in addition to the cell cycle-related functional genes RUNX3, c-myc, and cyclin-dependent kinases (CDKs). In contrast, the levels of p21 and p53 were downregulated compared with control cells (Fig. 1D). The Ago2-induced self-renewal and survival-related gene cluster interactome revealed that ectopically-expressed Ago2 was highly associated with the interactomes of P38MAPK, telomerase reverse transcriptase, c-myc, and KLF4 (Fig. 1E). Following Ago2 overexpression, the expression of genes controlling self-renewal and development was significantly increased, and the expression levels of ESC pluripotency-related genes, chromatin remodeling genes, and development-related genes were also upregulated (Fig. 1F). In cells expressing high levels of Ago2, the CpG dinucleotides within the region of −425 to +188 in each gene was prominently demethylated compared with cells expressing lower levels of Ago2 (Ago2+: 10% vs. control: 80%; Fig. 1G). The regulatory regions of Ago2 and Oct4 genes in Ago2-overexpressing ATSCs were also highly demethylated compared with control ATSCs (Fig. 1G).
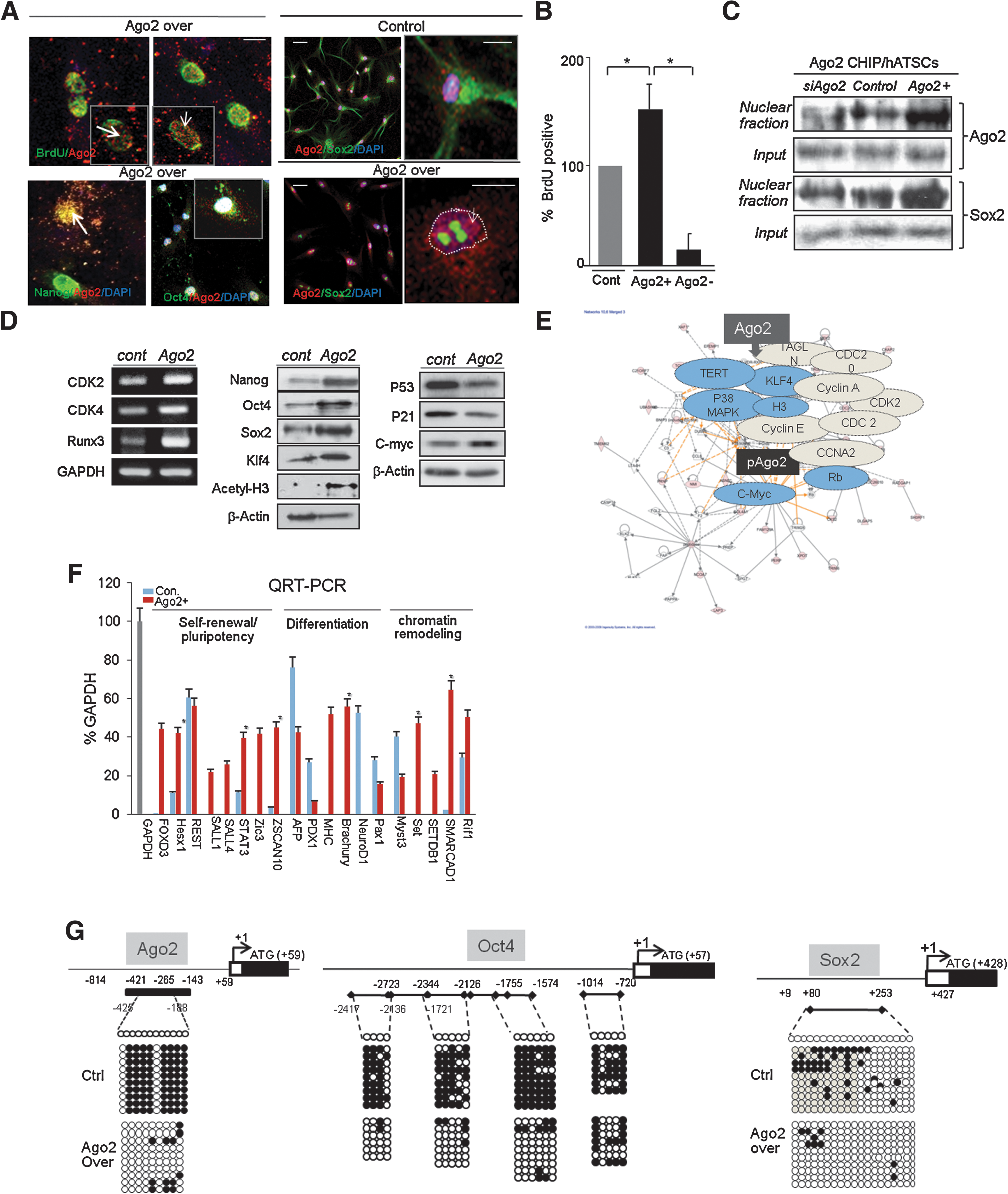
Nuclear Ago2 efficiently enhances ATSC self-renewal and development-related gene expression via demethylation of Ago2 and stemness genes.
The involvement of K+ channel activation in Ago2-mediated ATSC self-renewal
Next, we investigated the DNA binding frequency of Ago2 before and after overexpression in ATSCs. In the case of Ago2 overexpressing cells, an almost 14-fold increase in Ago2 DNA binding frequency (n=1,727) was observed compared with control ATSCs (n=122; data not shown). The Ago2-occupied genes were functionally categorized as playing roles in the following processes: transcriptional regulation, neural development, and cell proliferation (data not shown). Next, we investigated which genes are specifically regulated by Ago2 in the nucleus through ChIP-on-chip analysis and ChIP-PCR confirmation. Ago2, Sox2, and c-myc were identified as binding to the regulatory regions of calcium-activated potassium channel 3 (KCNN3), voltage-gated potassium channel 1, and voltage-gated potassium channel 2 (KCNA2). Cells expressing Ago2 also showed a high K+ channel action potential that was effectively inhibited by the K+ channel-specific inhibitor TEA (5 mM; Fig. 2A). The mean frequency of electrophysiologically-active channel-carrying cells was 12 out of the 14 total examined in Ago2-overexpressing ATSCs and 8 out of the 12 control ATSCs (Fig. 2B). Both Ago2-mediated K+ channel activation and the Ago2-specific subtype K+ channel protein pKV1.3 play important roles in ATSC self-renewal (Fig. 2C). In the nucleus, Ago2 bound to the promoter regions of KCNN3, potassium channel subfamily K member 1 (KCNK1), and KCNA2 in addition to the regulatory region of Ago2 itself. We determined that the expression levels of KCNA1, KCNA2, KCNN3, Kras, and KLF3 were upregulated at the transcriptional level (Fig. 2D, E). We also identified an active K+ channel that plays a critical role in Ago2-mediated ATSC self-renewal (Fig. 2F–I). Ago2-mediated cell self-renewal was inhibited by the RNA interference-induced knockdown of KCNA1 expression (Fig. 2F, G). Additionally, knockdown of KCNA1 led to the downregulation of CDK2, CDK4, and RUNX3 expression (Fig. 2H). We determined that pKv1.3 expression interfered with the expression of cell self-renewal-related genes at the protein level, as we showed that a knockdown of pKv1.3 expression led to a downregulation in pKv1.3 and c-myc expression, whereas p53 and p21 expression levels were significantly increased (Fig. 2I).

The involvement of the K+ channel in Ago2-mediated ATSC self-renewal.
The blockage of K+ channels affects the capacity of ATSCs for cell self-renewal and survival via the control of stemness gene expression and protein kinase C δ activation
To determine the effect of K+ channel expression on cell proliferation in Ago2-expressing ATSCs, we exposed cultured Ago2-expressing ATSCs to various concentrations of the K+ channel inhibitor TEA (0, 50, 100, 500 mM). We found that there was a dose-dependent effect on cell proliferation and cell death, as both cell proliferation activity and survival gradually decreased following drug treatment (Fig. 3A). We also determined the effect of TEA treatment over various time intervals and found that the overexpression of Ago2 induced the expression of its receptor, protein kinase C α, β, or δ, in cultured ATSCs (Fig. 3B). Pharmacological blockage of K+ channels significantly induced PKC α, β, and δ phosphorylation. In contrast, TEA treatment negatively affected Ago2- and Oct4-induced stemness gene expression in ATSCs, whereas the expression levels of p53 and p21 were significantly upregulated at each time point. Phosphorylation of the potassium channel-related signal mediators MEK, ERK1/2, AKT, β-catenin, and STAT3 was prominently decreased (Fig. 3C). One of the adaptor molecules of the K+ channel, PKC δ, is inhibited by Rottlerin, a drug that also effectively inhibited Ago2-mediated ATSC survival (Fig. 3D). When we inactivated K+ channels and PKC δ together via a combinational exposure to TEA and Rottlerin, cell growth and survival were almost completely blocked (Fig. 3D). The activation of additional related signal mediators was also affected by TEA/Rottlerin treatment in Ago2-expressing ATSCs (Fig. 3E). The combinational treatment of TEA and Rottlerin significantly downregulated Ago2 and c-myc expression (Fig. 3E). Additionally, K+ channel blockage by TEA effectively inhibited ectopic Ago2-induced self-renewal and overexpression of both cell checkpoint genes and stemness genes (Fig. 3E). When we treated cells with Rottlerin, TEA, or Rottlerin and TEA together, we found that either treatment with Rottlerin alone or Rottlerin in combination with TEA significantly inhibited Ago2-mediated ATSC self-renewal and caused an inactivation of cell proliferation-related signal mediators (Fig. 3E).
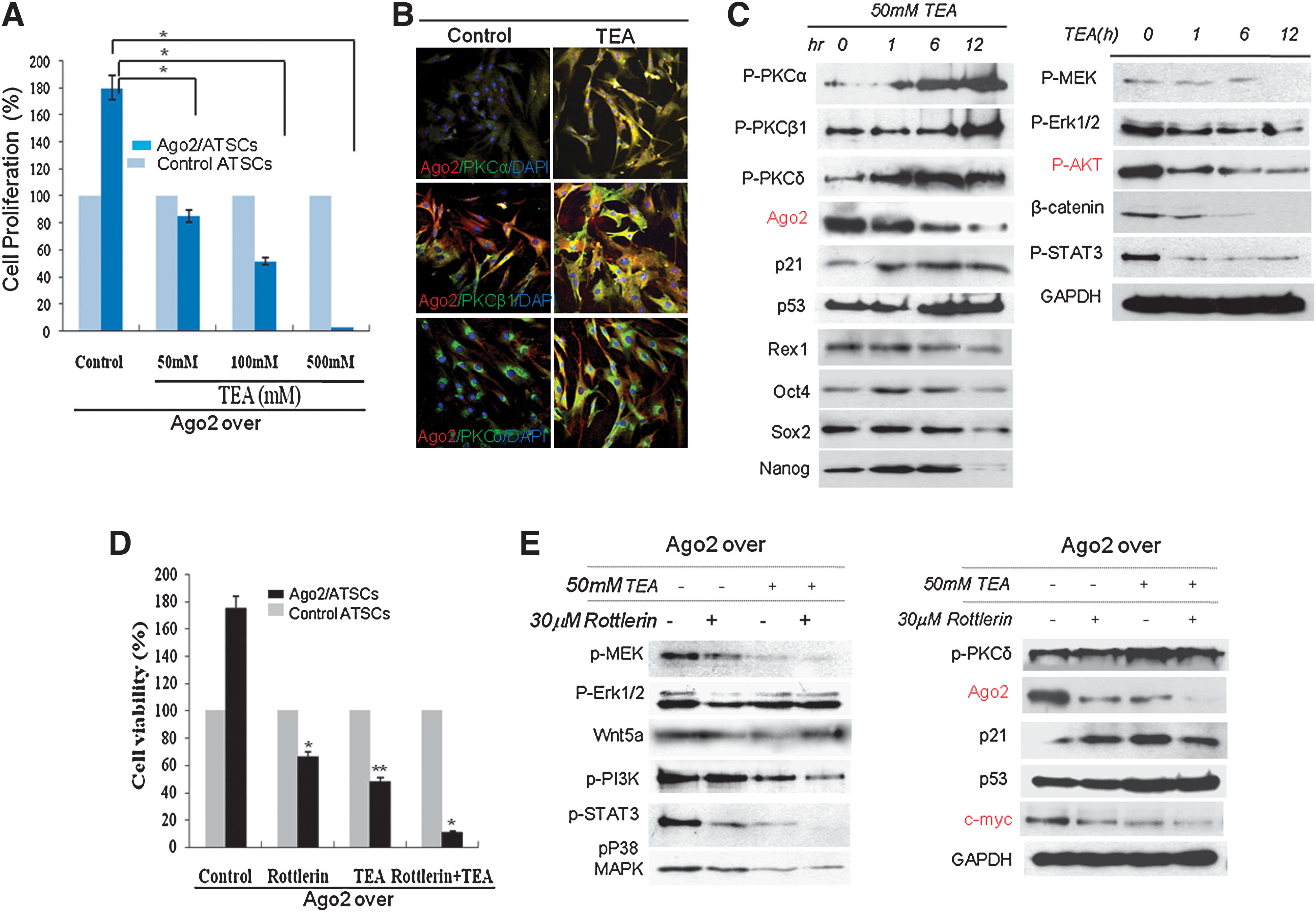
During Ago2/K+ channel-mediated ATSC self-renewal, the PKC δ-mediated signaling pathway induces stemness expression.
Stemness is directly controlled by Ago2 in ATSCs
Our ChIP/PCR results demonstrated that Ago2 directly regulates stemness gene expression following its binding to the regulatory regions of functional genes (Fig. 4A). We also determined that, along with Ago2, Oct4 also directly regulates KCNA1 and KCNN3 expression via binding to the regulatory regions of these genes (Fig. 4B). To define the role of Oct4 regulation in K+ channel expression, we assessed KCNA1, KCNN2, and KCNN3 expression levels after RNAi-induced interference knockdown of Oct4 expression in ATSCs. Blockage of Oct4 expression significantly downregulated the expression of the K+ channel subtypes KCNA1, KCNA2, and KCNA3 in cultured ATSCs (Fig. 4C). To determine the role of the expression of stemness genes in Ago2-overexpressing ATSCs, following interference with stemness gene expression, we examined the relative self-renewal activity of ATSCs via tryphan blue exclusion viable cell counting. As shown in Fig. 4D, knockdown of Nanog, Sox2, and Oct4 significantly blocked the capacity of Ago2-expressing ATSCs for self-renewal. Moreover, in Ago2-overexpressing cells, RNA-induced knockdown of the stem cell genes Ago2, c-myc, CDK2, and CDK4 negatively regulated cell cycle progression and the capacity of ATSCs for self-renewal. In contrast, stemness gene knockdown effectively induced the upregulation of p53 and p21 expression (Fig. 4E). Collectively, we showed that Ago2 and K+ channel expression control a gene expression regulatory network (Fig. 4F).
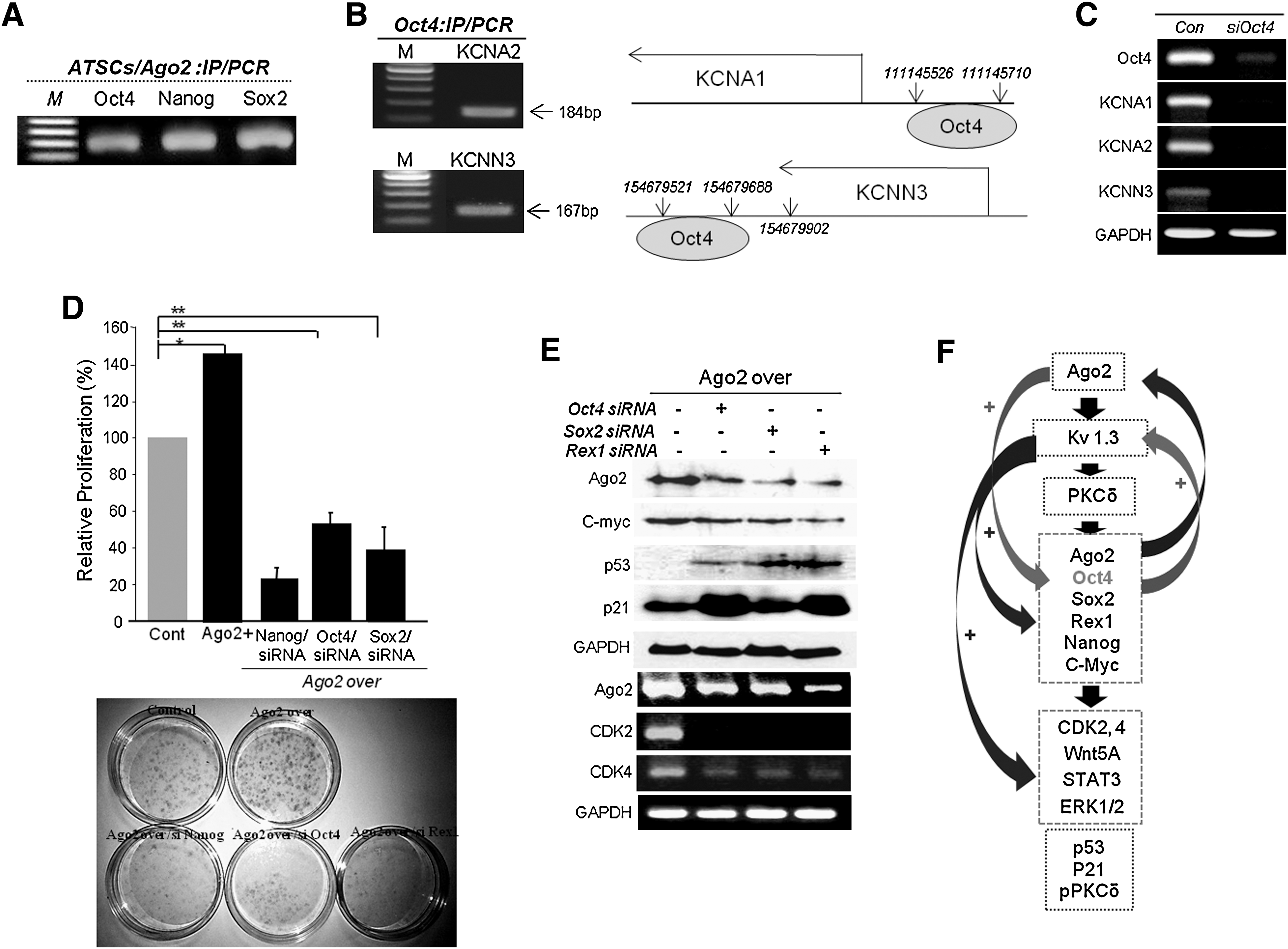
Ago2 indirectly controls the regulation of KCNA2 and KCNN3 gene expression via Oct4 regulation.
The silencing of K+ channel genes results in a significant downregulation of stemness gene expression and the self-renewal of ATSCs
To determine the nucleotide sequence required for Ago2 binding in specific genes, we performed single nucleotide or multiple nucleotide mutagenesis of the KCNA1 gene in Ago2-overexpressing ATSCs or control cells. As shown in Fig. 5A, the binding affinity of Ago2 for each mutant KCNA1 gene was significantly decreased compared with control cells (Fig. 5A). Moreover, the expression of mutated KCNA1 caused attenuated cell self-renewal activity, and KCNA mutant 1-transfected cells entered cell senescence processes, were not stably attached, and exhibited increased cell spreading (Fig. 5B, C). When we transfected KCNA1 mutant 1 into Ago2-expressing ATSCs, the expression levels of stemness genes Nanog, Oct4, Sox2, and RUNX3 were prominently downregulated compared with control ATSCs (Fig. 5D). Additionally, we identified the nucleotide sequence bound by Ago2 in both Ago2-overexpressing ATSCs and control ATSCs. These cell types shared consensus sequence motifs in the Ago2-binding genes Oct4, Nanog, Sox, and KCNA1. Through the overlapping of multiple ChIP DNA fragment sequences and through qualitative analysis of mutated putative Ago2 binding sites via ChIP-qPCR, we also detected several miRNAs bound by Ago2 (data not shown). Site-directed mutagenesis of the Ago2 binding sequence in KCNA1 resulted in the attenuation of cell self-renewal and the downregulation of stemness and cell cycle-related gene expression (Fig. 5C, D). Figure 5E shows the Ago2 binding region on the KCNA1 gene, with positive KCNA1 gene expression regulation confirmed at the transcriptional level. Finally, we obtained an Ago2 composite element consisting of an Ago2 binding site consensus sequence (5′-CTTCCTC(G)-3′ or 5′-CTTCTC(G)-3′) adjacent to Sox2 and c-myc binding elements on a specific chromosome (Fig. 5A, B). Thus, we identified the specific nucleotide sequence that mediates Ago2 binding to the regulatory region of the KCNA1 gene (Fig. 5F).
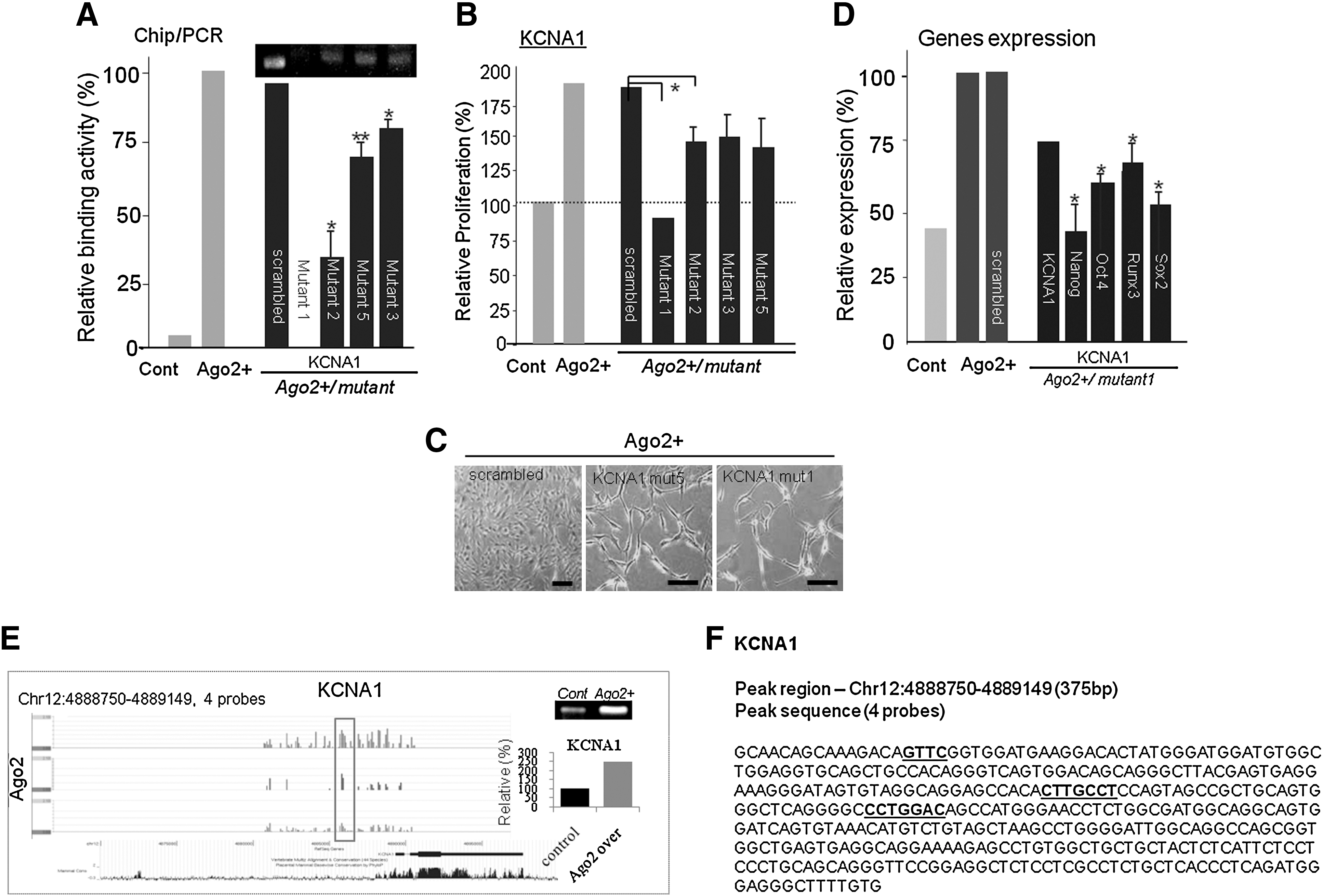
The molecular role of KCNA1 during ATSC self-renewal and stemness gene expression.
The involvement of Ago2 and KCNA in ATSC aging and self-renewal
We determined the differential expression patterns of Ago2, Oct4, and KCNA1 at various passages of ATSCs. Ago2 and stemness gene expression gradually increased in early passage (P10) cells and significantly decreased in late passage (P21) cells (Fig. 6A, B). Cell proliferation activity and self-renewal-related gene expression patterns were similar to Ago2 expression patterns, with stemness and KCNA1 expression increased in early passages and decreased in later passages (Fig. 6B, C). KCNA1 expression at various cells passages was also related to cell self-renewal ability, with KCNA1 expression gradually increasing up to P10 and gradually decreasing after P10 (Fig. 6D). Additionally, the nuclear localization of Ago2 increased at P10 and decreased at P21 (Fig. 6E). The DNA binding affinity of Ago2, as determined by ChIP/IP, revealed that the Ago2 DNA binding affinity gradually increased in later cell passages (Fig. 6F).
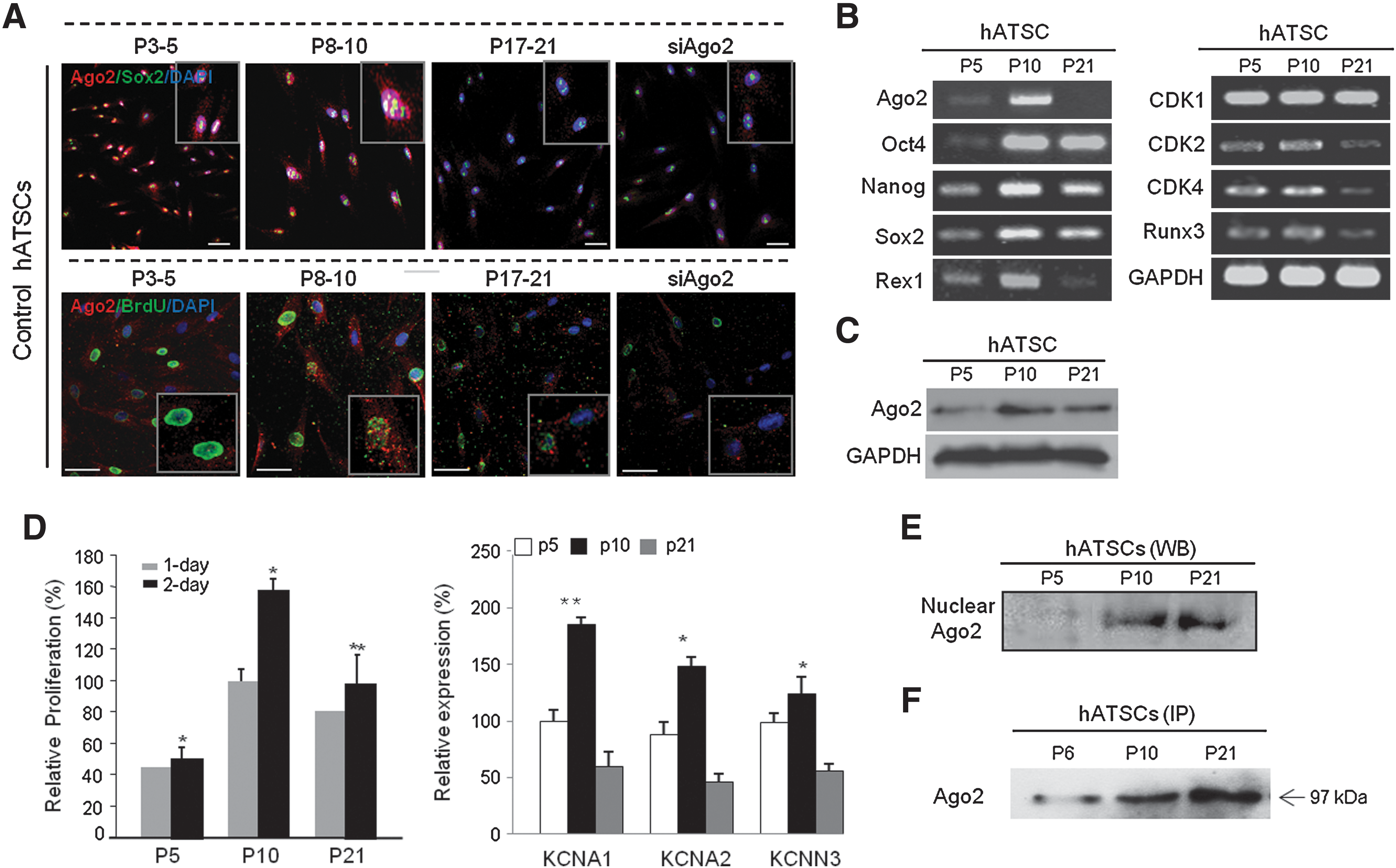
Analysis of the Ago2-mediated differential expression of stemness and self-renewal related genes in early and late passages of ATSCs.
The role of KCNA during Oct4-mediated ATSC differentiation
Stem cells isolated from mesodermal tissue, ATSCs, will preferentially differentiate into cells of mesodermal lineage, such as bone, fat, or cartilage, in differentiation induction media. In our ATSCs, calcium deposits and lipid droplets were produced in cells after 2 to 4 weeks of differentiation induction. We studied the role of one of K+ channel, KCNA, in ATSC differentiation. Knockdown of KCNA1 expression in ATSCs caused the accumulation of large calcium deposits. Lipid droplet formation frequency was also different between the control ATSCs and ATSCs treated with the K+ channel blocker TEA. Figure 7 shows that a nearly 450% increase in lipid droplets and calcium nodules was detected in TEA-treated cells compared with control cells. Our staining results are consistent with our gene expression analysis of the transcription factors osteopontin, osteonectin, PPAR gamma, and AP (Fig. 7A). Our previous study showed that KCNA1 expression is controlled by Oct4 expression in ATSCs. Therefore, we also determined the effect of Oct4 expression on mesodermal differentiation in ATSCs. Knockdown of Oct4 expression significantly increased adipogenesis, chondrogenesis, and osteogenesis and additionally downregulated lineage-specific gene expression (Fig. 7B).
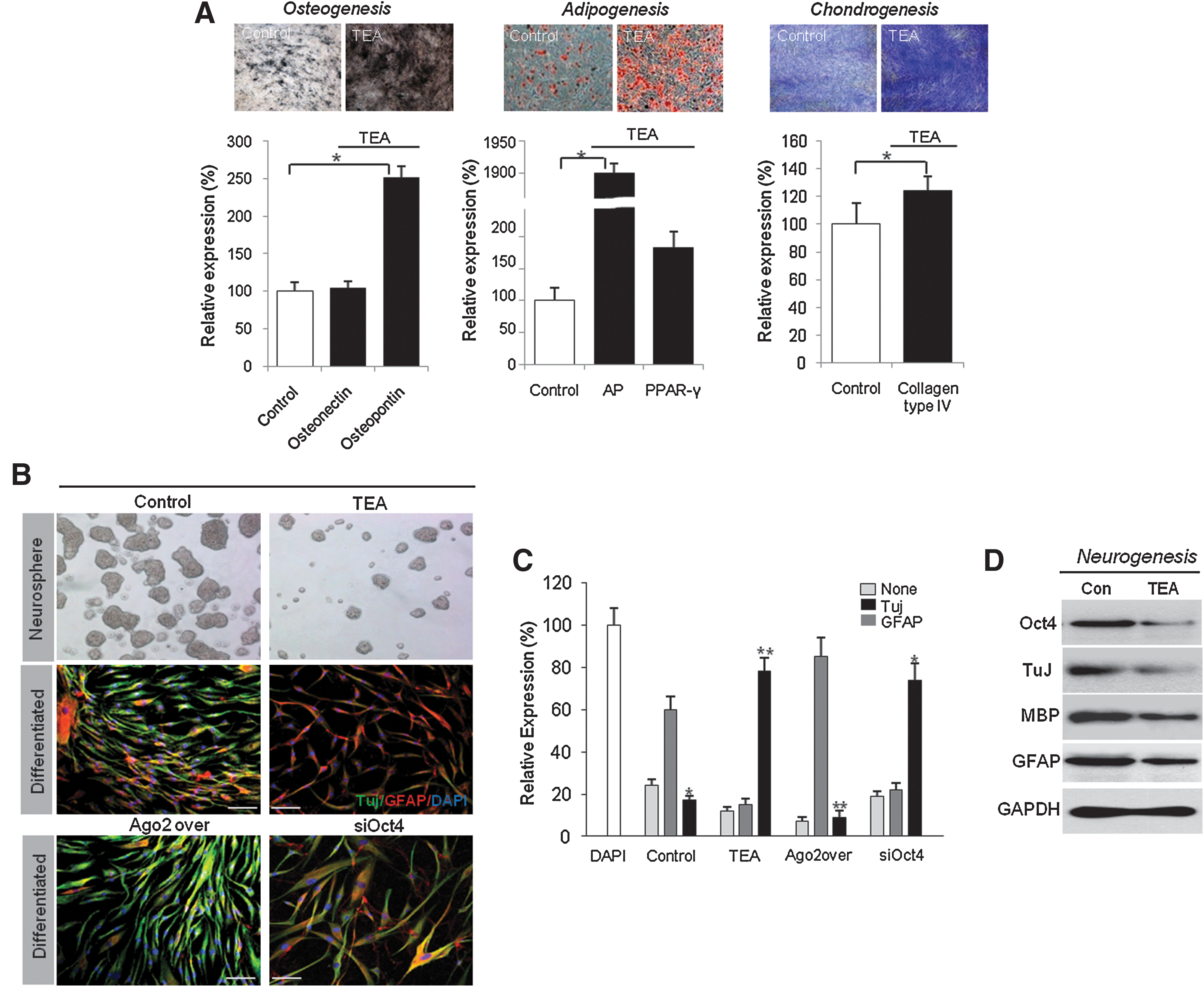
The crucial role of K+ channels in ATSC differentiation.
Moreover, we determined the neurogenic potency of KCNA1-expressing or silenced cells. After neural differentiation of KCNA1-expressing control cells or Ago2-overexpressing cells, KCNA1 was upregulated in ATSCs for 7 days. We evaluated neural differentiation potency by immunocytochemistry and western blot using the neural-specific antibodies TuJ and GFAP. Our in vitro results showed that KCNA1-expressing ATSCs exhibited a prominent upregulation of the neuronal markers Tuj, myelin basic protein, and GFAP in comparison with control ATSCs (Fig. 7C, D).
Discussion
The crucial role of K+ channels in the proliferation of several cell types, including immune and cancer cells, has been reported [35,36]. Additionally, a putative role for these channels in apoptosis and proliferation in many cell types has been suggested [37 –39]. Voltage-gated channels (Kv) control repolarization following action potentials and the electrical excitability of nerves and muscles [37]. These channels also have a function in apoptosis and proliferation in mammalian cells [40]. Several reports have confirmed functions for Kv channels in cell differentiation and cell cycle control [38,40]. For instance, protein expression patterns of K+ channel subtypes were reported to vary with the cell cycle phase in adult stem cells, and K+ channels were found to contribute to cell cycle progression during stem cell self-renewal. Additionally, the general K+ channel blockers 4-AP and TEA effectively induce cell cycle arrest. Although neither direct evidence nor a molecular mechanism exist, reports on TEA-mediated cell self-renewal inhibition suggest that Kv channels may be inhibited by CDK inhibitors, such as P21 and P27 [12,36,41 –43]. A recent study also reported the identification of mutations in K+ channel genes in glioblastoma, breast cancer, and colorectal cancer and showed that the mutations changed conserved residues in or near voltage-regulating segments [44 –48]. The report also showed that endocrine tumors, which can cause severe hypertension, had uncontrolled hormone secretion and unrestrained cell proliferation. The authors argue that abnormal aldosterone-producing adrenal adenomas were generated by recurrent somatic mutations in the K+ channel gene in the human samples [45].
Previously, we showed that Ago2 expression was crucial for ATSC differentiation and self-renewal. Analysis of the cell cycle profile of Ago2-knockdown cells showed a reduced S phase cell population, whereas Ago2-expressing cells showed a significantly expanded S phase cell population with the upregulation of CDK, RUNX3, and c-myc genes. Here, our experiments done in cells expressing Ago2 revealed that Ago2, Sox2, and c-myc collaboratively bind to the regulatory regions of KCNN3, KCNN1, and KCNA2 (Fig. 2). We also show that Ago2-expressing cells had a significantly higher frequency of DNA binding activity. Ago2 typically binds cooperatively with c-myc and Sox2 to the regulatory region of Ago2 itself, and Ago2 was previously suggested to either directly regulate specific gene expression or to affect miRNAs to elicit new cellular functions [49]. In this study, we show that Ago2 binding to the regulatory regions of DNA plays a significant role in cell reprogramming by regulating the expression levels of functional genes, particularly the K+ channel genes KCNN1, KCNN3, and KCNA2. These channel genes also play important roles in the self-renewal of ATSCs through PKC δ, JAK/STAT3, and P38MAPK phosphorylation and stemness expression (Fig. 3C). Experimental knockdown of the KCNA1 gene significantly attenuated Ago2-mediated self-renewal and resulted in a decreased population of S phase cells. In contrast, it was reported that knockdown of miR10b actively induced cell self-renewal with increased S phase cell density and, additionally, resulted in significantly increased self-renewal-related and stemness gene expression [49]. When we inhibited KCNA1 expression, stemness genes and cell self-renewal-related genes were significantly downregulated (Fig. 3A, C). The functions of Ago2 in adult tissues and stem cells have been increasingly explored in our various studies. Along with miRNAs, which are themselves part of the miRNA processing machinery, Ago2 is essential for proliferation and the ability of cells to transdifferentiate into most tissue and cell types, including the endodermal and mesodermal lineages. Functional disruption of the miRNA processing machinery or Ago2 is sufficient to reduce the differentiation and proliferation ability of stem cells. Our results suggest that the specific functional miRNAs and genes modulated by nuclear Ago2 play critical roles in stem cell pluripotency and that Ago2 expression can induce a cell reprogramming event that results in a more pluripotent stem cell status [49].
In conclusion, we suggest an expanded model of nuclear Ago2 function that involves the stem cell transcriptional regulatory circuitry and its molecular role. Nuclear Ago2 coordinates the transcriptional regulation of specific functional genes and post-transcriptional regulation by overloaded miRNAs. The discovery that nuclear Ago2 plays a very important role as a key regulator for K+ channel/Oct4-mediated stem cell behaviors allows for an increased understanding of the molecular events involved in stem cell pluripotency and escape of differentiation (Fig. 5F, 7). Our current study revealed that Ago2 and the miRNA processing machinery actively induce cell reprogramming by cooperating with Oct4, Sox, and c-myc to modulate transcription. Following escape from the aging process, these proteins participate in a feed-forward regulation event that modifies the gene expression program to obtain stem cell self-renewal and differentiation abilities.
Footnotes
Acknowledgments
This work was supported by the National Research Foundation of Korean (NRF) grant funded by the Korea government (MEST, 2010-0020265).
Author Disclosure Statement
The authors have no competing financial interests to disclose.