
Abstract
Human mesenchymal stromal cells (MSCs) have been successfully utilized for the treatment of refractory graft-versus-host disease (GvHD). Despite the large number of in vitro and in vivo models developed for clarifying their immunomodulatory properties, the mechanism of action of MSCs remains elusive and their efficacy controversial. Here, we tested the ability of cord blood-derived MSCs to alleviate the symptoms of GvHD induced by the injection of human peripheral blood mononuclear cells into NOD/SCID/γc− mice. In this in vivo xeno-GvHD model, we demonstrate that a single MSC injection is able to inhibit GvHD in terms of clinical signs and related mortality. We also show that in this model MSCs act by both immunomodulating T-cells and fostering recovery after irradiation. The translational impact of these findings could provide a reliable preclinical model for studying the efficacy, dosage, and time of administration of human MSCs for the prevention of acute GvHD.
Introduction
A
Methods
Cord blood-derived MSCs
Cord blood samples were obtained from the CHU Sainte-Justine Research Cord Blood Bank following approval by the ethics committee. Human cord blood-derived MSCs were obtained as previously described [20,21], with the modification that cord blood-derived PBMCs (106 cells/75-cm2 flask) were initially plated in a conditioned medium [alpha-minimum essential medium (MEM) supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin; all purchased from Invitrogen, Burlington, Ontario, Canada]. Conditioned medium was collected after 24 h incubation over an established bone marrow-derived MSC population and changed twice a week until cells adherent to plastic formed visible colony-forming units. Cells were then trypsinized and expanded in regular alpha-MEM and characterized for the expression of common MSC markers by flow cytometry. As expected, MSCs were negative for CD45 and CD31 and positive for CD90, CD44, and CD105 (see Supplementary Fig. S1; Supplementary Data are available online at
Mice
NOD/LtSz-scidIL2rγ−/− (NSG) mice were obtained from the Jackson Laboratory and housed in the animal care facility at the CHU Sainte-Justine Research Centre under specific pathogen-free conditions in sterile ventilated racks. All procedures were previously approved by the institutional committee for good laboratory practices for animal research (GLPAR), as per our research protocol (SST09-22). Irradiated mice were given 0.08 mg/mL ciprofloxacin (Sandoz, Boucherville, Québec, Canada) in their drinking water.
Xenogenic model of GvHD
Leukocytes, collected by leukapheresis from healthy donors following informed written consent, were used in the isolation of huPBMCs by Ficoll-Hypaque separation. HuPBMCs were then frozen in FBS with 10% DMSO and stored in liquid nitrogen until needed. Following a 3 Gy irradiation, 6–8-week-old mice were injected intraperitoneally with 107 huPBMCs at day 0 and treated on the same day, with either 100 μL of phosphate-buffered saline (PBS) or 106 MSCs, intravenously. CsA-treated mice (Sandimmune I.V.; Novartis Pharmaceutical, Dorval, Québec, Canada) were injected intraperitoneally daily (0.375 mg/mouse). Both the number of injected MSCs and the dose of CsA were decided based on data previously reported [25,26].
Assessment of GvHD
Treated mice were evaluated every other day in a blinded fashion for clinical signs of GvHD and physical appearance. The GvHD assessment scale, inspired by the grading system established by Cooke et al. [27], refers to weight loss, general appearance of the fur, and mobility to evaluate the development of GvHD. Mice were diagnosed with GvHD based upon a minimum of 10% weight loss, appearance of ruffled fur, and limited mobility [18]. Survival was also noted for each group. As requested by our GLPAR committee, mice were sacrificed when the clinical endpoints were reached (weight loss >20% and appearance of either limited mobility or disruption of general appearance of the mice).
Flow cytometry
For immunophenotypic analysis of circulating lymphocytes, 100 μL of peripheral blood was drawn weekly from the saphenous vein of xeno-GvHD mice, and human and murine cell populations were analyzed by flow cytometry. Anti-murine CD45-fluorescein isothiocyanate (FITC) and anti-human-CD56-phycoerythrin (PE), -CD45-Allophycocyan (APC), and -CD3-APC-Cy7 antibodies (all purchased from BD Biosciences, Mississauga, Ontario, Canada) were used to identify cell populations, along with 7-amino-actinomycin D (7-AAD) for exclusion of nonviable cells from the assay (BD Biosciences). CountBright beads (Invitrogen) were added before fluorescence activated cell sorting (FACS) acquisition to determine the absolute number of cells in each sample. Samples were acquired on a FACSAria flow cytometer and analyzed using FACSDiva software (BD Biosciences).
Histology and immunohistochemistry
Organs (liver, spleen, colon, and skin) were fixed in 3.7% formaldehyde immediately after collection and embedded in paraffin. Five-micrometer-thick sections were obtained and stained for further analysis. Examination was performed after hematoxylin–eosin staining and evaluated for signs of GvHD by a trained pathologist blinded to the clinical data. For human T-cell identification, indirect immunoperoxidase staining was performed with an anti-CD3 antibody (Cellmarque, Rocklin, CA). Apoptotic cells were detected by morphological analysis and both CD3+ cell and apoptotic cell numbers were quantitatively evaluated per 45 high-power fields (400×).
Ex vivo human cell phenotype and cell cycle analysis
We sacrificed animals injected with PBMCs and treated with either MSCs, CsA, or PBS at day 14 and performed immunophenotype and cell cycling analysis on cells isolated from blood, spleen, and liver. For activation marker analysis, cells were stained with anti-human-CD3-PE-Cy7, -HLA-DR-FITC, -CD69-PerCp-Cy5.5, and -CD8-APC. Cell cycle analysis was performed by surface staining followed by intracellular staining for Ki67-FITC (marker for cells out of G0 phase). Regulatory T cells (Tregs) were analyzed for cell surface markers (CD4, CD25, CD127) on blood samples, whereas for organ samples, intracellular staining for forkhead box P3 (FoxP3-APC) was added (fixation and permeabilization solution was used according to the manufacturer's instructions; eBioscience, San Diego, CA). All antibodies were from obtained from BD Biosciences.
Human dendritic cell differentiation
To differentiate human dendritic cells (DCs) from monocytes, CD14+ cells were isolated with magnetic beads by positive selection (Miltenyi Biotec, Auburn, CA) and plated in 6-well plates (106 cells/well). Cells were cultivated for 5 days in the presence of human recombinant granulocyte-macrophage colony-stimulating factor (50 ng/mL; R&D Systems, Minneapolis, MN) and interleukin 4 (IL-4; 10 ng/mL; eBioscience). The phenotype of both monocytes (at day 0) and immature DCs (at day 5) was tested by staining cells with the following antibodies: anti-CD14-FITC, -CD1a-APC, -CD80-PE, -CD83-PE, -CD86-PE, and -HLA-DR-FITC.
In experiments requiring murine peptide loading into human DCs, 2×105 splenocytes were isolated from NSG mice and resuspended in 50 μL PBS, sonicated, and freeze-thawed and the immature human DCs were loaded for 2 h at 37°C. Cells were then irradiated (5,400 rad) and used as targets for T-cell stimulation.
Human T-cell isolation and stimulation
T cells were purified by magnetic bead negative selection (Miltenyi Biotec) and 2×105 cells were cultivated for 6 days with autologous DCs pulsed or not with NSG antigens (ratio T:DC=40:1) in 96-multiwell round-bottomed plates. Alternatively, 2×105 huPBMCs were cocultured with 2×105 NSG-irradiated (5,400 rad) splenocytes. To test the immunomodulatory effect of MSCs in vitro, each condition was also tested in the presence of 2×104 irradiated MSCs (ratio of effector cells:MSCs=10:1). Human cell proliferation was tracked by flow cytometry using the fluorescent dye, carboxyfluorescein succinimidyl ester (CFSE; Invitrogen). Briefly, at day 0, effector cells were resuspended in PBS and stained with CFSE (final concentration: 0.625 μM) for 7 min at room temperature. CFSE incorporation was blocked by adding 1 mL of pure FBS and incubating for 1 min on ice followed by 2 washes in complete culture medium (RPMI, 10% FBS, 100 U/mL penicillin/streptomycin, and 0.1 mM glutamine). All tested conditions were done in duplicate, one well with CFSE-stained cells (to test for proliferation) and one with unstained cells (to stain for markers of T-cell activation). As read out for the mixed leukocyte reaction (MLR), cells were stained on day 6 with anti-CD3-PE-Cy7, -CD4-APC, -CD8-PE, -HLA-DR-FITC, -CD69-PerCp-Cy5.5, -CD25-PE, and 7-AAD (cell viability dye).
Statistics
Results were analyzed using GraphPad Prism version 5.0 (GraphPad Software, Inc., La Jolla, CA) and shown as mean±SEM. Survival and incidence of GvHD statistics were determined using the Mantel-Cox (log-rank) test, which was stratified according to time as needed. Elsewhere, either the t-test or Mann–Whitney test was used, as specified in each figure legend. Significance was set at P≤0.05.
Results
MSC treatment increases survival and reduces GvHD incidence upon PBMC injection
We induced xeno-GvHD by injecting 2 groups of sublethally irradiated NSG mice (n=16 per group) with 107 huPBMCs. The control group was treated with PBS and the other with a single dose of 106 MSCs. As shown in Figure 1A, almost all control mice died within a period of 42 days, whereas mice treated with MSCs at day 0 demonstrated a significant increase in survival (P<0.01). Furthermore, injection of MSCs was also associated with a significant reduction of GvHD in terms of clinical signs (weight loss, general appearance of the fur, and mobility) when compared with PBS-treated mice (P<0.01; Fig. 1B).

MSCs increase survival following induction of xeno-GvHD in NSG mice. NSG mice were injected with 107 huPBMCs and treated intravenously with PBS (filled line, n=16) or with 106 MSCs (dotted line, n=16). In the MSC-treated group, both the mortality
MSCs protect mice from radiation-induced weight loss in vivo and reduce human T-cell proliferation
In different in vivo models, MSCs were able to inhibit T-cell activation and T-cell proliferation [28,29]. MSCs were also shown to foster the healing process [30 –36] with a major regenerative effect on irradiation-induced tissue damage [37]. As both these 2 properties could explain the inhibition of GvHD in our model, we tested the effect of MSCs on tissue following sublethal irradiation of NSG mice and treating or not treating them with a single dose of 106 MSCs. As shown in Figure 2, MSCs significantly prevented radiation-induced weight loss at the early time points.
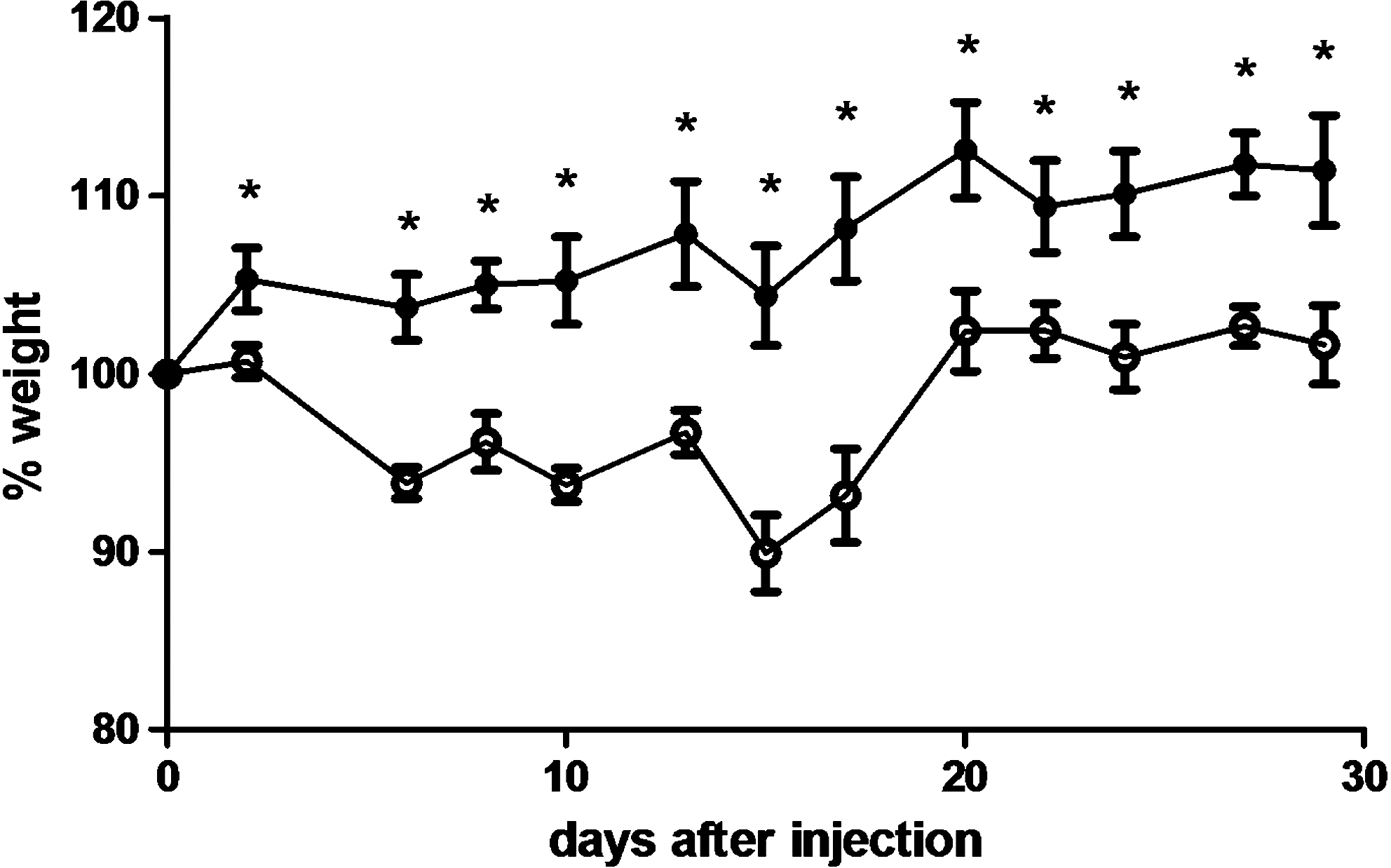
MSCs have a protective effect against radiation-induced damages. NSG mice were treated with MSCs (n=3, filled circles) or PBS (n=5, empty circles) upon irradiation (3 Gy). Animal weight was recorded every other day. *P≤0.05, Mann–Whitney one-tailed test.
To evaluate a possible effect of MSCs on human T-cell proliferation, we initially compared in blood the proportion of human cells over the total number of circulating CD45+ cells (murine and human) at different time points in the PBS- and MSC-treated groups. At every time point (ranging from 7 to 42 days postinjection), all human leukocytes (CD45+ cells) were T lymphocytes (CD3+ cells; data not shown). We observed a significant decrease in the proportion of human cells at days 7 and 14 in the MSC-treated group in comparison with the PBS-treated group (Fig. 3A). Nevertheless, the absolute count of human CD45+ cells in the PBS-treated group was lower than in the MSC-treated group (Fig. 3B). The apparent discrepancy between the results based on percentage compared with absolute number was due to the higher number of murine cells in the MSC-treated group (Fig. 3C).

MSCs reduce the percentage but not the absolute number of human cells after xeno-GvHD induction. Upon injection with 107 huPBMCs, circulating human and murine CD45+ cells were counted by flow cytometry using counting beads. Results are expressed as a percentage of human CD45+ cells on total CD45+ cells (hCD45/hDC45+mCD45)
As immunomodulators, either MSCs could inhibit the effects of human T-cells, consequently allowing the circulation of a higher number of human T-cells, or they could actively inhibit T-cell proliferation. According to the latter possibility, one would have expected the amount of human cells to be higher in the PBS-treated group than in the MSC-treated group. Yet, our results showed that the absolute number of human CD45+ cells was not higher in the PBS-treated mice, suggesting that human MSCs do not inhibit T-cell proliferation in this context. Nevertheless, it is also possible that cord blood MSCs inhibit T-cell proliferation but that the effect is not detected because of the higher incidence of GvHD leading to early death in PBS-treated mice. To verify this hypothesis, we injected huPBMCs into nonirradiated NSG mice and tested the effect of MSCs compared with PBS. We observed that in absence of radiation-induced damage, MSCs were able to significantly decrease both the percentage and the absolute number of human cells in mice up to 42 days postinjection (Fig. 4). Moreover, we observed that in nonirradiated mice the absolute number of human cells in the peripheral blood of injected animals reached levels that were much higher (about 30-fold more) than those observed in irradiated mice, without killing mice. This indicates that without irradiation, animals are more readily able to tolerate the proliferation of human T-cells (Fig. 4).

MSCs reduce both the percentage
To test the antiproliferative effect of MSCs in our xeno-GvHD model, mice were sacrificed 2 weeks after the injection of huPBMCs to analyze GvHD before mortality occurred. Using this strategy we measured the effect of MSCs compared with PBS or CsA on human T-cell proliferation and GvHD lesions in the liver, a major target organ in xenogenic GvHD [19,38]. CsA was used as control, as this drug is routinely used for GvHD prophylaxis in humans [39], and we have already demonstrated its efficacy in inhibiting GvHD and GvHD-related mortality in our xeno-GvHD model in NSG mice [18]. Liver sections were stained for morphological evaluation of the number of both human T-cells and apoptotic murine hepatocytes. The number of human CD3+ cells was significantly reduced in both CsA- and MSC-treated mice when compared with PBS-treated mice, but no significant difference was observed between MSC- and CsA-treated mice (Fig. 5A, B). We also observed a reduced number of apoptotic bodies in the liver sections obtained from MSC- and CsA-treated mice when compared with PBS-treated mice (Fig. 5C, D), although this decrease was not statistically significant. Consistent with the histopathology data, analysis of Ki67 expression revealed that MSC treatment decreased the percentage of cycling human CD4+ cells in the liver in comparison to PBS treatment, although not to the same extent as CsA (Fig. 5E). We also analyzed the expression of activation markers on the surface of human T-cells (HLA-DR, CD69, and CD25) in both blood and liver. We observed that MSC treatment did not induce a significant reduction of these markers, whereas CsA was able to significantly decrease HLA-DR expression in both CD4+ and CD8+ cells and to induce a moderate, although not significant, increase of CD69 expression in CD4+ cells (Fig. 6). Moreover, we did not find any increase of Tregs in MSC- and CsA-treated mice when compared with PBS-treated mice (data not shown).

MSC treatment affects human T-cell proliferation and infiltration and murine cell apoptosis in the liver of NSG mice upon xeno-GvHD induction. Animals were injected with 107 huPBMCs, treated with CsA (n=5), PBS (n=5), or MSCs (n=4), and sacrificed 2 weeks later for histopathologic analysis. Infiltrating human CD3+ cells were detected by immunohistochemistry and were counted in the different treatment groups
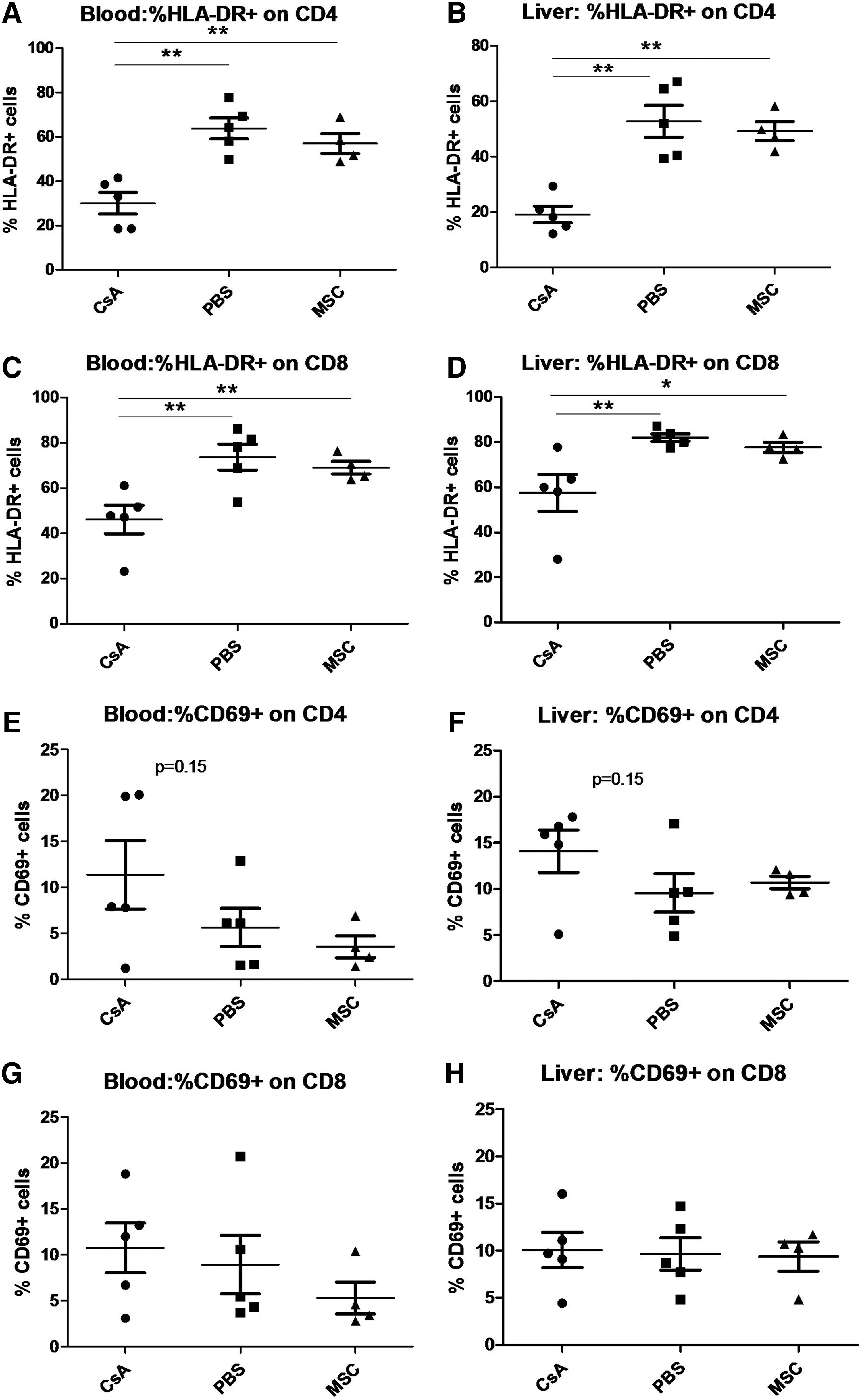
MSC injection does not modify the activation marker profile of human T-cells. Activation marker expression in human CD4+
Taken together, these data suggest that MSCs ameliorate GvHD by protecting the animal from radiation-induced damage and inhibiting T-cell proliferation.
Acute human T-cell activation against NSG cells is mainly driven by human APCs
We performed in vitro experiments aimed to determine whether human T-cells were able to trigger xeno-GvHD by interacting with human or murine APCs. As shown in Figure 7A–D, NSG splenocytes failed to stimulate huPBMCs, suggesting that murine major histocompatibility complex could not trigger efficiently the activation of human cells. However, we observed that human DCs loaded with lysates from NSG mice splenocytes could induce autologous human CD4+ T-cell activation and proliferation (Fig. 7E–H). As expected, CD8+ cells were not stimulated in this experimental setting, because the T-cell activation occurs as a result of HLA class-II presentation by autologous APCs (data not shown). These data indicate that human APCs are able to sample murine antigens and to efficiently present them in the context of human HLA molecules. In this in vitro model of xenogenic stimulation, MSCs strongly inhibited T-cell proliferation, and unlike our in vivo findings, we observed a significantly decreased expression of the activation marker HLA-DR in the presence of MSCs (Fig. 7F). We also observed that MSCs induced a very moderate, although not significant, increase of CD69 expression (Fig. 7H). Moreover, MSCs were unable to induce an immune response or to present NSG antigens to T cells, confirming their status as immunoprivileged cells despite their third-party donor origin (Fig. 7E–H). Taken together, and as already suggested by Wilson et al. [40], these data suggest that the activation of human T-cells in our xenogenic model of GvHD is mainly due to the presentation of murine antigens by human APCs and that this process can be strongly inhibited by the presence of MSCs.
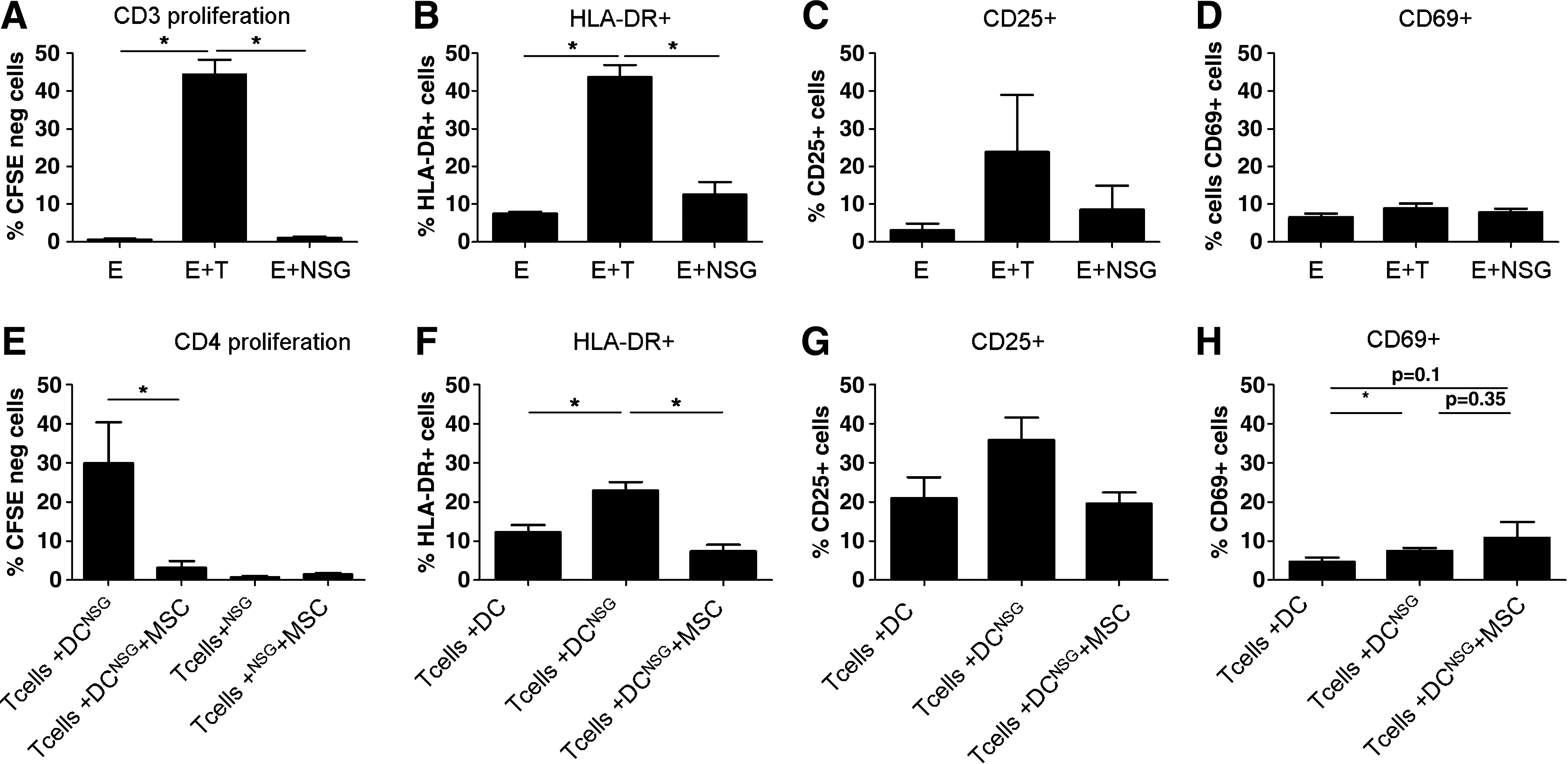
Human APCs are able to present murine antigens and trigger T-cell activation.
Discussion
Based on their immunomodulatory properties, MSCs have been proposed as a potential treatment for acute GvHD in humans. Several clinical trials have demonstrated their potential efficacy and lack of side effects in patients receiving third-party donor MSC infusion [7,8,11]. To study the best dosage, administration strategy, and mechanism of action of MSCs, various murine models have been developed [41 –43]. In the present study, we show that human MSCs are able to inhibit the occurrence of GvHD induced by huPBMC injection in sublethally irradiated NSG mice.
Thus far, the immunomodulatory properties of MSCs in vivo have been studied mostly in immunocompetent mice transplanted with mismatched murine bone marrow cells and treated with either murine [42] or human [44] MSCs. The advantage of our model is that it allows one to study in vivo both effector cells (T cells) and MSCs of human origin, and this is an important consideration given the known differences between human and murine MSCs in terms of immune modulation [45,46]. A possible drawback of the NSG xeno-GvHD model is that GvHD, although triggered by human cells, develops in a murine environment and cannot reflect exactly what occurs in the clinical setting after HSCT. Moreover, huPBMCs are injected without any previous bone marrow transplant. Nevertheless, this approach has the important advantage of modeling the effect of MSCs alone as GvHD treatment, which would be ethically impossible in humans, because for clinical reasons MSC infusion is always accompanied by other immunosuppressive therapies, rendering the interpretation of the clinical results difficult.
To our knowledge the only previous study using human MSCs in immune-deficient mice has been performed with NOD/SCID animals [41]. These mice, although severely immunocompromised, still present circulating murine natural killer cells and indeed require double the number of PBMCs to induce GvHD. In addition, the disease develops considerably later with respect to our model. Moreover, in the aforementioned study, the authors considered the variation of human cells only in terms of percentage, but in the present study we demonstrate that it is possible to observe a discrepancy between the percentage and absolute number of human CD45+ cells, depending on the effect that MSCs have on murine hematopoietic cells.
Another advantage of our model is that one single MSC injection at day 0 was sufficient to observe clinical efficacy, whereas repeated injections were required in the NOD/SCID model. Regarding the mechanism of GvHD occurrence in PBMC-injected NSG mice, human T-cells could be activated by both murine cells and human APCs present in the graft. Data reported in the literature underline the necessity for the presence of a donor's DCs in the transplant to trigger acute GvHD [40]. Our in vitro data support this perspective, as murine splenocytes were not able to induce human T-cell proliferation or activation in an MLR, whereas human autologous DCs pulsed with NSG lysate were potent APCs.
MSCs can be derived from various adult and fetal tissues [47]. We performed our experiments using an immortalized cord blood-derived MSC line. Use of an immortalized cell line can be seen as a potential caveat, as in the clinical setting only freshly derived and expanded MSCs have so far been accepted for cellular therapy. Nevertheless, our cell line has been repeatedly tested for quality control and has never given rise to a cellular mass (either in vitro or in vivo). Further, in the context of setting up a humanized mouse model to study the mechanism of action of MSCs to prevent GvHD, the importance of standardizing the quality of the MSCs cannot be overstated. Moreover, it has been shown that although displaying a higher self-renewal and proliferative capacity [48], cord blood-derived MSCs appear to exhibit similar progenitor activity compared with bone marrow-derived MSCs.
In our NSG xeno-GvHD model, MSC-treated mice presented a better clinical picture in several respects; they displayed fewer clinical signs, had less weight loss, and had reduced human T-cell infiltration in the liver (the main GvHD target organ [19,38]). Such clinical improvement occurred despite a lack of downmodulation in the expression of activation markers in blood- and liver-derived human T-cells. Therefore, from these data, it is difficult to determine whether MSCs are acting as immunomodulators or as reparative agents. The importance of MSCs as protective agents has been already underscored in humans [49], but this aspect has so far not been taken into consideration in the proposed animal models. Using irradiated mice not injected with PBMCs, we have shown that MSCs can inhibit weight loss, thereby demonstrating that MSCs could reduce side effects induced by irradiation. Also, we observed that in irradiated mice injected with PBMCs, MSC treatment was associated with a rise in murine CD45+ cell count that likely accounts for the observed inhibition of radiation-induced bone marrow damage.
To isolate the immunomodulatory component of MSCs in the treatment of xeno-GvHD inhibition, we injected huPBMCs into nonirradiated mice. As expected, in this setting, MSC treatment decreased both percentage and absolute number of human CD45+ cells. Another indication of the effect of MSCs on human cells is that in mice irradiated and injected with huPBMCs, MSCs lowered the human cell numbers in the liver and their Ki-67 expression. This suggests that the decreased hepatic damage could also be related to the inhibition of proliferation of human T-cells. Altogether, our data strongly suggest that MSCs inhibited GvHD in our model by acting as both an immunomodulator and reparative agent.
Data reported in the literature suggest that an important component of MSC-induced inhibition of GvHD in vivo is mediated by Tregs [42,44]. In our model we did not find the induction of a T-regulatory phenotype in the effector cell pool upon MSC infusion. This discrepancy can be explained by the different animal models used in the different studies. Indeed, the intriguing aspect of our study is that both MSCs and T cells are of human origin. Notwithstanding a few case reports [50], very little in vivo data are available in humans that are statistically reliable, because these studies report a highly variable increase in Tregs after MSC treatment [51,52]. Interestingly, in vitro we observed that MSCs induced a moderate, although not significant, increase of CD69 expression on CD4+ cells. We studied CD69 as an activation marker but also as a marker expressed by regulatory T-cells expressing or not CD25 and FoxP3 [53], and it was shown that MSCs are able to promote the sustained expression of this marker [54]. Based on our in vitro results we cannot exclude that MSCs induce tolerance by fostering the expansion of this particular T-cell regulatory subset.
Concerning the antiproliferative effect of MSCs on human lymphocytes, concomitant with a lack of effect on activation marker expression, this phenomenon, although surprising, has been previously noted [25]. It could be argued that despite their attenuated proliferation, T cells that still express activation markers could trigger GvHD. Indeed, GvHD is significantly delayed but not abolished in MSC-treated mice.
Unlike other studies [55,56], the decrease in human T-cell proliferation induced in vivo by MSC injection was not associated with a decreased Th1/Th2 ratio in comparison to the PBS-treated group. Indeed, the analysis of 9 circulating human cytokines [IL-2, IL-4, IL-6, IL-8, IL-10, IL-15, IL-17, tumor necrosis factor-α, and interferon-γ (IFN-γ)] performed using a customized Bio-Plex Pro Human Cytokine 9-plex Assay (Bio-Rad Laboratories, Hercules, CA) did not show any significant difference between PBS- and MSC-treated mice (data not shown). This finding could indicate that in our model MSCs can directly modulate T-cell proliferation in target organs without necessarily modifying the “cytokine storm” observed in the peripheral blood. Interestingly, Christensen et al. [56] showed a modulation of IFN-γ during the first week after GVHD induction, whereas our first time point analyzed was at day 6, so it is possible that an earlier test would have revealed some effect due to MSC injection. Nevertheless, the difference between the models (we used a humanized NSG model injected with human MSCs) could justify discrepancies and does not allow further comparisons.
The NSG mouse model is also suitable for comparing MSCs with different established immunosuppressor drugs, such as CsA. In our xeno-GvHD model, CsA treatment resulted in the expected outcome by strongly reducing T-cell proliferation and activation marker expression. We have already shown that CsA is able to significantly reduce GvHD-related mortality and clinical signs [18]. Here we have confirmed our previous results and added information regarding the strong downregulation of Ki-67 in human cells in the liver. Compared with CsA, MSC treatment showed various differences, indicating a different modality of T-cell inhibition. Indeed, CsA inhibited both T-cell proliferation and activation, whereas MSCs inhibited only T-cell proliferation. Interestingly, despite the absence of inhibition of T-cell activation and despite a lower inhibition of T-cell cycling, as suggested by the Ki67 test, the absolute T-cell counts in the liver of mice from the 2 treatment groups were not significantly different. This suggests that MSCs may inhibit the infiltration of human T-cells in GvHD target organs by another mechanism in addition to the inhibition of T-cell proliferation; however, the existence and nature of the other potential mechanism remains to be demonstrated.
Taken together, our data confirm the NSG mouse as a suitable model for xeno-GvHD development and for testing human MSC infusion as a treatment for human cell-induced GvHD. We suggest that MSC infusion can inhibit GvHD through both reducing T-cell proliferation and fostering healing after irradiation. The translational impact of this animal model could help in determining the best treatment strategies to test in future clinical trials in humans undergoing allogeneic HSCT.
Footnotes
Acknowledgments
This work was funded by La Fondation CHU Sainte-Justine and La Fondation Charles-Bruneau. J.G.G. received a scholarship from La Fondation CHU Sainte-Justine and La Fondation des Étoiles. S.S. received a scholarship from La Fondation CHU Sainte-Justine.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.