
Abstract
Bone marrow–derived mesenchymal stromal cells (MSCs) have been used successfully as a source of stem cells for treating neurodegenerative diseases. However, for reasons that are not clear, autologous MSC transplants have not yielded successful results in human trials. To test one possible reason, we compared the migratory ability of MSCs from amyotrophic lateral sclerosis (ALS) patients with those of healthy controls. We found that MSCs derived from ALS patients (ALS-MSCs) had a reduced ability to migrate, which may explain why autologous transplantation is not successful. We also found that expression of one of the intracellular factors implicated in migration, β-PIX, was significantly reduced in ALS-MSCs compared with healthy stem cells. Restoration of β-PIX expression by genetic manipulation restored the migratory ability of ALS-MSCs, and inhibition of β-PIX expression with shRNA reduced the migration of healthy MSCs. We suggest that transplantation of allogeneic or genetically modified autologous stem cells might be a more promising strategy for ALS patients than transplantation of autologous stem cells.
Introduction
I
We hypothesized that MSCs from patients might lack the therapeutic efficacy of those from healthy controls because adult MSCs derived from different tissues [6] and different human sources might vary in their ability to function correctly and so to correct individual disorders. Hence if the MSCs from patients with neurodegenerative diseases lacked the capability to provide neuroprotection or to migrate, they would be less effective therapeutically, and a universal donor approach might be required instead.
p21-activated protein kinase (PAK)-interacting exchange factor-β (β-PIX; also known as ARHGEF7) is an intracellular protein implicated in T cell chemotaxis across reactive barriers, cancer cell migration, neurite outgrowth, and virus migration [7 –12]. β-PIX lies downstream of Ras-related C3 botulinum toxin substrate 1 (Rac1) and regulates the activation of focal adhesion kinase and p38 mitogen-activated protein kinase, and thus plays a pivotal role in cell migration [13]. However, its role in stem cells has not been established.
In this study, we compared the migratory capacity of MSCs from different tissues and human sources, including ALS patients, and identified intracellular factors important for stem cell migration. We also genetically modified β-PIX expression to clarify its role in stem cell migration because expression of this protein and its mRNA was significantly lower in MSCs from ALS patients than in those from healthy controls.
Materials and Methods
Isolation and culture of ALS-MSCs, C-MSCs, hUC-MSCs, and UCB-MSCs
Ethical approval for the current study was obtained from the Hanyang University Hospital in Seoul, Korea (HYU-IRB-2004-643 and HYU-IRB-2005-452). The isolation and culture of MSCs derived from the endothelial/subendothelial layer of human umbilical cords (hUC-MSCs, n=4) were carried out as previously described [14]. Human MSCs derived from the bone marrow of ALS patients (ALS-MSCs, n=7) and healthy donors (control healthy MSCs [C-MSCs], n=5), and human MSCs derived from the umbilical cord blood (UCB) of normal healthy infants (UCB-MSCs, n=4) were isolated and cultured as previously described [4,15,16]. ALS-MSCs were obtained from probable and definite ALS patients (based on the El Escorial criteria) undergoing autologous therapy. The demographic and clinical features of the ALS patients are shown in Table 1.
M, male; F, female; ALSFRS-R, amyotrophic lateral sclerosis functional rating scale-revised.
Cells were labeled with the following antihuman antibodies: CD45-phycoerythrin (PE), CD44-fluorescein isothiocyanate (FITC) (DakoCytomation), CD73-PE (BD Pharmingen), CD34-PE, CD29-FITC, CD49C-PE, CD54-FITC, CD105-FITC, CD106-FITC, HLA-DR-FITC, and PE- and FITC-conjugated isotype controls (Serotec). After labeling, cells were analyzed by flow cytometry (Calibur).
The antigens expressed by the various MSCs at their 5th passage were analyzed (Supplementary Table S1; Supplementary Data are available online at
Cell migration assay
Cell migration was examined with a QCM chemotaxis (8-μm pore size) 96-well migration assay (Chemicon). The migration chambers were coated with 50 μL of hemagglutinin (5 mg/mL) and air-dried overnight. MSCs (5×104 in 100 μL of serum-free medium) were then placed in the migration chamber. The lower chamber contained 150 μL of serum-free medium with 10% bovine serum albumin (BSA). The plates were incubated for 24 h at 37°C in 5% CO2. Thereafter MSCs suspended in the medium in the migration chamber were removed gently by pouring off the medium. Cells adhering to the upper side of the membrane were removed by scraping with a cotton applicator, and the migration chamber plate was then placed onto a new 96-well feeder tray containing 150 μL of prewarmed cell detachment solution (Accutase, Innovative Cell Technologies, Inc.) in the wells. After 30 min at 37°C, 50 μL of a lysis buffer/dye solution was added to the feeder tray, and the tray was incubated for 15 min at room temperature. The mixture (150 μL) was then transferred to a new 96-well plate and the plate was read with a fluorescence plate reader, using a 480/520 nm filter set (HTS 7000 Bioassay reader), according to the manufacturer's instructions [17].
Quantitative polymerase chain reaction array
Two plates of the RT2 Profiler polymerase chain reaction (PCR) array for human tumor metastasis (PAHS-028; SuperArray Bioscience Corporation) were used to compare quantitative PCR (Q-PCR)–validated cDNA samples of normal- and ALS-MSCs (Supplementary Table S2). cDNA equivalent to 1 μg of total RNA was used for each plate. The cDNA was mixed with the RT2 SYBR Green/ROX Q-PCR Master Mix, and 25 μL was added to wells containing the different primer sets. The plate was run under the same conditions as described in the following section for real-time PCR. The cycling parameters were as follows: initial denaturation at 95°C for 10 min, 40 cycles of 15 s at 95°C for denaturation, and 1 min at 60°C for primer annealing and extension. After applying the amplification protocol, a dissociation curve was constructed by ramping the temperature from 60°C to 90°C, and the outcome was normalized against the set of reference genes used for the Q-PCR. Analysis using the reference genes present on the array yielded a similar outcome.
Real-time PCR and reverse transcription-PCR
To measure β-PIX mRNA levels in MSCs, the cells were harvested near confluence and RNA was extracted with Trizol reagent (Invitrogen). Five micrograms of RNA was reverse-transcribed using RevertAid™ M-MuLV reverse transcriptase (MBI Fermentas), 0.2 μg random primer (Invitrogen), 1 mM dNTPs, and the buffer supplied. First-strand cDNA was amplified using Taq DNA polymerase (MBI Fermentas) with 5′-AAGCGCAAACCTGAACGGAA-3′ (upstream) and 5′-TCA CCTCAGAACTGGTCTTCA-3′ (downstream) as primers for β-PIX, and 5′-TGCTATCCCTGAAAGCCTCTG-3′ (upstream) and 5′-AGCTGGGGTGATGAAGCTGTA-3′ (downstream) for β-actin. To clone the human β-PIX gene, the first-strand cDNA from wild-type MSCs was amplified with Pyrobest™ DNA polymerase (TaKaRa) with 5′-CACCATGACCGATAATAG CAACAA-3′ (forward) and 5′-TCACCTCAGAACTGGTCT TCA
For quantification of gene transcripts, we performed real-time PCR in 96-well plates, with a final volume of 20 μL/well, using an SYBR Green PCR kit (Applied Biosystems, Inc.). Each reaction volume contained 10 μL of SYBR Green mix (2×concentrated), 6 μL of H2O, 1 μL of cDNA sample, and 3 μL of primer mix (sense and antisense primers, each 2 pmol/μL). Cycling parameters were as follows: initial denaturation at 95°C for 10 min, 40 cycles of 15 s at 95°C for denaturation, and 1 min at 60°C for primer annealing and extension. After completing the amplification protocol, a dissociation curve was constructed by ramping the temperature from 60°C to 90°C. The resulting concentration values were converted to absolute amounts of cDNA present in the sample (E-Ct). To correct for differences in cDNA quantity between samples, we normalized the target PCR to the geometric mean values of PCRs in the set of reference genes.
Immunoblotting and immunocytochemistry
To assess β-PIX protein levels, we performed immunoblotting and immunocytochemistry with a specific antibody for β-PIX (Cell Signalling). First we carried out immunoblotting with the antibody (1:1,000), according to previously described procedures [17], using aliquots of 5×106 MSCs, cultured for 24 h. All data are derived from at least 5 independent experiments.
For immunocytochemistry, cells were washed with phosphate buffered saline (PBS) and fixed with 4% paraformaldehyde in PBS for 20 min at 4°C. Following several washes, the cells were permeabilized with 0.5% Triton X-100 for 20 min. After incubation in 5% BSA in PBS for 1 h, they were incubated with anti-β-PIX monoclonal antibody (1:100) overnight at 4°C. They were then washed 3 times for 5 min each to remove nonbinding antibodies and incubated with the appropriate TRITC-conjugated secondary antibody (DakoCytomation) for 20 min at room temperature. Unbound antibody was removed by 3 rinses of 10 min each, and coverslips were overlaid with Vector Shield mounting medium. As a negative control, the above procedures were carried out using mouse IgG antibody (Kamiya Biomedical) instead of antibody specific for β-PIX.
Production and propagation of the recombinant lentivirus
To demonstrate the role of β-PIX in the migration of stem cells in vivo, we used lentiviral DNAs bearing β-PIX-specific shRNA or cDNA. Lentiviral DNA containing β-PIX-specific shRNA was purchased from Open Biosystems with the Trans-Lentiviral™ GIPZ packaging System (Open Biosystems). Viral stocks were produced according to the manufacturer's instructions. For analysis of β-PIX gene expression we subcloned β-PIX and GFP cDNAs into pLenti6/V5-D-TOPO (Invitrogen), and confirmed the construct by sequencing. The GFP cDNA was synthesized from a pEGFP vector with the following primers: 5′-CACCATGGTGAGC AAGGGCGAGGAG-3′ and 5′-TTACTTGTACAGCTCGTC CAT-3′. Human β-PIX cDNAs were isolated from adult human mesenchymal stromal cells (C-MSCs), using the following primers: 5′-CACCATGACCGATAATAGCAACAA-3′ and 5′-TCACCTCAGAACTGGTCTTCA-3′. Both synthesized cDNAs were subcloned into pLenti6/V5-D-TOPO (Invitrogen) and confirmed by sequencing. To determine the concentrations of the viral stocks, we serially diluted the viral supernatants and infected them into hBM-MSCs or HT1080 cells with 6 μg/mL Polybrene (Sigma) and selected expressing cells with 6 μg/mL Blasticidin (Invitrogen) for 10 days [18]. The remaining cells were stained with crystal violet and colonies were counted under a light microscope. To analyze lentiviruses carrying the GFP gene, the virus was infected into hBM-MSCs as described previously. GFP-infected hBM-MSCs were grown for 3 days, fixed in 1% paraformaldehyde, and analyzed for GFP expression by flow cytometry. We obtained 6×105 TU (transduction unit)/mL of β-PIX gene-carrying virus particles in HT1080 cells, 2×105 TU/mL in hBM-MSCs, and 1×105 TU/mL of shRNA-carrying virus particles in hBM-MSCs. To obtain high concentrations of recombinant virus, virus-containing supernatants were harvested by ultracentrifugation at 28,000×g for 90 min. Supernatants were stored at −80°C.
Lentiviral infection
hBM-MSCs were seeded at 8×105 cells per 75 cm2. They were infected at multiplicities of infection of 0, 2, or 5 infectious virus particles containing the β-PIX shRNA, the GFP gene, or the β-PIX gene in 15 mL of DMEM at 37°C overnight. The following day, the medium was removed and the cells were washed once with DMEM. They were then incubated for 4 days with normal medium, and migratory capacity was evaluated in vitro and in vivo.
Animal preparation and ischemic surgery
All animal procedures were performed in accordance with the Hanyang University guidelines for the care and use of laboratory animals, and were approved by the Institutional Animal Care and Use Committee (IACUC) of the Hanyang University. Sprague-Dawley (SD) rats, weighing 243 to 265 g, were purchased from Biogenomics, Inc. After periods of adaptation and pretraining over 7 days in preparation for neurological examination, the left middle cerebral arteries (MCA) of 90 SD rats weighing 295 to 313 g (Supplementary Table S3) were occluded for 2 h using the intraluminal filament technique described previously [19,20]. The rats were anesthetized by intraperitoneal injection of tiletamine (25 mg/kg), zolazepam (25 mg/kg, Zoletil; Yuhan Corp.), and xylazine (10 mg/kg, Rompun; Bayer), and rectal temperature was maintained at 36.6°C±0.5°C with a thermistor-controlled heating pad (Supplementary Table S4). Physiological data (pH, pCO2, pO2, and hematocrit) were measured in 0.1 mL aliquots of arterial blood obtained from a right femoral catheter using a blood analysis system (International Technidyne) (Supplementary Table S4). Arterial pressure was monitored from the arterial catheter with a strain-gauge transducer (LIFE KIT DX-360; Nihon Kohden) and amplifier (MacLab Bridge Amplifier; AD Instruments Pty Ltd.). Phasic pressure, mean arterial pressure, and heart rate were recorded at a sampling rate of 200/s, using a data acquisition system and laboratory computer (MacLab 8 analog-to-digital converter and Macintosh computer) (Supplementary Table S4). For the cerebral blood flow study, a wire-type probe (0.3-mm diameter; Unique Medical) connected to a Laser Doppler flow meter (ALF21; Advance) was inserted 6 mm through a small burr hole placed 2 mm lateral to the bregma, such that the probe lay against the dural surface overlying the frontal cortex. Measurements were taken at a depth of 6 mm from the cortex to evaluate the deep ischemic core regions (caudate and putamen) of the affected hemisphere (Supplementary Table S4). After the 2-h occlusion, reperfusion was performed as described previously [19,21]. Sham surgery was performed on an additional 10 rats, by introducing and immediately withdrawing a thread into the left common carotid artery. Other procedures in the sham group were identical to those in the ischemic surgery.
Labeling of MSCs with ferumoxides and protamine sulfate
Commercially available ferumoxides (FE) (TAEJOON Pharmaceutical Co., Ltd., Mfd by Advanced Magnetics, Inc.) have a total iron content of 11.2 mg/mL (11.2 iron μg/μL). Protamine sulfate (Sigma) was prepared as a fresh stock solution of 1 mg/mL in distilled water at the time of use. FE, at a concentration of 25 μg/mL, were dispensed into a tube containing serum-free DMEM (Gibco Invitrogen) containing 100 U/mL penicillin and 100 mg/mL streptomycin. Protamine sulfate (Pro) of 1 μg/mL was then added. The solution (FE–Pro complexes) containing FE and protamine sulfate was mixed for 60 min, and then added to adherent hMSC cultures. FE–Pro complexes were added directly to the cells, incubated for 2 h, and an equal volume of complete medium was added. The cells were then incubated overnight [22].
MSC grafting in ischemic stroke model rats
We chose 4 different types of MSCs to test whether the migratory activity of implanted MSCs varies with their origin: ALS-MSCs (n=7), C-MSCs (n=5), UCB-MSCs (n=4), and hUC-MSCs (n=4). Each type of MSCs (6×105) was injected into 10 SD rats (a total of 40 experimental animals) by stereotaxic surgery 2 weeks after intraluminal MCA occlusion. An equivalent volume of PBS was similarly injected into an additional 10 rats (PBS group). The mean weights of the groups were not significantly different (from 307.3±22.0 g to 314.3±14.1 g) and neurological deficit scores were also similar immediately before MSC implantation (Supplementary Fig. S1). All animals were anesthetized with sodium pentobarbital (50 mg/kg, IP). We implanted 5 μL of either 6×105 MSCs, or PBS into a site contralateral (AP=+ 0.7, R=+ 2, V=−5.5) to the lesion. The suspension was delivered for 2 min, with the syringe remaining in place for an additional 2 min. The rats in all groups received daily immunosuppression with subcutaneous cyclosporine A (10 mg/kg body weight; Sandoz) starting 2 days before cell transplantation until the end of the study [20].
In vivo MRI
For the MRI studies the rats were anesthetized with sodium pentobarbital (50 mg/kg, IP) and fixed to a Taoka rat cradle. All were able to maintain respiration without assistance. A 3-inch-diameter circular receive-only surface coil (Shanghai Chenguang Medical Technologies Co., Ltd) was placed under the head of each rat, with the center of the coil at the midpoint of the midline between the ear-ear and eye-eye lines. Body temperature was maintained at 37°C with a heating pad. The temperature of the MRI room was controlled to roughly 27°C, and MRI was performed with a 3T clinical instrument (Philips). To assess the extent of ischemic foci, we obtained fluid attenuated inversion recovery images using the spin-echo technique (TR=11,000 ms and TE=125 ms) between the vertex of the head and the bottom of the brain. Other imaging parameters were 0.7-mm slice thickness, 284×286 μm in-plane resolution (voxel size is 0.0569 mm3), and number of acquisitions is 1. To obtain T2*-weighted images of a rat brain with multiple planar gradient-recalled (MPGR) pulse sequences, we used the following parameters: TR=596 ms, TE=16 ms, section thickness=0.7 mm, in-plane resolution: 292×290 μm (voxel size of 0.0593 mm3), and number of acquisitions=1.
Immunohistochemistry of human mitochondria
To confirm whether the low signal intensities seen in the MPGR images were due to the implanted MSCs, we killed 3 rats from the C-MSC group 35 days after implantation and performed mitochondrial immunofluorescence studies. Antihuman mitochondria monoclonal antibody (1:100, Chemicon) was used as the primary antibody. Coronal sections (20-μm thickness) of the brain were prepared and incubated with the primary antibody for 72 h at 4°C. The sections were washed 3 times for 5 min each to remove unbound antibody and incubated for 24 h with the appropriate TRITC-conjugated secondary antibody (DakoCytomation). Unbound secondary antibody was removed by 3 rinses of 5 min each. After air drying, coverslips were applied to the slides with Vector Shield mounting medium (Vector Laboratories). As a negative control, the primary antibody was omitted and no cell staining was observed. We used a laser-scanning confocal microscopy system, mounted on a Leica DMIRE2 microscope. The red (TRITC) fluorochrome was excited at 557 nm, and emissions were acquired sequentially with a photomultiplier tube through 576-nm emission filters [20].
Statistical analysis
All data are represented as means±SD of 5 or more independent experiments. Differences were analyzed by the Wilcoxon rank sum test or by the Kruskal-Wallis test followed by Tukey's test using ranks (SPSS). P values<0.05 were considered statistically significant.
Results
Differences in migratory activity of MSCs depending on their origin
Ischemic stroke was induced by the transient occlusion of the left MCA in all the rats used in this study. To confirm whether the migration of MSCs labeled with FE and protamine sulfate [22] can be detected by MRI, C-MSCs labeled with them were injected into the nonaffected contralateral hemispheres (each n=5) 2 weeks after the onset of ischemic stroke via stereotaxic surgery. We performed MRI to evaluate the in vivo migration of MSCs in live rats and immunohistochemistry using human-specific mitochondrial antibodies to confirm whether the hypointense signals in MPGR images are due to C-MSCs labeled with FE and protamine sulfate. The MPGR images (top panels in Fig. 1) and the immunohistochemical staining (bottom panels in Fig. 1) show that the hypointense signals in the MPGR images are due to C-MSCs labeled with FE and protamine sulfate and that we can detect the C-MSCs that migrated from the injection site to the contralateral ischemic hemisphere. The migratory paths of the C-MSCs were easily tracked by observing the hypointense voxels in T2*-weighted images (i.e., dark regions) (Fig. 1).

Migration of implanted MSCs assessed by MRI and immunohistochemistry. The multiple planar gradient-recalled (MPGR) image and immunohistochemical staining show that the hypointense signals in MPGR images are MSCs labeled with ferumoxides and protamine sulfate, and that these MSCs have migrated from the injection site to the contralateral lesion site. *1, area inside the injection track; 2, area around the injection target; Is-1, periinfarct area without hypointense voxels (i.e., dark spots) in T2*-weighted MRI; Is-2 and Is-3, periinfarct areas containing hypointense voxels. MSCs, mesenchymal stromal cells. Arrows indicate the primary injection route. Color images available online at
To measure the differences in the in vivo migratory activity of MSCs depending on their origins, we labeled 4 kinds of MSCs (ALS-MSCs, C-MSCs, UCB-MSCs, and hUC-MSCs) with FE and protamine sulfate and injected them into the nonaffected contralateral hemispheres (each n=10) 2 weeks after the onset of ischemic stroke via stereotaxic surgery. We performed serial in vivo MRI to follow the migration of ferumoxide-labeled MSCs in rat brains. We found that most of the MSCs derived from the bone marrow of ALS patients (ALS-MSCs) lacked migratory capability, while C-MSCs, hUC-derived MSCs, and UCB-derived MSCs could migrate well to the infarcted areas (Fig. 2A). We detected hypointense voxels in the left periinfarct area, which is the contralateral site of the initial injection site, 35 days after the injection of MSCs in 7 of the 10 rats of the C-MSC group, 8 of the 10 of the hUC-MSC group, 9 of 10 of the UCB-MSC group, but only 1 of the 10 of the ALS-MSC group. This finding disproves the common assumption that MSCs from all sources possess equal cerebral migratory capabilities. We serially followed the migratory behavior of ALS-MSCs and C-MSCs in MSC-implanted rat brains for 35 days posttransplantation; after 35 days, the ALS-MSCs showed significantly less migratory activity than the C-MSCs (or none) (Fig. 2B). We performed the Kruskal-Wallis test followed by Tukey's test using ranks to determine whether the migratory activity of ALS-MSCs is significantly lower than those of C-MSCs, hUC-MSCs, and UCB-MSCs and found that the migratory activity of MSCs differed depending on their origin (P<0.001 by the Kruskal-Wallis test) and that the migratory activity of ALS-MSCs was significantly reduced compared with the other MSCs (Fig. 2C).

The migratory capacities of MSCs depend on their origins.
In vitro transwell chemotaxis assays showed that almost all ALS-MSCs had less migratory capacity than the other MSCs in agreement with the MRI data (Fig. 2D and Supplementary Fig. S2).
Differences in expression of migration-related genes in MSCs depending on their origin
We employed quantitative PCR to identify genes that might play important roles in the migration of MSCs. The expression levels of VEGFA, CTSK, β-PIX, and MTSS1 were markedly lower in ALS-MSCs than in C-MSCs (Fig. 3). VEGFA was used as an internal positive control as it is known to be downregulated in ALS patients [23]. Expression of β-PIX, whose function in stem cell migration is not yet known, was nearly 8-fold lower in ALS-MSCs than in C-MSCs (Fig. 3). Levels of both β-PIX mRNA and protein measured by reverse transcription-PCR, immunoblotting, and immunohistochemistry were significantly higher in C-MSCs, UCB-MSCs, and hUC-MSCs than in ALS-MSCs (Fig. 4 and Supplementary Fig. S3).

Expression of migration-associated factors in MSCs of different origin. Expression levels of some migration-associated genes, notably β-PIX, were lower in ALS-MSCs than C-MSCs as assessed by quantitative PCR array and real-time PCR. PCR, polymerase chain reaction. Color images available online at
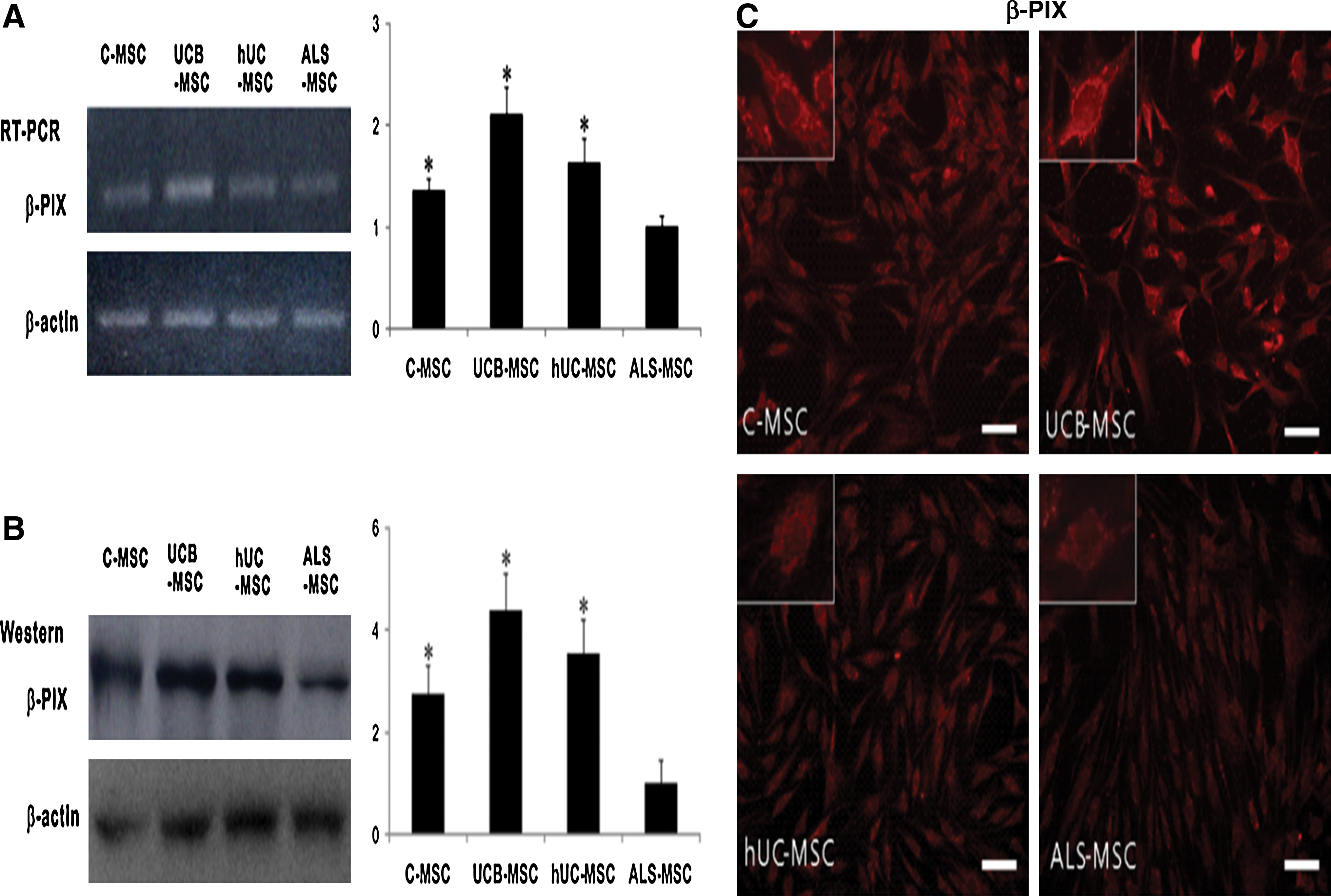
Expression levels of mRNA and protein of β-PIX in MSCs of different origin. Expression levels of mRNA
The importance of β-PIX in MSC migration
To determine whether β-PIX plays a critical role in MSC migration, we knocked down β-PIX in C-MSCs, which have good migratory activity, with lentivirus-transduced shRNA, and constructed ALS-MSCs, which have poor migratory activity, ectopically expressing β-PIX. The knockdown C-MSCs contained reduced levels of β-PIX mRNA and protein (Fig. 5A), while β-PIX was overexpressed in the ALS-MSCs transduced with lentivirus DNA carrying β-PIX cDNA (Fig. 5B). These changes in β-PIX expression level did not affect the surface markers CD45−, CD34−, CD29+, CD73+, CD105+, CD44+, and HLA-DR (Supplementary Table S5). β-PIX knockdown in the C-MSCs drastically reduced their migratory capacity in in vitro transwell chemotaxis assays, while β-PIX overexpression in ALS-MSCs restored their migratory capacity (Fig. 5C).
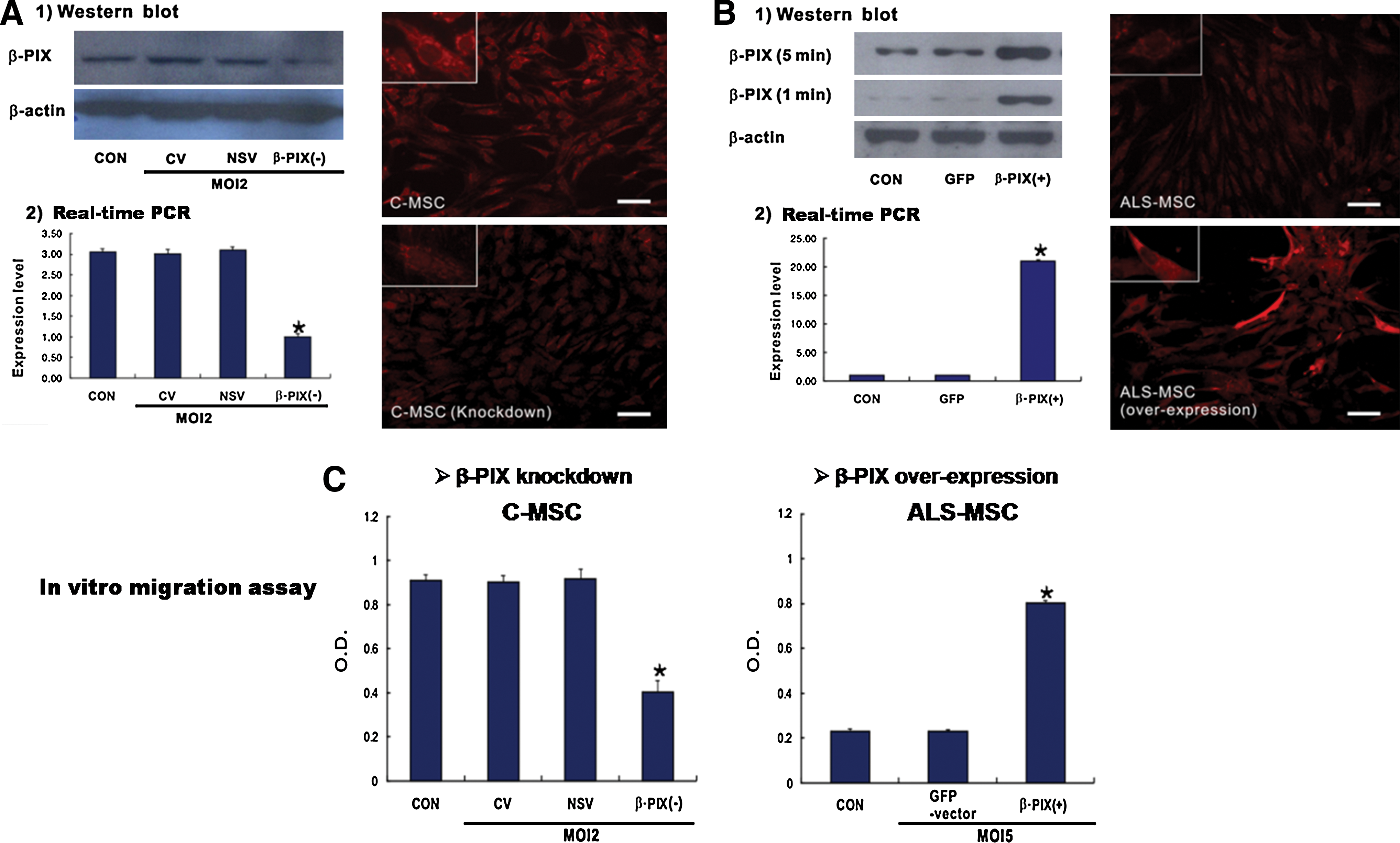
Alteration of β-PIX expression affects migratory capacity.
We also investigated the in vivo migration of the β-PIX–overexpressing ALS-MSCs and the β-PIX knockdown C-MSCs to confirm the importance of β-PIX in in vivo migration of MSCs. Both types of MSCs were implanted into experimental rat brains 2 weeks after the induction of ischemic stroke. T2*-weighted images of the rat brains showed that the β-PIX knockdown C-MSCs displayed markedly reduced levels of migration (migration was confirmed in 2 out of 10 rats) compared with normal C-MSCs (8 of 10) (Fig. 6A) and that migration was more robust in the ALS-MSCs overexpressing β-PIX (7 of 10) than in the controls (1 of 10) (Fig. 6B). This finding confirmed that β-PIX plays very important roles in the in vivo migration of MSCs to ischemic foci (Fig. 6A, B).
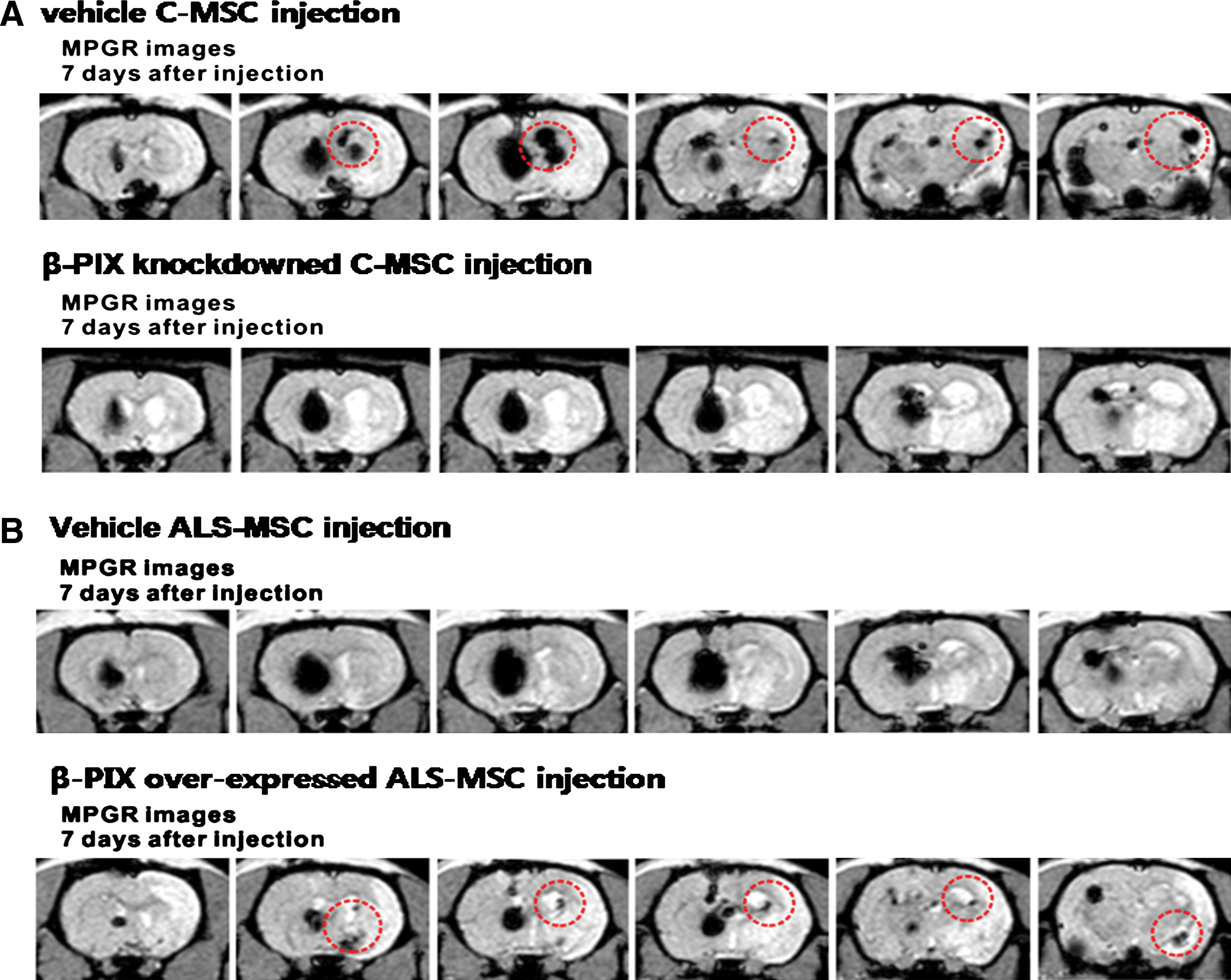
In vivo migratory capacity of MSCs depends upon the expression level of β-PIX. The migration of human MSCs to lesion sites in the rat stroke model was assessed by MRI.
Discussion
There have been many clinical trials using MSCs for the treatment of neurological diseases including ALS, ischemic stroke, and Parkinson's disease [3 –5,24,25]. However, the trials have shown that, although MSC transplantation is safe, the outcomes are not satisfactory despite the positive effects reported in vitro and in animal models. Recently, there is increasing interest in the factors responsible for the unsatisfactory results, and it has been reported that endothelial progenitor cells from patients with coronary artery disease are impaired in CXCR4 signaling and have reduced neovascularization capacity [26]. Although endothelial progenitor cells are quite different from MSCs, the finding suggested to us that MSCs from ALS patients might be less able to migrate to the sites of lesions and to secrete neurotrophic factors than MSCs from healthy controls.
We have shown previously that the origin of MSCs does indeed affect their migratory activity as well as their capacity to restore neurological function (Fig. 2 and Supplementary Figs. S1 and S2), and that levels of expression of genes related to migration vary depending on the origin of the MSCs (Fig. 3). The expression level of β-PIX, a protein associated with migration, was low in the ALS-MSCs from 7 patients (Fig. 4 and Supplementary Fig. S3), indicating that the migratory ability of ALS-MSCs is impaired regardless of severity and family history (Supplementary Figs. S2 and S3). Moreover overexpression and knockdown of β-PIX altered the migratory activities of ALS-MSCs and C-MSCs, respectively (Figs. 5 and 6). These observations indicate that β-PIX is critical for the migration of implanted MSCs to ischemic lesions. Migration to the sites of lesions may be important in treating neurological diseases because they should secrete neurotrophic factors near the lesion sites.
PAK-interacting exchange factor-β (β-PIX; ARHGEF7) is a rho guanine nucleotide exchange factor that facilitates polymerization of actin, induces protrusion of the cell membrane, and promotes cell migration [27,28]. Since overexpression of β-PIX in ALS-MSCs dramatically increased their migratory activity when implanted in the rat model of ischemic stroke (Figs. 5 and 6), we expected that the neurobehavioral functions of the rats would also be improved. However, this was not the case (Supplementary Fig. S4). Therefore it appears that migration of MSCs to the sites of ischemic lesions is necessary but not sufficient for restoring neurological function. This may be because although expression of β-PIX is restored, production of neurotrophic factors may be defective. This idea is supported by our previous finding that secretion of angiogenin, basic fibroblast growth factor-2, insulin-like growth factor-1, and vascular endothelial growth factor (VEGF) is lower in ALS-MSCs than in MSCs from healthy controls [16]. Moreover we showed in the current study that the mRNA and protein levels of many other trophic factors, including stromal cell–derived factor-1α (SDF-1α), hepatocyte growth factor, and placental growth factor, are also reduced in ALS-MSCs (see also Supplementary Figs. S5–S8).
Our results suggest that autologous MSCs of patient origin may not be the best option for stem cell therapy. The low levels of secretion of migratory and trophic factors that we observed may account for the poor efficacy of autologous therapy, despite the normal morphology of the stem cells [4,29]. These results add fuel to the debate over whether well-characterized, commercially available, and allogeneic MSCs harvested from healthy individuals might be preferable to autologous MSCs from individuals with underlying disease.
One limitation of our study was that the migratory capacities of the MSCs were assessed in a rat model of ischemic stroke. We acknowledge that it would have been more appropriate to employ an animal model of ALS if the purpose of this study was only to compare the therapeutic potential of ALS-MSCs and non-ALS-MSCs in ALS. However, the reason why we needed to use ischemic rats was as follows. In our previous study to evaluate the dose-dependent efficacy of MSCs in SOD1-G93A ALS mice [30], we performed histology to evaluate the migration of MSCs into the brain and the spinal cord and found that few of MSCs injected into the cistern magna were detected in the brain and the spinal cord. We did not decide whether this finding might be due to poor migratory activity of MSCs originated from ALS patients or due to the fact that we could not investigate all tissues from the brain to the spinal cord because of the limitation of the procedure of histology. Therefore, we thought that other methods were needed to detect the in vivo migration of MSCs injected into the cistern magna. So, we decided to use MRI for the detection of in vivo migration of MSCs. However, our preliminary study showed that we could not discriminate migrated MSCs from nonmigrated ones by in vivo MRI because of the small size of the spinal cords and brains of ALS mice and the paramagnetic effect of the FE used to label the MSCs. These findings suggested that the ALS mouse was not appropriate for examination by MRI and needed to use larger animals. We tried to use the rat model of ALS, but that was not possible so we turned to ischemic rats. Therefore, we used ischemic rats, which can be easily evaluated by MRI and would permit us to detect the migration of MSCs by MRI.
A second limitation concerns the time when the MSCs were injected. As shown in the brain MRIs (Fig. 2), we used rats with large infarcts to evaluate the migratory activity of the MSCs. Rats with large infarcts are very unstable and tend to die, and it is well known that many cytokines and inflammatory cells that can affect MSC migration and survival increase during the acute stage of ischemic infarction. Therefore we thought that the acute stage would not be appropriate for evaluating the intrinsic migratory activity of the MSCs. In a preliminary study to decide the appropriate time of injection, almost all the rats that received early injections stereotactically in the brain within 24 or 48 h after the onset of infarct died. Therefore, we decided to inject MSCs 2 weeks after ischemic infarction, even though treatment for stroke is not effective at this time. To try to avoid this limitation, we measured the concentration of SDF-1α, which is known to be a chemoattractant for MSCs, in normal areas and periinfarct areas 2 weeks after the infarct. We found that SDF-1α was significantly higher in the periinfarct areas than in the normal areas (Supplementary Fig. S9). Anyway, to overcome this limitation completely, further studies using the rats with smaller infarcts will need to be performed.
The third limitation concerns the injection route. The delivery route used in this study was not clinically relevant considering that IV injection of MSCs is being employed in the ongoing clinical trials and in most preclinical animal studies using MSCs in neurological diseases [31,32]. However, it was not possible to evaluate the differences of the in vivo migratory activities of MSCs depending on their origins after IV injection because few MSCs reach the injured brain or spinal cord after IV injection due to the filtering effect of the reticuloendothelial system, liver, lung, and so on. As we pointed out in the Introduction, the aims of this study were to compare the migratory capacity of MSCs from different tissues and human sources, to identify intracellular factors important for stem cell migration, and to investigate whether genetical modification of β-PIX expression affected the in vivo migratory activity of MSCs. Therefore, we injected the MSCs into the hemispheres opposite the ones with the infarcts to evaluate the difference of in vivo migratory activity of MSCs depending on their origins.
In conclusion, we have shown that the migratory capacity of MSCs differs depending on their tissue of origin and the underlying condition of the donor. In addition, we have found that β-PIX facilitates migration of MSCs to the sites of lesions. Therefore, selecting allogeneic stem cells producing high levels of β-PIX and neurotrophic factors could be an effective strategy for stem cell therapy in patients with neurodegenerative diseases such as ALS.
Footnotes
Acknowledgments
This work was supported by the NanoBio R&D Program of the Korea Science and Engineering Foundation, funded by the Ministry of Education, Science and Technology (2007-04717), and a grant from the Korea Healthcare Technology R&D Project, Ministry for Health, Welfare & Family Affairs, Republic of Korea (A101712).
Author Disclosure Statement
The authors have declared that no conflict of interest exists.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.