
Abstract
The epithelial cell rests of Malassez (ERM) are odontogenic epithelial cells located within the periodontal ligament matrix. While their function is unknown, they may support tissue homeostasis and maintain periodontal ligament space or even contribute to periodontal regeneration. We investigated the notion that ERM contain a subpopulation of stem cells that could undergo epithelial–mesenchymal transition and differentiate into mesenchymal stem-like cells with multilineage potential. For this purpose, ERM collected from ovine incisors were subjected to different inductive conditions in vitro, previously developed for the characterization of bone marrow mesenchymal stromal/stem cells (BMSC). We found that ex vivo-expanded ERM expressed both epithelial (cytokeratin-8, E-cadherin, and epithelial membrane protein-1) and BMSC markers (CD44, CD29, and heat shock protein-90β). Integrin α6/CD49f could be used for the enrichment of clonogenic cell clusters [colony-forming units-epithelial cells (CFU-Epi)]. Integrin α6/CD49f-positive-selected epithelial cells demonstrated over 50- and 7-fold greater CFU-Epi than integrin α6/CD49f-negative cells and unfractionated cells, respectively. Importantly, ERM demonstrated stem cell-like properties in their differentiation capacity to form bone, fat, cartilage, and neural cells in vitro. When transplanted into immunocompromised mice, ERM generated bone, cementum-like and Sharpey's fiber-like structures. Additionally, gene expression studies showed that osteogenic induction of ERM triggered an epithelial–mesenchymal transition. In conclusion, ERM are unusual cells that display the morphological and phenotypic characteristics of ectoderm-derived epithelial cells; however, they also have the capacity to differentiate into a mesenchymal phenotype and thus represent a unique stem cell population within the periodontal ligament.
Introduction
T
Given that Hertwig's epithelial root sheath plays an important role in tooth root formation and that periodontal regeneration is a re-enactment of the development process [4], it is possible that ERM populations, as the descendants of Hertwig's epithelial root sheath, inherit some biological functions from the Hertwig's epithelial root sheath. Although ERM have been described in the adult periodontium since 1885, their function is still a matter of conjecture [5]. It has been reported that ERM cells actively participate in the homeostasis of the periodontium by paracrine secretion of a variety of soluble factors, including cytokines, chemokines, growth factors, and related proteins [6]. In addition, ERM cells demonstrate a functional role in maintaining periodontal ligament space and preventing dento-alveolar ankylosis [7]. Furthermore, recent evidence suggests that ERM cells may contribute to cementum or enamel formation and repair [8,9]. Interestingly, despite the ectodermal nature of epithelial cells, ERM also express a number of mesenchymal-associated proteins [5].
Since periodontal tissues have a poor regenerative capacity and outcomes of current periodontal regeneration therapies are clinically unpredictable, alternative strategies are being investigated to treat damaged tissues after trauma or periodontal disease. Stem cells represent an important potential for developing cell-based tissue engineering owing to their capacity of self-renewal and differentiation into multiple cell lineages. The clinical success of hematopoietic stem cells has encouraged further characterization of stem cells derived from various tissues [10,11]. To date, at least 6 different dental stem/progenitor cells have been identified [12], including dental pulp stem cells [13], stem cells from exfoliated deciduous teeth [14], periodontal ligament stem cells [15], stem cells from apical papilla [16,17], dental follicle progenitor cells [18], and recently gingiva-derived mesenchymal stem cells [19]. However, there is no direct evidence that a putative stem-cell population exists within the ERM population. With this in mind, the aim of the present study was to investigate whether ERM cells contain a population of multipotent stem cells that are capable of undergoing an epithelial–mesenchymal transition, with the capacity to form osteoblasts, adipocytes, chondrocytes, and neuron-like cells in vitro and mineralized bone/cementum in vivo.
Materials and Methods
Cell isolation and culture
Periodontal ligament cells were isolated from ovine incisors following approved guidelines set by the Institute of Medical and Veterinary Science (IMVS, South Australia, Australia) Animal Ethics Committee (#130/06). Periodontal ligament was enzymatically digested to generate single-cell suspensions as previously described [15]. Selective trypsinization [20,21] was used to separate periodontal fibroblasts from ERM cells using 0.05% trypsin/0.02% ethylenediaminetetraacetic acid (EDTA) (SAFC; Sigma-Aldrich Biotechnology, Lenexa, KS; #59418C). Briefly, after 5 min treatment in trypsin, all detached cells were collected and the culture flasks washed twice before adding fresh media. Stringent multiple enzymatic digestion was repeated until no cells were detaching within 5 min. Selective trypsinization was also performed whenever cells became confluent. ERM cells were cultured at 8×103–12×103 cells/cm2 in Oral Keratinocyte Medium (OKM; ScienCell Research Laboratories, Carlsbad, CA; #2611) supplemented with oral keratinocyte growth supplement (ScienCell Research Laboratories; #2652), and 100 U/mL penicillin and 100 μg/mL streptomycin solution (P/S; ScienCell Research Laboratories; #0503). To isolate individual colonies, primary ERM cultures were plated into 10-cm culture dishes at cell low density (5×103 cells/cm2) and incubated at 37°C, 5% CO2 for 14 days. Individual colonies were isolated using colony rings and cultured in OKM with additives for further expansion in individual vessels as previously described [22,23]. Periodontal ligament fibroblasts were cultured at 5×103 cells/cm2 as previously described [24]. Unless mentioned otherwise, the experiments were repeated for 3 donors.
Ovine tooth preparation
Fresh ovine incisors were collected and fixed in 10% buffered formalin for 7 days. The tissues were then decalcified at room temperature in 10% EDTA (pH 7.4) solution where decalcification was determined by radiography. The tissues were then embedded in paraffin and cut into 5 μm sections. Hematoxylin and eosin (H&E) staining was performed every 5 sections to check for the presence of ERM cells before staining for immunohistochemistry.
Immunohistochemistry
Chamber slides (Nalge-Nunc Lab-Tek, Rochester, NY; #177445) were seeded with 8×103 cells/cm2 in OKM with additives for 2 days. The slides were fixed with 4% paraformaldehyde for 20 min at room temperature. After washing in phosphate-buffered saline (PBS; Sigma-Aldrich), endogenous peroxidase activity was inhibited using 0.5% H2O2 in methanol at room temperature for 30 min. The sections were then washed in PBS and blocked for nonspecific antibody binding using 3% goat serum in Tween PBS for 3 h before incubating with primary antibodies or isotype control antibodies (Table 1) overnight at 4°C. After washing, the slides were incubated with secondary antibodies (Table 1) for 1 h at room temperature. After washing, slides were incubated with Vectastain ABC reagents (Vector Laboratories, Burlingame, CA) according to the manufacturer's recommendations and then developed with diaminobenzidine (Dako, Campbellfield, VIC, Australia) for 5–10 min. The slides were then washed and counterstained with hematoxylin. For ovine incisor sections, pretreatment with 0.1% trypsin (Sigma-Aldrich, #T9201) for 20 min at 37°C was performed for anti-integrin α6/CD49f.
Flow cytometric analysis
Flow cytometric analysis was performed as previously described [24]. Primary antibodies or corresponding isotype control antibodies and FITC-conjugated secondary antibodies are outlined in Table 1. Samples were washed, fixed with FACS Fix, and analyzed using an Epics-XL/MCL flow cytometer and Analysis Software (Beckman Coulter, Miami, FL).
Fluorescence-activated cell sorting
Fluorescence-activated cell sorting was performed as previously described [25]. Ex vivo-expanded ERM cells were liberated from the culture plates. Single-cell suspensions of 9×106 ERM cells were blocked for 45 min and incubated with anti-integrin α6/CD49f antibody or corresponding isotype control antibody (Table 1) [26,27]. Cells were then sorted using an Epics Altra HyPer Sort FACS machine (Beckman Coulter). The brightest 30% of integrin α6/CD49f-positive cells were selected using Expo 32 Multi-comp software. Selected cells were collected in 2 mL of OKM media with additives and then centrifuged, resuspended in OKM with additives, and plated at 1×104 cells/cm2.
Immunomagnetic selection
Immunomagnetic selection was performed as described previously [25]. Briefly, ∼5.44×105 periodontal ligament cells were blocked and then incubated with integrin α6/CD49f antibody or corresponding isotype control antibody (Table 1). Cells were washed and then incubated with sheep anti-rat IgG-conjugated magnetic Dynabeads (Table 1) (Dynal, Oslo, Norway). Bound cells were collected using MPC-1 magnetic particle concentrator (Dynal).
Colony forming assay
To assess colony-forming efficiency, integrin α6/CD49f positive, negative, and unfractionated cells were seeded in 6-well plates as previously described [25]. Integrin α6/CD49f-positive cells were seeded at 3,450 cells/well and integrin α6/CD49f-negative and unfractionated cells at 1×104 cells/well. Clusters of more than 50 cells were counted as colonies [25].
Differentiation assays
Mineralization assays were performed as previously described [28]. Briefly, ERM cells and periodontal ligament fibroblasts at passage 2 were seeded in 96-well plates at 1×104 cells and 5×103 cells/cm2, respectively, for 28 days in osteogenic media changed twice weekly. Mineral deposits were identified by Alizarin Red (Alizarin Red S; Sigma-Aldrich) staining. ERM cells were also plated in 6-well plates as described above and RNA was isolated with TRIzol (Invitrogen, Carlsbad, CA) at day 14 after osteogenic induction.
For adipogenic induction, ERM cells and periodontal ligament fibroblasts at passage 2 were seeded in 96-well plates at 1×104 cells and 5×103 cells/cm2, respectively, and cultured for 4 weeks in adipogenic media changed twice weekly [28,29]. Lipid deposits were observed after staining with Oil Red O (MP Biomedicals, Solon, OH). ERM cells were also plated in 6-well plates as described above and RNA was isolated with TRIzol at day 14 after adipogenic induction.
Chondrogenesis induction was performed as previously reported [30]. Cell pellets from 2×106 ERM cells and periodontal ligament fibroblasts at passage 2 were spun at 600 g and cultured in polypropylene tubes in chondrogenic media changed twice a week for 28 days. Pellet cultures designated for histological assessment were fixed, paraffin-embedded, sectioned, and stained with H&E and toluidine blue. Immunohistochemical staining was performed with anti-collagen type II monoclonal antibody (Table 1) as described previously. Pellet cultures designated for real-time reverse transcriptase–polymerase chain reaction (RT-PCR) analysis were washed and digested with collagenase I (3 mg/mL; Worthington Biochemical, Lakewood, NJ) and dispase II (4 mg/mL; Roche Diagnostics, Indianapolis, IN) and RNA was isolated with TRIzol.
For neuronal differentiation, a method based on a protocol for neurogenesis of human dental pulp stem cells was used [31]. ERM cells were seeded at 1×104 cells/cm2 onto polyornithine and laminin-coated plates. Cultures designated for immunocytochemical analysis were liberated after 3 weeks of neuronal induction and seeded at 6×103 cells/cm2 in coated chamber slides. Cells were fixed after overnight adhesion. The staining protocol was used as following for all the other antibodies, with the exception of anti-glial fibrillary acidic protein (GFAP) antibody. Cultures were blocked and then incubated with primary antibodies or isotype control (Table 1) overnight at 4°C. After washing, sections were incubated with secondary antibodies (Table 1) for 30 min at room temperature. After washing, the slides were incubated with streptavidin Alexa Fluor 488 (Invitrogen, #S32354) for 1 h at room temperature in the dark. Finally, cultures were washed and co-stained with Prolong gold anti-fade with DAPI (Invitrogen, #P36931) for 10 min at room temperature, washed, and then coverslipped with fluorescence mounting medium (DAKO, #S3023). Sections for anti-GFAP antibody were blocked and incubated in anti-GFAP antibody or isotype control (Table 1) as described above. After washing, sections were incubated with donkey anti-rabbit Cy3 (Jackson Immunoresearch, West Grove, PA; #711-165-152) for 1 h at room temperature, washed, co-stained with DAPI, and cover-slipped. Cultures designated for real-time RT-PCR analysis were lysed with TRIzol.
Real-time PCR analysis
Total cellular RNA was isolated using TRIzol (Invitrogen) extraction according to the manufacturer's instructions. Complementary DNA was generated using the SuperScript III Reverse Transcriptase kit (Invitrogen). PCR primers were manufactured by GeneWorks (SA, Australia) and are outlined in Table 2. The expression of genes of interest was assessed by real-time PCR as previously described [23,24]. All mRNA quantification data represent the mean±standard error of the mean (SEM) of triplicate experiments normalized to the house-keeping gene β-actin. Statistical differences of P<0.05 was determined using the unpaired t-test.
In vivo transplantation of ERM cells
Expanded integrin α6/CD49f-positive ERM cells were trypsinized at passage 4 in preparation for transplantation. Approximately 5×106 ERM cells (integrin α6/CD49f-positive) were then mixed with 40 mg of hydroxyapatite tricalcium phosphate (HA/TCP) ceramic powder (Zimmer, Warsaw, IN) and transplanted subcutaneously into immunocompromised NOD/SCID mice (IMVS animal facility, South Australia, Australia) according to an approved animal protocol with institutional animal ethics approval (IMVS, South Australia, Australia, Animal Ethics Committee # 33/05) [28]. Immunohistochemical staining using mouse anti-ovine CD44 antibody (H9H11), which does not cross-react with murine tissue or murine mesenchymal stem cell populations, was performed to identify the origin of the bone tissue formed in the transplants. Moreover, staining of anti-osteocalcin and anti-CD44 was performed in serial sections. Anti-CK-8 antibody was used to trace the fate of ERM cells in the transplants. Immunohistochemistry for anti-CD44, anti-osteocalcin, and CK-8 antibodies (Table 1) was performed as described previously. Stained sections were imaged using NanoZoomer Digital Pathology system (Hamamatsu, Hamamatsu City, Shizuoka, Japan).
Gene expression of epithelial–mesenchymal transition markers by RT-PCR
To examine whether ERM cells undergo epithelial–mesenchymal transition under osteogenic conditions, various epithelial–mesenchymal transition-associated gene expression was examined by real-time RT-PCR. ERM cells were cultured in osteogenic media as described previously and total cellular RNA was collected after 1, 2, 3, and 4 weeks. Real-time RT-PCR was performed as described above with primers outlined in Table 2.
Statistical analysis
All values are expressed as mean±SEM and statistical significance of P≤0.05 was determined using an unpaired Student t-test.
Results
ERM cells express both epithelial and mesenchymal markers
To characterize the immunophenotype of ERM cells, immunohistochemical studies were performed using a panel of antibodies associated with epithelial and mesenchymal phenotypes. Ex vivo-expanded ERM cells were found to express epithelial cell markers cytokeratin-8 (CK-8), E-cadherin, and epithelial membrane protein-1 (EMP-1) (Fig. 1A), consistent with the epithelial nature of these cells. Interestingly, ERM cells also demonstrated positive expression of mesenchymal stem cell-associated markers, CD44, CD29, and heat shock protein90β (HSP90β) (Fig. 1A), suggesting that ERM cells have characteristics of both epithelial and mesenchymal cells. Immunochemical analysis showed that hematopoietic cell markers CD14 and CD45 were not expressed by ERM cells (Fig. 1A). Confirmatory studies using flow cytometric analysis demonstrated that ERM cells exhibited cell surface expression of CD44, CD29, and HSP90β while lacked expression of CD14 and CD45 (Fig. 1B). However, the epithelial cell markers failed to be analyzed by flow cytometry due to the intracellular position of CK-8 and availability of antibodies for ovine cells that are only reactive with the intracellular domain of E-cadherin and EMP-1. Localization of ERM cells in decalcified ovine teeth was demonstrated by H&E staining (Supplementary Fig. S1A; Supplementary Data are available online at
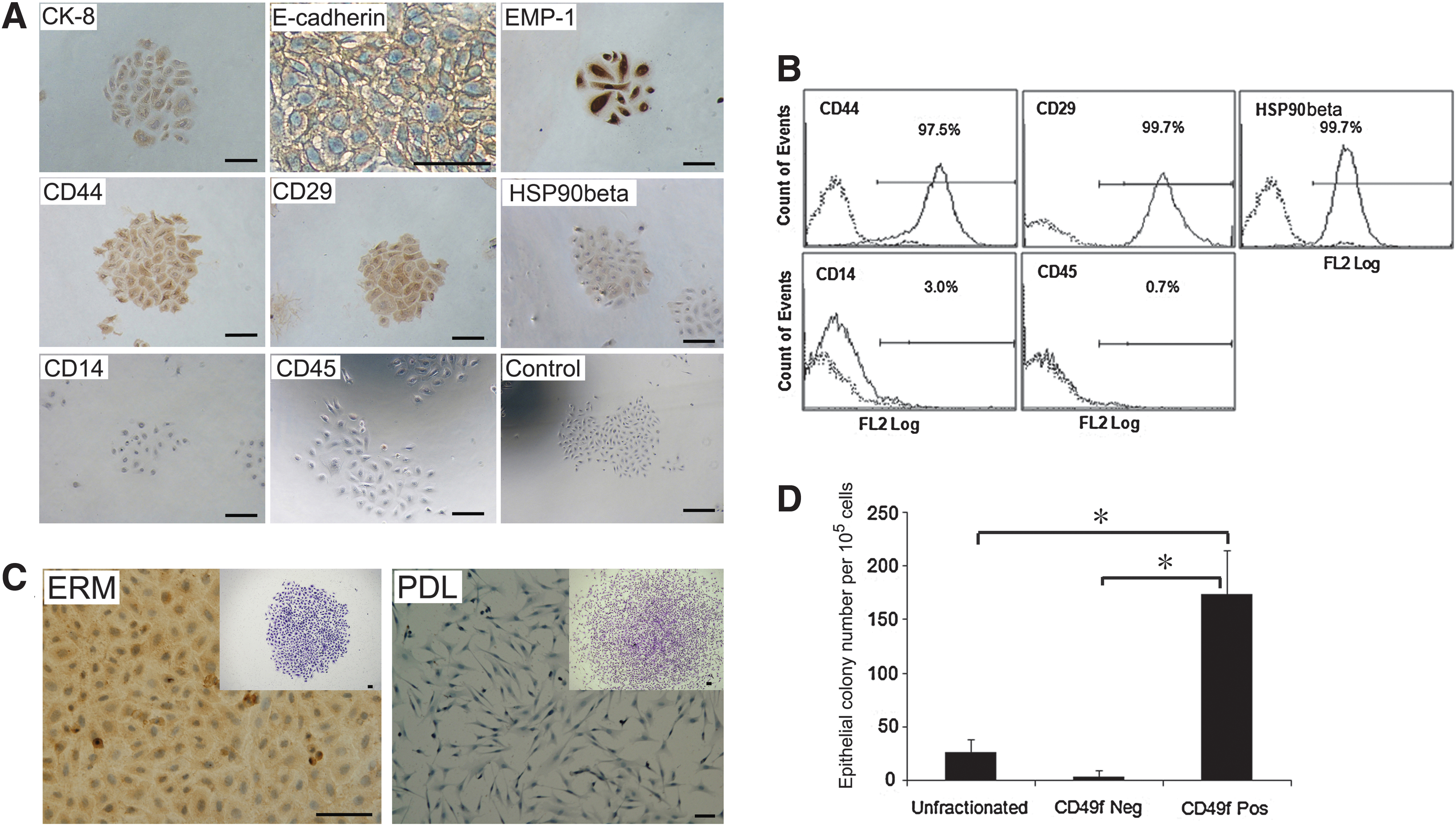
Epithelial cell rests of Malassez exhibit an epithelial progenitor phenotype.
Integrin α6/CD49f can serve as a surface marker to purify ERM cells
Previous studies have reported that human keratinocytes expressing high levels of integrin α6 subunit (laminin ligand) and low levels of CD71 (transferrin receptor) possess numerous stem cell features, including small cell size, quiescence, and skin regenerative capacity [27]. Given that ERM cells appear to display a cell surface expression profile overlapping with periodontal ligament fibroblasts (data not shown), we examined whether surface expression of integrin α6/CD49f could be used as a potential marker of epithelial cells within a heterogeneous periodontal ligament cell population. Ex vivo-expanded ERM cells were found to be positive for integrin α6/CD49f, while periodontal ligament fibroblasts were negative (Fig. 1C). Immunomagnetic bead selection was used to determine the efficiency to enrich ERM cells from freshly collected digested periodontal ligament tissues based on their expression of integrin α6/CD49f. The integrin α6/CD49f-positive fraction represented ∼2% of the total freshly isolated periodontal ligament cell population (data not shown). The presence of clonogenic cell populations was demonstrated in ERM cells and periodontal ligament fibroblasts. Cells within epithelial clones were characterized by typical cobblestone morphology (Fig. 1C inserted), while periodontal ligament fibroblasts clones showed a typical spindle-shaped fibroblastic morphology (Fig. 1C inserted). The colony-forming capacity of integrin α6/CD49f-positive selected epithelial cells (colony-forming unit epithelial cells) at day 14 showed a high level of enrichment of over 50- and 7-fold (173.91±40.99 per 105 cells plated) greater than the integrin α6/CD49f-negative cells (3.33±5.77 per 105 cells plated) and unfractionated cells (26.67±11.55 per 105 cells plated), respectively (Fig. 1D). Taken together, these results demonstrated that integrin α6/CD49f can be used as a surface marker to enrich and purify clonogenic ERM cells from a heterogeneous periodontal ligament cell population.
Multilineage differentiation potential of ERM cells in vitro
To investigate whether ERM cells are capable of undergoing an epithelial–mesenchymal transition, we assessed the differentiation capacity of ERM cells into 3 lineages of mesoderm origin: osteocytes/cementocytes, adipocytes, and chondrocytes.
The potential of ERM cells to undergo osteogenic/cementogenic differentiation in vitro was examined in the presence of media supplemented with dexamethasone, ascorbate, and inorganic phosphate. After 4 weeks of osteogenic induction, Alizarin Red positive mineralized nodules formed in ERM cultures akin to periodontal ligament fibroblast cultures (Fig. 2A), in vitro. Furthermore, when ERM cells were cultured under osteogenic inductive conditions, a significant increase in mRNA expression levels was seen for the osteogenic-associated genes, Runx2 and bone sialoprotein (BSP-II) (Fig. 2B) (n=3, P<0.05), as detected by real-time RT-PCR.

Multilineage differentiation potential of ERM cells in vitro.
Similar studies were conducted to assess the adipogenic potential of ERM cells. After 4 weeks of adipogenic induction supplemented with hydrocortisone, indomethacin, and 3-isobutyl-1-methyl-xanthine (IBMX), ERM cells developed into Oil Red O-positive lipid-containing adipocytes, similar to that observed for periodontal ligament fibroblasts (Fig. 2C). Real-time RT-PCR analysis demonstrated the up-regulation of gene expression levels for adipogenic-associated markers, peroxisome proliferator activated receptor-γ2 and leptin (Fig. 2D) (n=3, P<0.05), after adipogenic differentiation.
We also examined the chondrogenic potential of ERM cells and periodontal ligament fibroblasts cultured in 3-dimensional cell aggregates in the presence of transforming growth factor (TGF)-β3, bovine serum albumin, dexamethasone, and ITS+Premix. While periodontal ligament fibroblasts formed tightly compact pellets, the chondrocyte pellets of ERM cells were less compact and formed a number of smaller aggregates. Sections of chondrocyte pellets were stained with H&E (Fig. 2Ei). Histological staining of the chondrogenic pellets with toluidine blue revealed that they were composed of cells embedded within a sulfated-proteoglycan-rich extracellular matrix (Fig. 2Eii) that was comprised of collagen type II (Fig. 2Eiii). Gene expression studies confirmed the up-regulation of chondrocyte-associated markers type II collagen and Sox9 (Fig. 2F) (n=3, P<0.05) at week 3 of chondrogenic culture shown by real-time RT-PCR.
To confirm the stem cell properties of ERM cell populations, individual colonies were isolated using colony rings, expanded, and subjected to multi-differentiation analyses. All of the clones assessed (7/7) produced Alizarin Red staining mineralized nodules and chondrocyte pellets showing positivity to anti-collagen type II antibody, while 43% of the clones (3/7) exhibited adipogenic differentiation potential, indicated by the formation of Oil Red O-positive lipid. The tri-differentiation potential of 3 clones is shown in Supplementary Fig. S2.
Studies were also performed to determine whether ERM cells could give rise to neural type cells under neurogenic inductive conditions previously described for dental pulp stem cells [31]. After 3 weeks of neuronal induction, a subpopulation of ERM cells developed to neuron-like cells (Fig. 3Ai, ii), compared to the typical cobblestone morphology of noninduced cells (Fig. 3Aiii). Immunocytochemical analysis demonstrated that ERM cells cultured in neurogenic media expressed various neural-associated markers, including neuronal markers [Nestin (Fig. 3Bi), neurofilament protein-heavy chain (NF-H) (Fig. 3Bii), Neuronal marker protein gene product 9.5 (PGP 9.5) (Fig. 3Biii), and Tau (Fig. 3Biv)] and Glial and Schwann cell marker, GFAP (Fig. 3Bv). Isotype-matched controls (Fig. 3Bvi) demonstrated levels of nonspecific antibody binding. Real-time RT-PCR analysis showed the up-regulation of the intermediate neural marker, β-III tubulin (n=3, P<0.05), which correlated to the down-regulation of the neuronal stem cell marker, nestin (n=3, P<0.05) (Fig. 3C). These findings indicated that ERM cells are capable of differentiating into neural cells under appropriate conditions.

Neurogenic potential of ERM cells in vitro.
Ex vivo-expanded ERM cells can generate bone, cementum-like and Sharpey's fiber-like structures in vivo
To examine the osteogenic/cementogenic potential of ERM cells in vivo, 5×106 integrin α6/CD49f-positive ERM cells were transplanted subcutaneously with HA/TCP particles into the dorsal surface of NOD/SCID mice. After 8 weeks, the ERM cells generated mineralized bone structures lined with osteoblast-like cells and contained encapulated osteocytes, at the surface of HA/TCP particles in all 5 transplants (Fig. 4A, B). Moreover, Sharpey's fiber-like structures were observed in 2 of 5 transplants (Fig. 4C, D). Furthermore, thin layers of bone/cementum-like structures formed in 3 of 5 transplants (Fig. 4E, F). Immunohistochemical staining showed that ERM cells had differentiated into osteocytes and osteoblasts as assessed by positive staining for ovine-specific CD44 (Fig. 4G, H). These CD44-positive ERM cells were also identified around blood vessels (Fig. 4I, J). Interestingly, ERM cells that differentiated into osteoblasts were positive for CK-8, while those that had differentiated into osteocytes lacked CK-8 expression (Fig. 4K, L). The co-localization of CD44 and osteocalcin antigens in osteocytes and bone-lining cells was demonstrated on the same area in serial sections (Fig. 4M, N).

Generation of bone, cementum-like and Sharpey's fiber-like structures in vivo by ERM cells.
ERM undergo epithelial–mesenchymal transition during osteogenic induction
To further examine whether ERM cells were capable of undergoing epithelial–mesenchymal transition, gene expression levels of various epithelial, mesenchymal, and epithelial–mesenchymal transition markers were measured by real-time RT-PCR after osteogenic induction after 1, 2, 3, and 4 weeks. ERM cells showed a decline of the expression of epithelial markers, CK-8, CK-14, and E-cadherin (Fig. 5A) over the time course, while the expression of mesenchymal-associated genes, fibronectin and N-cadherin, was up-regulated (Fig. 5B). However, the expression level of another cytoskeletal component of mesenchymal cells, vimentin, was down-regulated during osteogenic induction (Fig. 5B). Moreover, ERM cells expressed high levels of various transcription factors known to regulate epithelial–mesenchymal transition during embryogenesis and in different cancers, such as Twist, Zinc finger E-box-binding homeobox 1 (ZEB 1), ZEB 2, and SNAI 1 (Fig. 5C).
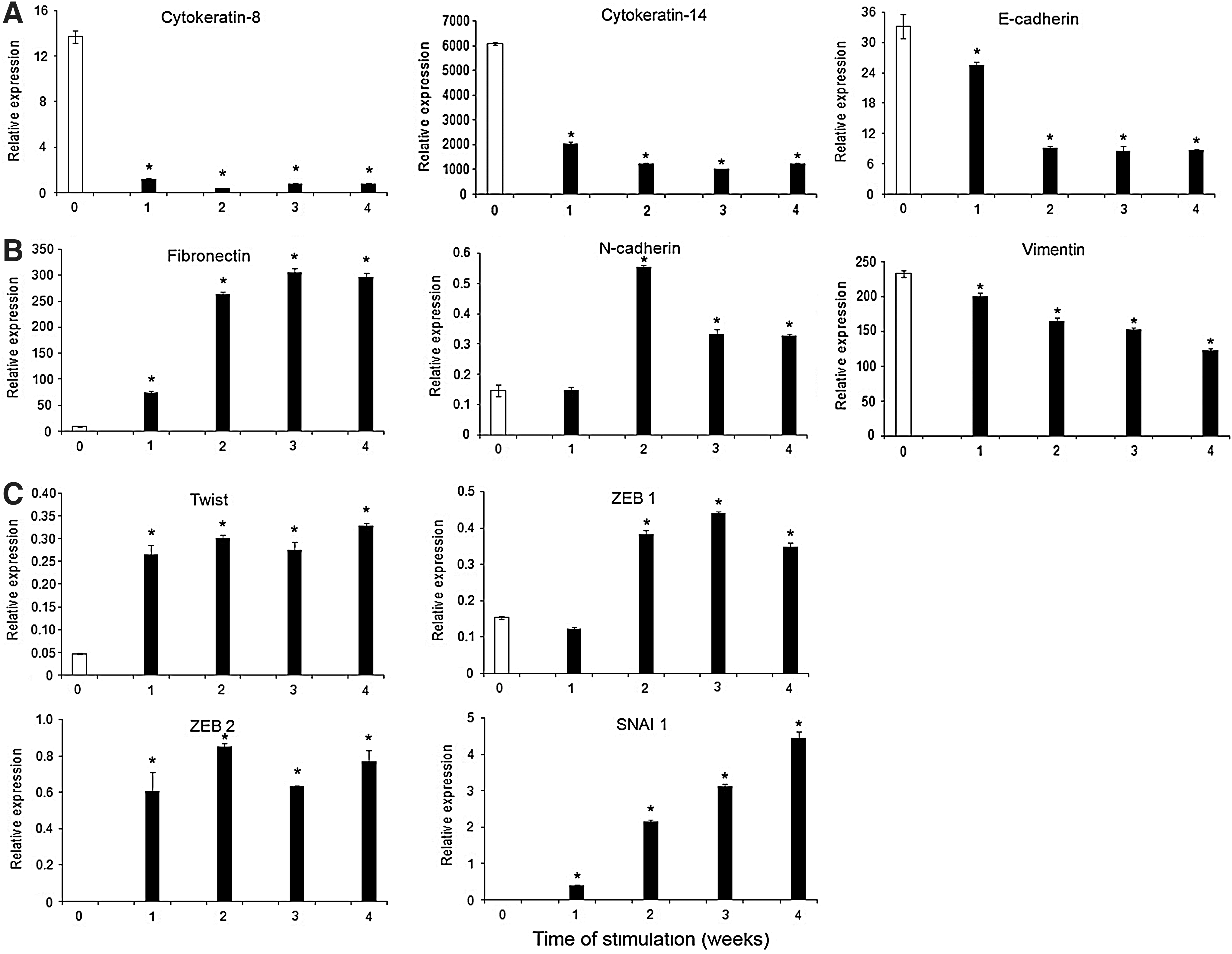
ERM cells are capable of undergoing epithelial–mesenchymal transition under osteogenic conditions. Real-time PCR analysis following osteogenic induction of ERM cells after 1, 2, 3, and 4 weeks for the expression of
Discussion
The results of the present study demonstrate that ERM cells share similar phenotypic and functional attributes with mesenchymal stem cells by their capacity to differentiate into diverse lineages indicative of mesodermal and ectodermal origin. While ERM cells are thought to be cell rests in postnatal life, our work supports the recent report that human ERM cells derived from the Hertwig's epithelial root sheath express embryonic stem cell markers such as Oct-4, Nanog, and SSEA-4, providing circumstantial evidence for the stem cell nature of ERM cells [32]. Therefore, ERM cells in postnatal tissue may function as a source of progenitor cells in periodontal ligament similar to that described for periodontal ligament stem cells [15] with the capacity for stem cell plasticity in terms of cross germ layer differentiation.
Stem cells are uncommitted cells capable of self-renewal and multi-lineage differentiation, where postnatal stem cells reside as subpopulations in various tissues responding to environmental stimuli to proliferate, migrate, and regenerate damaged tissues. Ever increasing evidence shows that adult stem cells have a broader differentiation capacity capable of forming cell types outside of the germ layer from which they were originally derived [33,34]. An important advance in characterizing adult stem cells has been the discovery that they can be pluripotent. For example, adult neural stem cells showed pluripotent capacity to give rise to cell types of all germ layers [35]. Similarly, pluripotency has been identified in adult bone marrow stem cells, termed as very small embryonic-like stem cells [36]. In addition, single bone marrow stem cells have been shown to differentiate into epithelial cells, which were traditionally thought to be exclusively of endoderm or ectoderm origin [37]. Collectively, these studies indicate that adult stem cells demonstrate broader plasticity than initially anticipated. However, it is still an open question under which circumstances adult stem cells demonstrate plasticity. Several alternative explanations are emerging, including the well-accepted theory of cell fusion and epigenetic changes when cells are exposed to extrinsic stimuli [36]. Equally important are the discovery of factors responsible for regulating stem cell plasticity [38 –40]. The establishment of induced pluripotent stem cells has helped us understand the mechanism for cell fate determination. A recent study demonstrated that mesenchymal–epithelial transition is crucial to induce pluripotency by converting fibroblasts into an intermediate epithelial cell stage [38,41]. This is consistent with the previous observations that morphologically embryonic stem cells resemble epithelial-like cells than mesenchymal-like cells, and epithelial cells can be reprogrammed more efficiently than fibroblasts [38,41,42]. Our study is in agreement with these previous studies that epithelial–mesenchymal transition, the reverse process of mesenchymal–epithelial transition, drives ERM cells to differentiate into multiple lineages.
Unlike some rapidly renewing tissues such as intestinal epithelia and skin epidermis that are in a process of constant regeneration, dental hard tissues, including enamel, dentin, and cementum, show limited, or no, capacity for regeneration. Since mesenchymal and epithelial components are 2 major parts in tooth regeneration, the improvement of purification of ERM cells is a matter of considerable interest in order to try to understand the role of this subpopulation in tissue homeostasis. Selective enzymatic digestion of periodontal ligament cell cultures that allows the less adherent periodontal ligament fibroblasts to be released and separated from the more adherent ERM has been used previously for the crude isolation of ERM cells [9,21,43]. However, to eliminate any residual periodontal ligament fibroblasts from the remaining adherent ERM cells, multiple selective enzymatic digestion steps that have the potential to introduce DNA damage to the cells are required [44]. Clearly, the availability of specific cell surface markers would greatly facilitate the isolation and characterization of ERM cells. Considering the lack of specific surface markers for ERM cells and in view of the cellular and molecular similarities of ERM cells with epithelial cells in other tissues, we utilized cell surface markers of skin keratinocyte stem cells. Keratinocyte stem cells were first isolated using β1 integrin as a cell surface marker using FACS [45]. However, it was subsequently found that cells with high β1 integrin expression contained both keratinocyte stem cells and transit amplifying cells [46]. The discovery of using the laminin receptor subunit, α6 integrin, as a key surface marker for keratinocyte stem cells was a further critical step for the purification of these epithelial progenitors [27]. Basal keratinocytes expressing high levels of α6 integrin or CD49f have been shown to display keratinocyte stem cell attributes, while low expressing α6 cells are post-mitotic differentiating keratinocytes [26,27,47]. Subsequently, oral keratinocyte stem cells have been isolated from oral mucosa using this marker [48] and also according to their relative cells size [49]. The present study has demonstrated that the enrichment of clonogenic ERM cells can be successfully achieved on the basis of integrin α6 expression, which was less expressed or lacking on clonogenic periodontal ligament stem cells. Interestingly, colonies generated from ERM cells displayed a distinct epithelial morphology and characteristic expression of CK-8, E-cadherin, and EMP-1, but also expressed the mesenchymal-associated markers CD29, CD44, and HSP90β [50]. The results of this study demonstrate that ex vivo-expanded α6-positive ERM cells are a multipotent epithelial stem cell population capable of developing into osteoblasts, adipocytes, chondrocytes, and neuron-like cells in vitro similar to that described for periodontal ligament stem cells [15,51].
As ERM cells co-habitate with periodontal ligament stem cells, it is critical to clarify that there is no contamination of periodontal ligament stem cells. First, stringent selective trypsinization was performed for the isolation of ERM cells. Second, serum-free keratinocyte medium (OKM) favoring epithelial over mesenchymal cell growth was used where previous studies have shown that mesenchymal cells do not survive in keratinocyte medium [52 –54]. We found that few periodontal ligament stem cells were capable of surviving in OKM, which appeared to lose the capacity to proliferate. Phenotypically, the sporadic periodontal ligament stem cells identified in these cultures expressed mesenchymal markers, CD44, CD29, and HSP-90β, but lacked the expression of epithelial markers CK-8 and integrin α6/CD49f (Supplementary Fig. S3). Third, any possible contamination of periodontal ligament stem cells has been minimized by using fluorescence-activated cell sorting to isolate integrin α6/CD49f high expressing epithelial cells. Additionally, to verify the stem cell properties of ERM cells, multi-lineage differentiation assays were performed at the clonal level (Supplementary Fig. S2). Individual epithelial colonies were isolated in low-density cultures using colony rings. Every subculture was expanded from 1 single epithelial colony, minimizing the contamination of other cell types. These data from single cell derived epithelial colonies support the notion that different somatic stem cells are a heterogeneous population of multi-potent stem cells and bi-potential progenitors similar to that described for the hematopoietic and mesenchymal cellular systems. Collectively, these studies indicate that periodontal ligament stem cells were unable to undergo mesenchymal–epithelial transition when grown under keratinocyte culture conditions.
While Hertwig's epithelial root sheath plays a pivotal role in tooth root development, the role of ERM cells in the adult periodontium remains unknown. However, it has been suggested that ERM cells may participate in postnatal tooth socket remodeling [55,56]. Other studies have reported the osteogenic potential of ERM cells using an experimental root resorption model, where ERM cells have been identified adjacent to root resorption lacunae and found to be immunoreactive to several bone-related markers such as bone morphogenetic protein-2, osteopontin, and ameloblastin [8]. Hence, it has been proposed that ERM cells may be involved in early cementum repair. Moreover, when ERM cells are combined with dental pulp cells, an enamel-like tissue can be produced in an in vivo transplantation assay [9]. Although it is widely accepted that dental follicle cells give rise to periodontal tissues, including cementum [8,57], Hertwig's epithelial root sheath might be another source of cementum-forming cells [52,57]. Recently, it has been reported that cementum formation by dental follicle cells can be increased by the combination of dental follicle cells and Hertwig's epithelial root sheath, which highlights a fundamental role of the epithelial cell population in periodontal regeneration [58]. In the present study, the osteogenic potential of ERM cells was further examined using an in vivo ectopic mineralization assay. Using ovine-specific antibodies, ovine ERM cells were demonstrated to be incorporated into the newly formed bone similar to osteocytes and bone-lining cells. This indicated that ovine ERM cells were responsible for the bone formation rather than endogenous mouse cells. Intriguingly, transplanted ERM cells that had differentiated into bone-lining cells showed immunoreactivity for CK-8, demonstrating the maintenance of some epithelial characteristics, while those ERM cells that had differentiated into osteocytes lacked expression of CK-8. While the present work indicates that ERM cells harbor mineral forming capacity in vitro and in vivo, it does not yet demonstrate their role in periodontal regeneration. Consequently, a more direct demonstration of this point will depend on transplantation of these cells into a periodontal defect model. The current study lends further support to the idea that ERM cells contribute, at least partially, to the production of bone/cementum-like mineral deposits and periodontal ligament-like structures or Sharpey's fibers.
It is of particular interest to note that the ERM cells expressed both epithelial and mesenchymal stromal/stem cell markers. This observation is consistent with other studies which showed that they expressed not only epithelial but also mesenchymal molecules [57,59]. We next explored whether this is an innate feature of these cells or the consequence of the epithelial–mesenchymal transition. Two recent studies provide evidence that Hertwig's epithelial root sheath, from which ERM cells are derived, can undergo epithelial–mesenchymal transition in the presence of TGF-β [52,59]. Hertwig's epithelial root sheath is able to regulate periodontal ligament stem cell differentiation and experience epithelial–mesenchymal transition to give rise to cementum-like tissues after being treated with TGF-β [52]. The present study demonstrated that ERM cells, which are descendents of Hertwig's epithelial root sheath, underwent epithelial–mesenchymal transition when exposed to osteoinductive signals over time. Interestingly, the expression level of vimentin, as a major cytoskeletal component of mesenchymal cells, was down-regulated over the time course of osteogenic differentiation in the present study in contrast with other mesenchymal markers such as fibronectin and N-cadherin. However, this is consistent with the previous report showing that vimentin inhibits osteocalcin transcription and osteoblast differentiation; thus, a down-regulation of vimentin expression may be essential for osteogenesis [60]. Epithelial–mesenchymal transition involves a phenotypic shift from epithelial cells to mesenchymal cells and allows epithelial cells to migrate to other locations of the body [61]. It can be induced by a variety of contextual signals, including the exposure to certain cytokines and chemokines such as TGF-β1 and exogenous expression of transcription factors such as the Twist or Snail families [61]. Moreover, epithelial–mesenchymal transition is believed to be involved in regulating cellular plasticity in normal adult tissues and in tumors [62]. This multipotency endowed by epithelial–mesenchymal transition [63] allows cells to migrate to injured sites and contribute to tissue repair or tumor progression, depending on their nature.
A direct link between experimentally induced epithelial–mesenchymal transition and the generation of cancer stem cells has been demonstrated [64]. The cells that had undergone epithelial–mesenchymal transition were induced to adopt the CD44high/CD24low antigenic phenotype and epithelial stem cell-like characteristics, as shown by the increased capacity to form mammospheres. This highlights that epithelial–mesenchymal transition is sufficient to generate cancer stem cells. In a recent study the same group has reported that, by ectopic expression of Twist or Snail, epithelial cells not only share antigenic profile typical of mesenchymal stromal/stem cells but also behave functionally similar with mesenchymal stromal/stem cells, including the capacity to differentiate into bone, fat, and cartilage and to invade and migrate toward tumor cells and wound sites [63]. These characteristics were not observed in the untreated epithelial cell fraction. However, in the present study, we have shown that ERM cells harbor mesenchymal stromal/stem cell-like multipotency without being forced to overexpress any epithelial–mesenchymal transition-associated genes. It therefore appears that ERM cells are more capable of adapting to the foreign environment than other epithelial cells. This higher plasticity of ERM cells may be correlated to the fact that these cells exist as epithelial islands within mesenchymal surroundings during postnatal life, while most epithelial cells in other tissues, if not all of them, are separated from adjacent tissues by a basal lamina [65]. The present study demonstrated an inverse correlation of epithelial and mesenchymal gene expression patterns during osteogenic differentiation that are generally used as markers of epithelial–mesenchymal transition during embryonic development and cancer metastasis [61,62,65,66]. Future studies will explore the underlying mechanisms that maintain ERM cells residing in the mesenchymal surroundings and whether these molecular signals are changed during times of tissue regeneration.
Conclusion
This study demonstrated that adult-derived ovine ERM cells harbor clonogenic epithelial stem cell populations that show similar properties to mesenchymal stromal/stem cells both functionally and phenotypically. This highlights that ERM cells, rather than being cell rests as indicated by their name in the literature, are an important stem cell source that might play pivotal role in periodontal regeneration. In addition, ERM cells might be a useful model to address some fundamental questions in the stem cell field such as stem cell plasticity and epithelial–mesenchymal transition.
Footnotes
Acknowledgments
We would like to acknowledge Dr. Naohisa Wada and Dr. Danijela Menicanin for technical assistance, Dr. Amy Li for advice on integrin α6 cell sorting and epithelial cell cultures, Mr. Victor Marino for ovine teeth collection, and Dr. Yeesim Khew-Goodall for providing the anti-E-cadherin antibody. We also acknowledge members of the Bone and Cancer Laboratory and the Periodontal Research Group for helpful discussions. This study was funded by National Health and Medical Research Council project grants (ID# 627143) and Australian Dental Research Foundation. Jimin Xiong was supported by the China Scholarship Council–University of Adelaide Joint Postgraduate Scholarship and Colgate Betty Fanning Postgraduate Scholarship. The authors thank the support of the School of Stomatology, Shandong University, China.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.