
Abstract
One of the earliest epithelial-to-mesenchymal transitions in mouse embryogenesis involves the differentiation of inner cell mass cells into primitive and then into parietal endoderm. These processes can be recapitulated in vitro using F9 teratocarcinoma cells, which differentiate into primitive endoderm when treated with retinoic acid (RA) and into parietal endoderm with subsequent treatment with dibutyryl cyclic adenosine monophosphate (db-cAMP). Our previous work on how primitive endoderm develops revealed that the Wnt6 gene is upregulated by RA, leading to the activation of the canonical WNT-β-catenin pathway. The mechanism by which Wnt6 is regulated was not determined, but in silico analysis of the human WNT6 promoter region had suggested that the GATA6 and FOXA2 transcription factors might be involved [1]. Subsequent analysis determined that both Gata6 and Foxa2 mRNA are upregulated in F9 cells treated with RA or RA and db-cAMP. More specifically, overexpression of Gata6 or Foxa2 alone induced molecular and morphological markers of primitive endoderm, which occurred concomitantly with the upregulation of the Wnt6 gene. Gata6- or Foxa2-overexpressing cells were also found to have increased levels in T-cell factor (TCF)-dependent transcription, and when these cells were treated with db-cAMP, they developed into parietal endoderm. Chromatin immunoprecipitation analysis revealed that GATA6 and FOXA2 were bound to the Wnt6 promoter, and overexpression studies showed that these transcription factors were sufficient to switch on the gene expression of a Wnt6 reporter construct. Together, these results provide evidence for the direct regulation of Wnt6 that leads to the activation of the canonical WNT-β-catenin pathway and subsequent induction of primitive extraembryonic endoderm.
Introduction
I
Wnts are secreted glycoproteins that are involved in a plethora of developmental processes [15 –18]. Wnt expression is first detected in the ICM and in cells surrounding the blastocele cavity, [19 –21]. In humans, 1 or more of the 19 different WNTs are expressed normally throughout gastrulation, organogenesis, and into adulthood, but they can also be expressed inadvertently, as evident in a variety of dissimilar cancers [22 –24]. Historically, WNTs have been classified based on their ability to signal through either the canonical β-catenin or noncanonical pathways. The canonical WNT pathway is activated when a WNT ligand binds to 1 of a group of 7-transmembrane Frizzled (FZD) receptors [3]. The pathway involves a complex of proteins that are regulated by post-translational modifications. In the absence of WNT, β-catenin is recruited to a destruction complex of proteins, including adenomatous polyposis coli (APC) and AXIN, where it is phosphorylated by GSK-3β and CK1γ. Phosphorylation primes β-catenin for ubquitination, leading to its degradation in a proteasome-dependent manner. When WNT is present, however, it binds to its FZD receptor and LRP5/6 co-receptor, which recruits the multidomain-containing protein Dishevelled (DVL) to the plasma membrane where it binds to FZD. GSK-3β and CK1γ now phosphorylate LRP5/6, which together with DVL facilitates the translocation and binding of AXIN to DVL and LRP5/6. AXIN can no longer participate as a part of the destruction complex, allowing β-catenin to accumulate in the cytoplasm. Subsequent translocation of β-catenin into the nucleus facilitates its interaction with T-cell factor/lymphoid enhancer factor-1 (TCF/LEF-1) transcription factors to impart changes in gene expression.
In the case of either the planar cell polarity (PCP) or WNT/Ca2+ noncanonical pathways, signaling occurs via WNT, FZD, and DVL, but further downstream events are independent of β-catenin [25]. In the PCP pathway, WNT binding to FZD recruits DVL to the plasma membrane, which results in the activation of the Rho-Rac-JNK pathway. This activation is necessary to induce the changes to the cytoskeleton that are needed for coordinated cell movements [26 –28]. In the WNT/Ca2+ pathway, the activation of FZD (Knypek and Ror2) and G-proteins triggers downstream effectors, including calcium-/calmodulin-dependent kinase IIα and PKC [25]. Much like the PCP pathway, the WNT/Ca2+ pathway influences cell polarity, cell adhesion, cell shape, as well as the nuclear factor of activated T-cells (NF-AT) [15,29,30]. The involvement of the WNT/Ca2+ pathway during ExE differentiation is not well understood, but there is evidence that p38 MAPK activation in F9 cells engineered to express rat FZD2 occurs in a DVL-independent manner after WNT5a stimulation [31]. In contrast, the canonical WNT and the PCP-signaling pathways are known to play an important role during PrE differentiation.
RA treatment of P19 cells activates RhoA, Rac1, and JNK in the PCP pathway, which is sufficient to induce PrE differentiation [32]. RA also upregulates the expression of Wnt6 in F9 cells, which activates the canonical β-catenin pathway, leading to PrE formation [13]. The same is true for WNT3a, when applied to F9 cells ectopically expressing rat FZD1 [33]. Although the link between RA and the differentiation of ExE in vitro is clear, that between RA and WNT is not. Toward that end and before implementing in vivo studies, we decided to elucidate the mechanism responsible for the RA-dependent induction of Wnt6 that initiates PrE formation in vitro. Numerous studies have provided possible candidates as regulators involved in PrE differentiation [8,34]. Gata6, a direct target gene of RA signaling [8], and Foxa2, a target gene of GATA6 [35], are well-known players in endoderm formation. In the mouse embryo, GATA6 is expressed in some embryonic stem (ES) cells of the ICM [2,3], which later become the cells of the ExE [36]. Gene-targeting experiments have revealed that Gata6-null mice die shortly after implantation [36]. Furthermore, in vitro studies show that Gata6 expression is upregulated when ES cells are treated with RA, and this is sufficient to downregulate the pluripotency marker Oct-3/4 and to induce ExE differentiation [35]. Interestingly, Gata6-null ES cells do not differentiate in the presence of RA, whereas transfection and expression of Gata6 in the absence of RA are sufficient to induce ExE differentiation [35 –37]. Although the evidence indicates that GATA6 is sufficient and necessary for RA-induced ExE differentiation of ES cells [35], and has also been proposed to bind to the rat and human WNT6 promoters [1], it is not known whether or not it signals directly or indirectly through WNT6 to induce ExE.
FOXA2 (HNF3β), initially identified as a liver-specific transcription factor [38], is another regulator of mouse visceral and definitive endoderm formation [39 –42]. The visceral endoderm, a derivative of PrE [43], has been classified as extraembryonic tissue required for supporting the proper growth of the embryo [44]. FOXA2 expression is also essential as Foxa2-null mice die between 6.5 and 9.5 days postfertilization due to a lack of a definitive node and notochord and severe constriction at the embryonic-extraembryonic junction [40,45]. Furthermore, these authors noted that mutant embryos often develop outside of the yolk sac, with defects in axial elongation and anterior development. Although FOXA2 is considered as a marker of visceral endoderm [46], it is also expressed in PrE as evident in F9 cells induced by RA [47], and like GATA6, may bind to the rat and human WNT6 promoters [1]. This information, together with that for GATA6, was enough to warrant further investigation on how Wnt6 is regulated during ExE formation.
Here, we provide new evidence for a role for GATA6 and FOXA2 regulating a canonical WNT-β-catenin-signaling pathway involved in the differentiation of F9 cells to primitive extraembryonic endoderm. In this study, we show that overexpression of Gata6 or Foxa2 in the absence of RA was accompanied by an increase in Wnt6 expression and corresponding changes in the cell shape that are hallmarks of PrE derived from F9 cells. Immunoblot and immunocytochemistry revealed that in the absence of RA, F9 cells overexpressing Gata6 or Foxa2 induced the appearance of TROMA-1 intermediate filaments characteristic of ExE. Gata6- or Foxa2-overexpressing F9 cells also had elevated levels of TCF-dependent transcription, indicative of active canonical WNT-β-catenin signaling. Chromatin immunoprecipitation (ChIP) analysis showed that GATA6 and FOXA2 could bind to the Wnt6 promoter, and when Gata6 or Foxa2 is overexpressed, there was increased activity in gene expression of a Wnt6 reporter construct. Furthermore, these Gata6- or Foxa2-expressing F9 cells, when treated with db-cAMP, were competent to complete the EMT, leading to parietal extraembryonic endoderm. Together, these results highlight a signaling hierarchy between RA, GATA6, FOXA2, and WNT6 during the specification of primitive endoderm.
Materials and Methods
Plasmids and reagents
pCMV-Gata6 was provided by Dr. E. E. Morrisey (University of Pennsylvania), pBS/KS-Foxa2 by Dr. K.H. Kaestner (University of Pennsylvania), pRL-TK by Dr. Rodney DeKoter (Western University), and pGL3-BARL by Dr. S. Anger (University of Toronto). All trans-RA and db-cAMP were from Sigma, and neomycin (G418) from Calbiochem.
Cell culture, transfection, and treatment
Mouse F9 teratocarcinoma cells (ATCCs) were cultured in the Dulbecco's modified Eagle's medium (Lonza) supplemented with 10% fetal bovine serum (Gibco), 100 units/mL penicillin, and 100 mg/mL streptomycin (Lonza). Cells were transfected with an empty vector, pcDNA3.1-Gata6, pcDNA3.1-Foxa2, pGL3-Wnt6, pGL3-BARL, and pRL-TK constructs using FuGENE6, according to the manufacturer's recommendations (Roche). Briefly, 3 μL of FuGENE6 reagent was mixed with a total of 1.5 μg of expression construct to transfect cells grown to 60% confluence in a 35-mm TC dish (BD Falcon); for co-transfection experiments, equal amounts of each of the constructs were used with the FuGENE6 reagent to a DNA ratio of 3:1.5. Cells transfected with pcDNA3.1-Gata6, pcDNA3.1-Foxa2, or the empty vector control were passed to 60-mm TC dishes (BD Falcon) 24 h post-transfection and treated with 400 μg/mL G418. Cells were treated with 0.05% dimethyl sulfoxide (DMSO; vehicle control), 10−7 M RA, or 10−7 M RA and 1 mM db-cAMP. All cells were incubated at 37°C and 5% CO2.
Reverse transcription–polymerase chain reaction
Oligodeoxynucleotide primers were designed to the mouse Wnt6 (Accession No. M89800), Gata6 (Accession No. AK142381), Foxa2 (Accession No. AL845297), and Thrombomodulin (TM) (Accession No. BC019154) nucleotide sequences. Wnt6 sense (5′ GCG GTA GAG CTC TCA GGA TG) and antisense (5′ AAA GCC CAT GGC ACT TAC AC); Gata6 sense (5′ CTC TGC ACG CTT TCC CTA CT) and antisense (5′ GTA GGT CGG GTG ATG GTG AT); Foxa2 sense (5′ ACC TGA GTC CGA GTC TGA GC) and antisense (5′ CAT GGT GAT GAG CGA GAT GT); and TM sense (5′ CCA GGC TCT TAC TCC TGT A) and antisense (5′ TGG CAC TGA AAC TCG CAG TT) primers were designed to amplify partial Wnt6, Gata6, Foxa2, and TM cDNAs. RNA was isolated from F9 cells treated with RA, RA and db-cAMP, or from F9 cells transfected with pcDNA3.1-Gata6, pcDNA3.1-Foxa2, or the empty vector, and converted into first-strand cDNA using SuperScript II reverse transcriptase (Invitrogen). The cDNAs were used as a template for polymerase chain reaction (PCR) under the following reaction conditions: Wnt6–35 cycles of 30 s at 94°C, 30 s at 62°C, and 45 s at 72°C; Gata6–35 cycles of 30 s at 94°C, 30 s at 55°C, and 30 s at 72°C; Foxa2–35 cycles of 30 s at 94°C, 30 s at 58°C, and 30 s at 72°C; TM–32 cycles of 30 s at 94°C, 30 s at 60°C, and 30 s at 72°C. Primers to L14 sense (5′ GGG AGA GGT GGC CTC GGA CGC) and antisense (5′ GGC TGG CTT CAC TCA AAG GCC) were used to amplify a constitutively expressed ribosomal gene. Samples were run on 1% agarose gels containing ethidium bromide and visualized using the FluorChem 8900 gel imaging station (Alpha Innotech). Amplicons from all PCRs were sequenced at the Robarts Research Sequencing Facility (London, ON) to confirm their identity.
Immunoblot analysis
Total cell lysates were prepared in a 1% sodium dodecyl sulfate lysis buffer containing 62.5 mM Tris–HCl, pH 6.8, 10% glycerol, 5% mercapto-2-ethanol, and 1×Halt Protease Inhibitor Cocktail (Thermo Scientific). Protein concentrations were quantified using a Bradford protein assay (Bio-Rad), and equal amounts were separated on denaturing 10% polyacrylamide gels and transferred to nitrocellulose membranes (Biotrace; Pall Corp.). The membranes were blocked in 5% skim milk and probed with antibodies, and the signals were detected using the SuperSignal West Pico Chemiluminescent Detection Kit (Pierce). The primary antibodies were directed against TROMA-1 (1:50; Developmental Studies Hybridoma Bank) and β-actin (1:10,000; Santa Cruz). Secondary antibodies were HRP-conjugated goat anti-rat and anti-mouse (1:10,000; Pierce).
Immunofluorescence and light microscopy
Cells were fixed in 4% paraformaldehyde in phosphate-buffered saline, blocked with 4% goat serum, and then incubated with a TROMA-1 antibody (1:50). After the incubation with the primary antibody, the cells were incubated in an Alexa488-conjugated anti-rat secondary antibody (1:200; Invitrogen). Cells were mounted on slides using the ProLong Gold antifade reagent (Invitrogen) and examined using a Zeiss Imager Z1 microscope, and images were captured using a Zeiss Axiocam MRm digital video camera. For light microscopy, cells were examined using a Zeiss Axio Observer A1, and images were captured using a QImaging Retiga digital video camera. All images were assembled as plates using Adobe Photoshop CS3 and Adobe Illustrator CS3.
TCF/LEF reporter assay
Cells transfected with pGL3-BARL and then treated with 0.05% DMSO (vehicle control) or 10−7 M RA, or co-transfected with pGL3-BARL and pcDNA3.1 empty vector, pcDNA3.1-Gata6, or pcDNA3.1-Foxa2 in equal amounts, were prepared 24 h post-treatment or post-transfection using the Dual Luciferase Assay Kit as per the manufacturer's instructions (Promega). Luciferase expression was quantified using the GloMax Multi Detection System (Promega). Cells were also co-transfected with pRL-TK to normalize luciferase levels.
Chromatin immunoprecipitation
ChIP assays were performed using the ChIP-IT kit (Active Motif), according to the manufacturer's protocol. Anti-GATA6 and anti-FOXA2 antibodies (sc-7244 and sc-6554, respectively) were from Santa Cruz. PCR analysis was performed on DNA isolated by ChIP, using the following primers to amplify the GATA6-binding and the FOXA2-binding regions within the mouse Wnt6 promoter: GATA6-F, 5′ TGT TCT CCG TTT CCA CTT CT; GATA6-R, 5′ AGT GCA GAG GGA CAG GTG; FOXA2-F, 5′ CAG TTG GAC AGC CTT CTA CC; FOXA2-R, 5′ CGG GAT GAA TAG TCG AGA AG. Cycling temperatures were as follows: GATA6–35 cycles of 30 s at 94°C, 30 s at 52°C, and 30 s at 72°C; FOXA2–35 cycles of 30 s at 94°C, 30 s at 58°C, and 30 s at 72°C. Samples were separated on 1.5% agarose gels containing ethidium bromide and visualized using the FluorChem 8900 gel imaging station. Amplicons were sequenced at the Robarts Research Sequencing Facility (London, ON) to confirm their identity.
Wnt6 promoter assay
The promoter region of the Wnt6 gene was cloned into the pGL3-luciferase vector (pGL3-Wnt6) and was co-transfected with pcDNA3.1-Gata6 or pcDNA3.1-Foxa2 in equal amounts. Cells were prepared using the Luciferase Assay Kit as per the manufacturer's instructions (Promega). Luciferase expression was quantified using the GloMax Multi Detection System (Promega). Cells co-transfected with pGL3-Wnt6 and pcDNA3.1 served as control.
Statistical analysis
Data from all experiments are representative of 3 independent biological replicates performed on separate occasions. Densitometry data were obtained using a FluorChem 8900 Chemiluminescence and Gel Image (Alpha Innotech). Analysis of data between control and treated or transfected groups was performed using a Student's t-test, assuming unequal variances (Excel; Microsoft Corp.). P values were 1-sided and considered statistically significant at the 0.05 level. Statistical data are presented as the mean±standard error.
Results
Gata6 and Foxa2 upregulation by RA leads to Wnt6 expression in F9 cells
Mouse F9 teratocarcinoma cells grown in monolayer differentiate into primitive endoderm (PrE) and parietal endoderm (PE) when exposed to RA or RA and db-cAMP, respectively. Although Gata6 and Foxa2 were previously reported to be upregulated in RA- and RA and db-cAMP-treated F9 cells, respectively [48,49], for our purposes, it was necessary to re-examine their expression profiles in PrE cells, which were induced by treating F9 cells with RA alone. Gata6 was not expressed in pcDNA3.1-containing cells (control), but was present in RA- and RA and db-cAMP-treated cells (Fig. 1). The expression of Foxa2 was detected in RA-treated cells, confirming an earlier report [47] and in RA and db-cAMP-treated cells. cDNA from Gata6-transfected cells was also used in a PCR with Foxa2 primers to test the notion that Foxa2 is regulated by GATA6 in F9 cells, as it is in ES cells [35]. Likewise, cDNA from Foxa2-transfected cells was used in a PCR with Gata6 primers. Results from these experiments revealed an amplicon corresponding to Foxa2 in Gata6-expressing cells (Fig. 1). Thus, the hierarchy at this point would indicate that RA upregulates Gata6 and Foxa2 expression, the latter either directly or indirectly via GATA6.
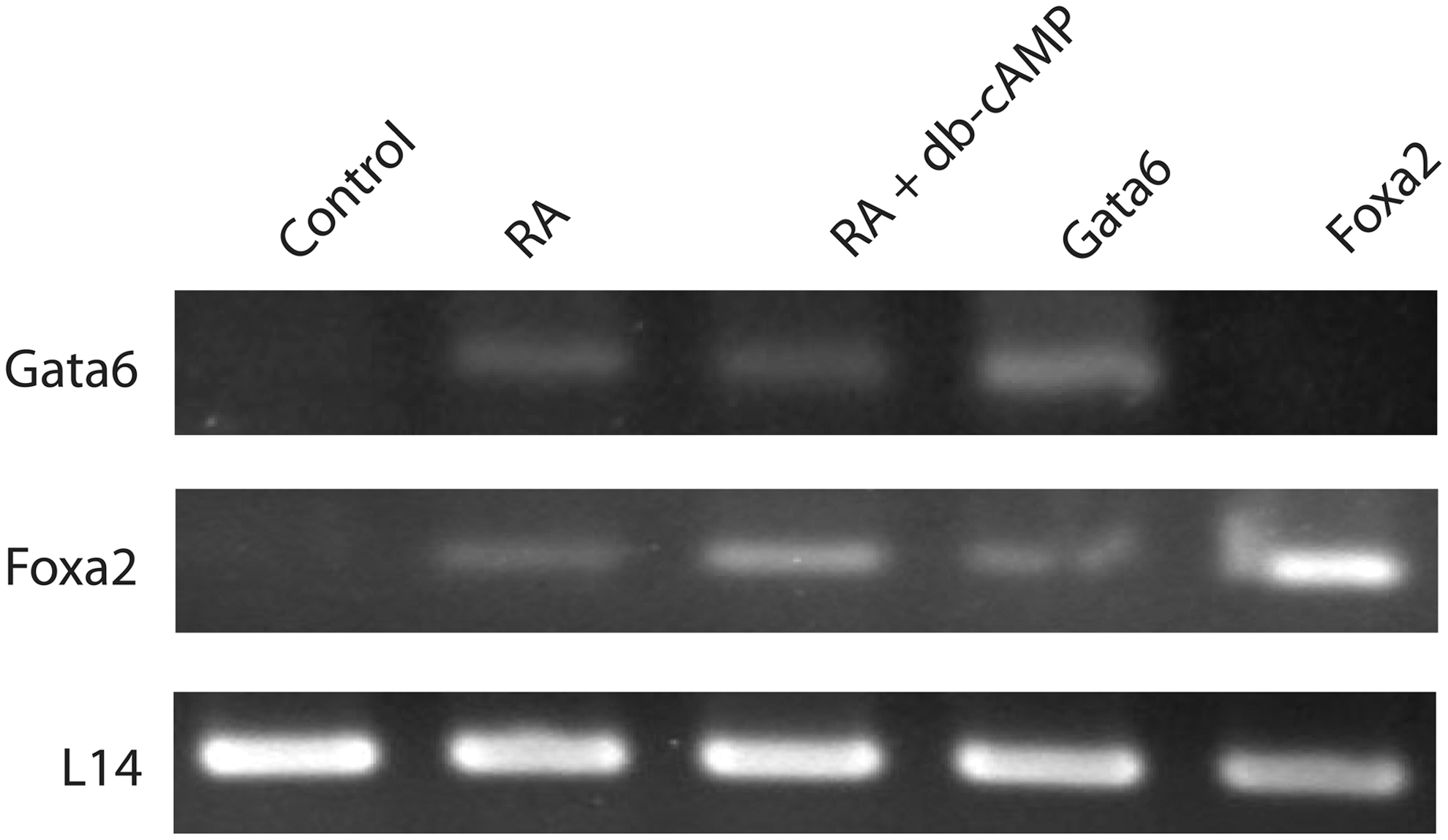
Gata6 and Foxa2 mRNA are upregulated during retinoic acid (RA)-induced differentiation. Total RNA from cells treated with RA or RA and dibutyryl cyclic adenosine monophosphate (db-cAMP) and cells transfected with empty vector (control), Gata6 or Foxa2 and selected with G418 were collected and reverse transcribed into first-strand cDNA for polymerase chain reaction (PCR). A Gata6 amplicon was seen in RA and RA and db-cAMP lanes, and as expected in cells ectopically expressing Gata6, but not in the control or cells ectopically expressing Foxa2. A Foxa2 amplicon was seen in RA and RA and db-cAMP lanes and in cells ectopically expressing Gata6 or Foxa2, but not in the control. The L14-positive control amplicon was seen under all conditions. Representative results from 5 independent experiments are shown.
With the evidence that Wnt6 is upregulated in F9 cells in response to RA and can promote PrE differentiation when overexpressed in F9 cells [13], and given the putative RA-responsive transcription factor- (GATA6 and FOXA2) binding sites in a region of the conserved human and rat WNT6 promoters [1], we hypothesized that in the absence of RA, the overexpression of Gata6 or Foxa2 would upregulate Wnt6 expression in F9 cells. To test this, total RNA was collected and reverse transcribed into cDNA from cells treated with RA and RA and db-cAMP and from cells transfected with pcDNA3.1-Gata6, pcDNA3.1-Foxa2, or the empty vector (negative control), the latter 3 following 7 days of G418 selection. PCR results with cDNAs and Wnt6 or L14 primers showed relatively equal levels of L14 expression under all treatments (Fig. 2). For Wnt6, no amplicon was seen in controls, but was as previously reported [13], present in cells induced to form PrE and to some extent in those treated with RA and db-cAMP to induce PE. Results also showed a Wnt6 amplicon of the expected size in F9 cells transfected with the Gata6 expression construct (Fig. 2). Experiments were repeated for the analysis of Foxa2-overexpressing cells, and results were similar to that seen for Gata6 (Fig. 2). Sequencing confirmed the identity of the amplicon as being Wnt6, and the appearance of the L14 amplicon confirmed that cDNAs were present under all conditions. Taken together, these results would indicate that in the absence of RA, Gata6 or Foxa2 overexpression was sufficient to upregulate the endogenous Wnt6 gene in F9 cells.

Overexpression of Gata6 or Foxa2 induces Wnt6 expression. Total RNA from cells treated with RA to induce primitive endoderm or RA and db-cAMP to induce parietal endoderm, and cells transfected with empty vector (control), Gata6 or Foxa2 and selected with G418 were collected and reverse transcribed into first-strand cDNA for PCR. Oligodeoxynucleotide primers for PCR were designed to detect Wnt6 or the constitutively expressed ribosomal gene L14. A Wnt6 amplicon was seen in the RA and RA and db-cAMP lanes, in cells transfected with Gata6 or Foxa2, but not in the control. The L14 amplicon was present in all lanes. Representative results from 3 independent experiments are shown.
Gata6 and Foxa2 expression is sufficient to induce ExE
Since Gata6 expression was RA-responsive and could upregulate a Wnt known for its ability to induce PrE [13], we hypothesized that Gata6 overexpression would induce PrE in the absence of RA. The appearance of TROMA-1, a cytokeratin A intermediate filament, was used as a definitive marker of ExE [50]. Immunoblot analysis showed that TROMA-1 was expressed in cells treated with RA and RA and db-cAMP, but not in empty vector-transfected controls (Fig. 3A). TROMA-1 was also expressed in cells transfected with the Gata6 construct, providing evidence for a role in ExE differentiation (Fig. 3A). Densitometry analysis confirmed that the relative levels of TROMA-1 induced by chemical treatment and by Gata6 overexpression were significantly higher than that in empty vector transfected F9 cells (Fig. 3B).
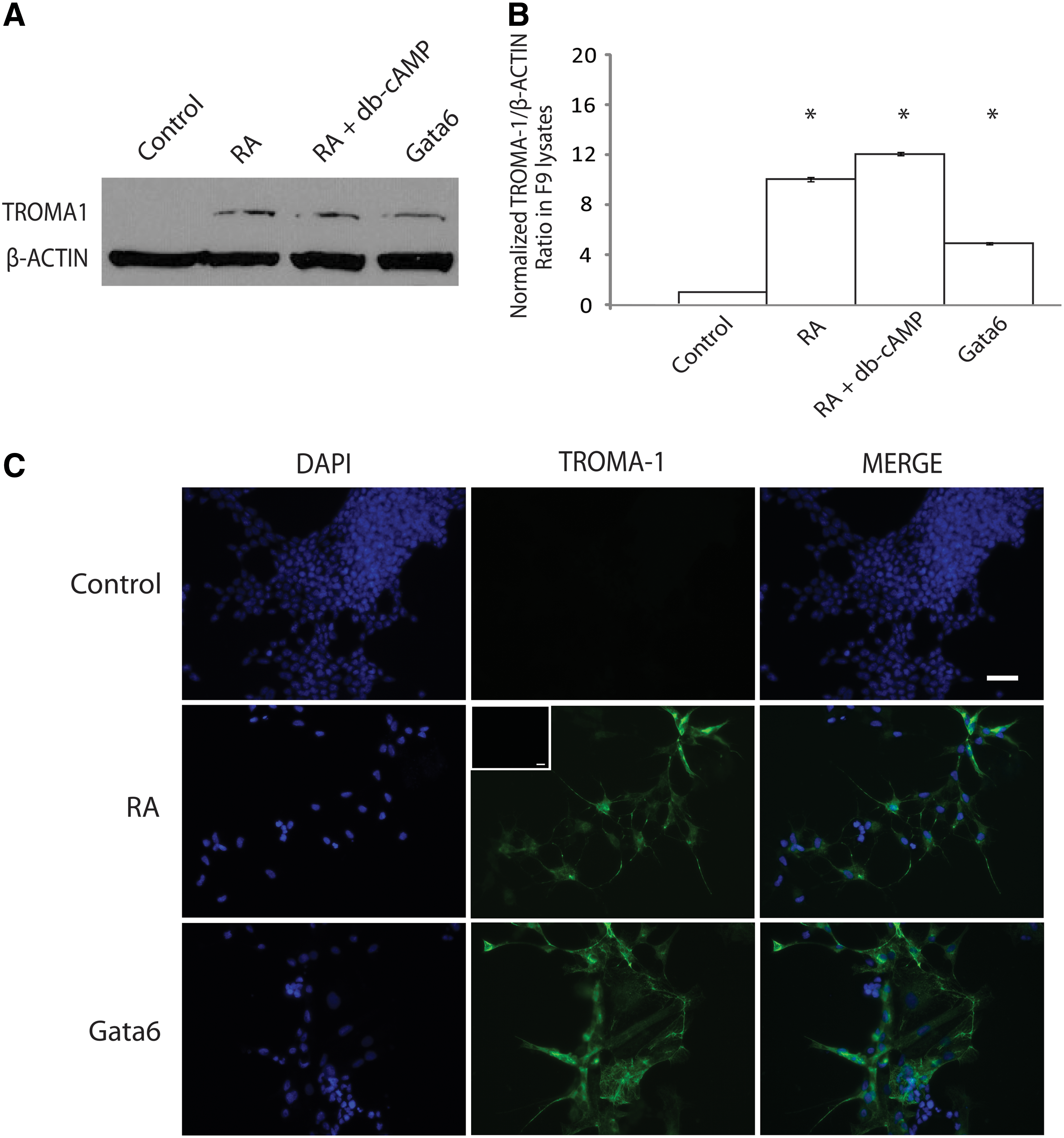
Overexpression of Gata6 induces extraembryonic endoderm (ExE).
The assembly of TROMA-1-positive cytokeratin A filaments was also used as a molecular readout of PrE and PE formation. To examine for the presence of TROMA-1-positive intermediate filaments, cells were treated with RA or transfected with either the Gata6 or the empty vector construct, selected with G418, and then processed for immunocytochemical analysis (Fig. 3C). The extensive network of intermediate filaments that formed when F9 cells were treated with RA corroborates the immunoblot data. A similar staining pattern was seen in cells transfected with the pcDNA3.1-Gata6 plasmid and selected using G418 (Fig. 3C). A control for the nonspecific binding of the secondary antibody alone showed no TROMA-1 staining (inset, Fig. 3C). Together, these results indicate that the overexpression of Gata6 alone is sufficient to increase the levels of cytokeratin A, which in turn gets assembled into intermediate filaments indicative of ExE.
Having established the expression pattern of Gata6 after RA treatment, the ability of Foxa2 overexpression to induce cells to form PrE was examined. Furthermore, since Gata6 overexpression induced markers of differentiation (Fig. 3), caused changes in cell morphology, and could induce the expression of Foxa2 (Fig. 1), then by inference, we predicted that Foxa2 overexpression would also induce PrE in the absence of RA. Immunoblot analysis showed that TROMA-1 was expressed in cells treated with RA and RA and db-cAMP, but not in empty vector-transfected controls (Fig. 4A). TROMA-1 was also expressed in cells transfected with the Foxa2 construct (Fig. 4A), and the densitometry analysis confirmed that the relative levels of TROMA-1 induced by chemical treatment and by Foxa2 overexpression were significantly higher than that in the controls (Fig. 4B). Immunocytochemistry was also used to confirm that the Foxa2-dependent increase in TROMA-1 levels would translate into the assembly of cytokeratin A intermediate filaments (Fig. 4C). Cells were treated with RA or transfected with either the Foxa2 or the empty vector construct, and then selected with G418 before being processed for immunocytochemistry with the TROMA-1 antibody. Results confirmed the immunoblot data, and the appearance of intermediate filaments in cells transfected with pcDNA3.1-Foxa2 was reminiscent of that in the positive control cells treated with RA (Fig. 4C). A control for the nonspecific binding of the secondary antibody alone showed no TROMA-1 staining (inset, Fig. 4C). These results indicated that the overexpression of Gata6 or Foxa2 alone was sufficient to induce a marker of primitive and parietal endoderm, and to cause changes in the morphology of these cells, both of which parallel the effects when F9 cells are treated with RA.
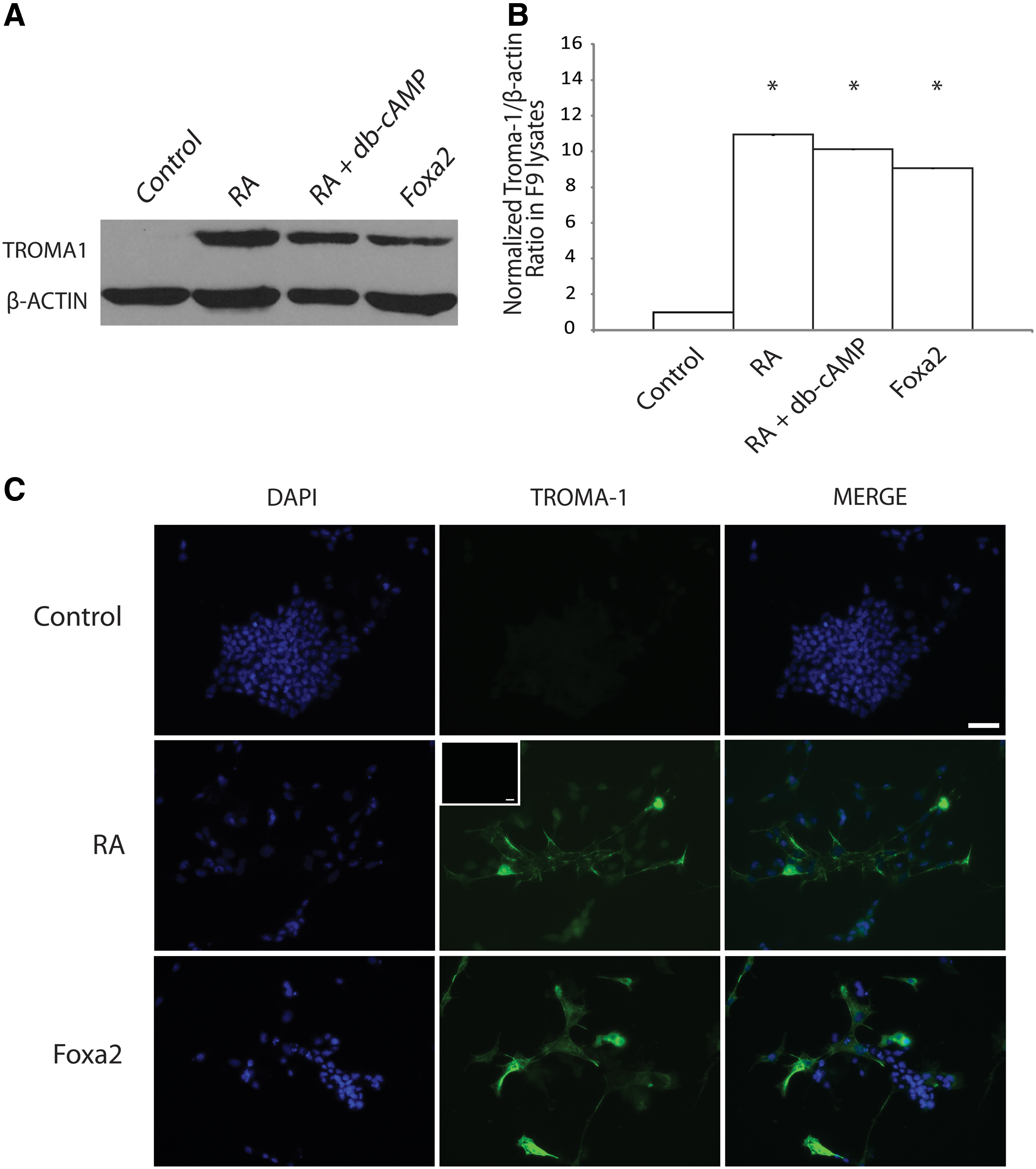
Overexpression of Foxa2 induces ExE.
GATA6 and FOXA2 signal through the canonical WNT-β-catenin-signaling pathway
Since GATA6 or FOXA2 was found to upregulate Wnt6 (Fig. 2), and WNT6 activates the canonical WNT-signaling pathway [13], then one would expect that Gata6 or Foxa2 overexpression should activate canonical WNT-β-catenin signaling. The activation of the canonical pathway and β-catenin/TCF/LEF-dependent transcription was tested directly using a pGL3-BARL reporter assay [51]. F9 cells co-transfected with pGL3-BARL and a Renilla luciferase construct (pRL-TK) and then treated with DMSO (vehicle control) had a 10-fold less luciferase activity relative to that in RA-treated cells (Fig. 5). F9 cells were also co-transfected with pGL3-BARL and pcDNA3.1-Gata6, pGL3-BARL and pcDNA3.1-Foxa2, or pGL3-BARL and pcDNA3.1 empty vector, and then assayed for luciferase activity after 24 h. Luciferase activity in cells overexpressing Gata6 or Foxa2 was significantly higher (12-fold) than in the controls. Together, this evidence allows us to place GATA6 and FOXA2 between RA- and β-catenin/TCF/LEF-dependent signaling in a hierarchy responsible for the induction of F9 cells to form ExE.
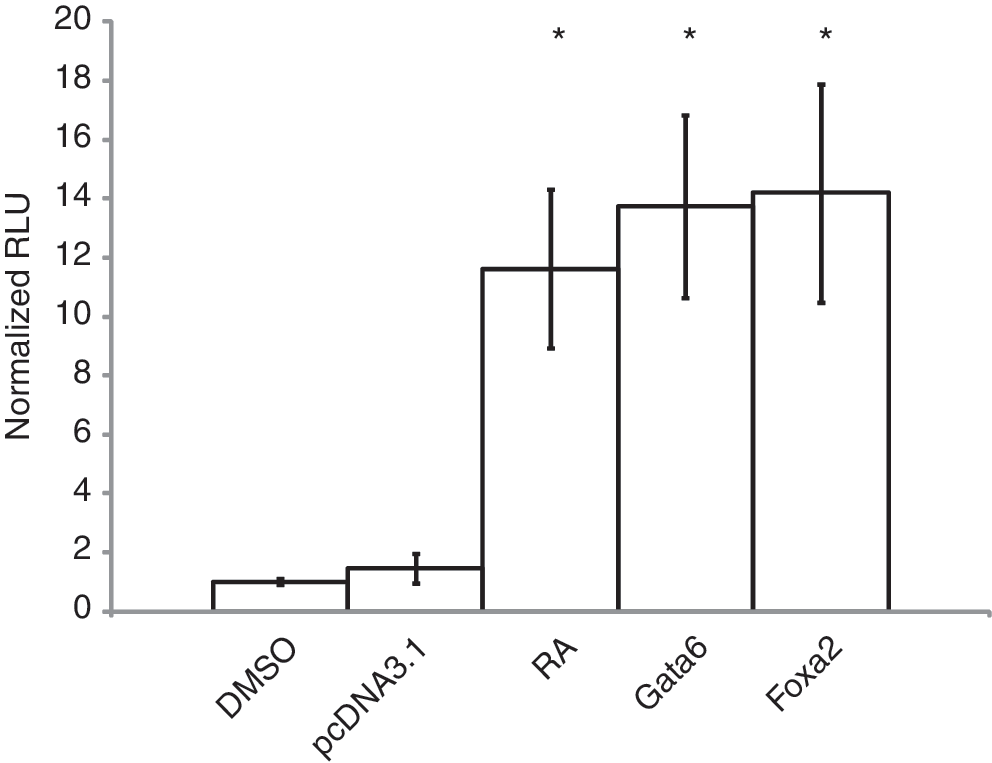
GATA6 and FOXA2 activate canonical WNT-β-catenin/TCF-dependent transcription. Cell lysates from cells transfected with pGL3-BARL, then treated with RA or dimethyl sulfoxide (DMSO) vehicle, and cells co-transfected with pGL3-BARL and the empty vector control, Gata6 or Foxa2, were collected and processed for luciferase activity 24 h post-RA treatment or transfection. Cells treated with RA had a 12-fold increase in luciferase activity relative to the DMSO-treated controls. Gata6- or Foxa2-transfected cells exhibited a 9-fold increase in luciferase activity relative to the transfected empty vector controls. Data are representative of 3 independent experiments. Bars represent the mean fold change in relative luciferase units (RLU)±standard error (SE), normalized for Renilla luciferase activity. *P<0.05. TCF, T-cell factor.
GATA6 and FOXA2 bind the Wnt6 promoter and regulate its activity, leading to primitive endoderm formation
To assign a direct link between GATA6 and FOXA2 and WNT6 expression during ExE differentiation, it was first necessary to demonstrate that GATA6 and FOXA2 could bind to the endogenous Wnt6 promoter. Once established, the next question was to ask whether or not binding was sufficient to drive the expression of a Wnt6 reporter construct. To begin, DMSO- and RA-treated F9 cells were processed for ChIP using antibodies against GATA6 and FOXA2. Immunoglobulin G (IgG) served as a negative control. PCR, using primers spanning the putative GATA6- and FOXA2-binding sites within the Wnt6 promoter [1], was first performed on sheared DNA before immunoprecipitation (input, Fig. 6A). Results revealed amplicons of the expected size in DMSO- and RA-treated cells, and sequencing confirmed their identity. PCR after immunoprecipitation with antibodies against GATA6 or FOXA2 produced similar results, but only in RA-treated cells (Fig. 6A). Amplicons were not seen in DMSO-treated cells, or when the immunoprecipitation was performed with IgG. Together, the data indicated that both transcription factors were capable of binding the Wnt6 promoter in F9 cells.
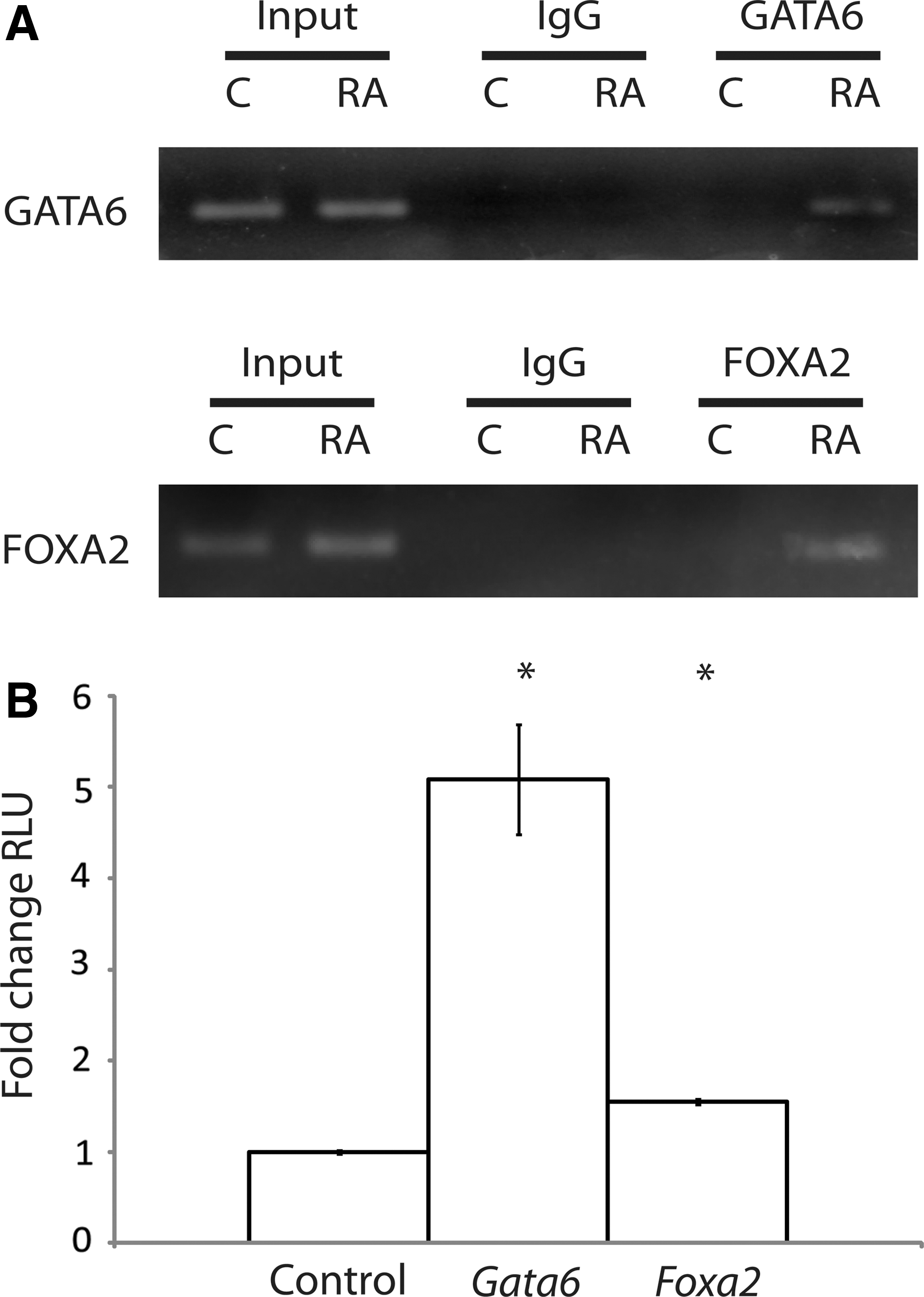
GATA6 and FOXA2 bind to and activate the Wnt6 promoter.
Satisfied with the data from the ChIP analysis, we next employed a luciferase reporter assay using ∼1.2 kb of sequence of the mouse Wnt6 promoter upstream of the ATG start site. COS-7 cells were co-transfected with the pGL3-Wnt6 reporter and either pcDNA3.1-Gata6 or pcDNA3.1-Foxa2, and then assayed for luciferase activity. The cell line was chosen, since it does not express Wnt6 (data not shown), Foxa2 [52], or any significant levels of Gata factors [53]. Furthermore, evidence also exists that Foxa2 expression is not enhanced in COS cells transfected with Gata4 [54], which has a similar consensus binding site found in GATA6 and plays a role during PrE differentiation [55,56]. Results revealed that when either the Gata6 or Foxa2 constructs were present, the luciferase activity was significantly higher relative to the empty vector control (Fig. 6B). Thus, GATA6 and FOXA2 acted directly on the Wnt6 promoter, and although reports indicate that the activation of these transcription factors is sufficient to induce cells to form PE [2,49], our previous work would argue that the upregulation of Wnt6 is only sufficient to induce cells to form PrE.
We demonstrated previously that WNT6 was sufficient to induce F9 cells to form PrE, but not PE [13], whereas the expression of constitutively active Gα13 permitted cells to proceed through to PE [10]. A previous study has also reported that Gata6 and Foxa2 expression is upregulated when F9 cells are induced to PE, and Gata6 overexpression in Sox7-silenced cells was also able to induce PE [49]. Thus, it is still not clear whether or not the overexpression of Gata6 or Foxa2 alone is sufficient to induce PE. To address this issue, cells were treated with RA or RA and db-cAMP, or transfected with pcDNA3.1-Gata6 or pcDNA3.1-Foxa2 and then treated with db-cAMP or left untreated, and then examined for changes in the morphology using phase-contrast microscopy (Fig. 7). Empty vector-transfected and DMSO-treated cells (controls) were morphologically similar as compact bodies (top panels Fig. 7). Morphologically, these cells were indistinguishable from cells treated with db-cAMP (data not shown). In contrast, RA-treated cells shared morphological similarities with those transfected with pcDNA3.1-Gata6 or pcDNA3.1-Foxa2. Specifically, cells had migrated away from the compact bodies and flattened out over the surface of the plate. Cells transfected with pcDNA3.1-Gata6 or pcDNA3.1-Foxa2 and then treated with db-cAMP resembled the RA and db-cAMP-treated positive control. Under these conditions, cells appeared to have lost their stellate shape and instead have rounded up, developed long slender filopodia, and became more refractile. These changes in morphology prompted further investigation using molecular markers to determine the fate of the cell resulting from the individual treatments. PCR analysis was performed with primers to TM, a PE marker that increases 4-fold over the barely detectable levels in PrE [10,57]. Results showed that TM was not detected in empty vector-transfected or DMSO-treated cells or when cells were treated with RA alone. As expected, TM was expressed in the positive controls, which were those cells induced to form PE by RA and db-cAMP. Interestingly, TM amplicons were not seen in Gata6- or Foxa2-overexpressing cells, although cDNA was available for amplification as evident by the L14 amplicons in all lanes. In contrast, TM amplicons were seen in Gata6- or Foxa2-transfected cells that were treated with db-cAMP. Thus, we are proposing that these transfected cells had developed into PrE and remained competent to form PE and complete the EMT following the appropriate stimulation to augment PKA activity. Taken together, our study provides new evidence that GATA6 and FOXA2 signal through a canonical WNT-β-catenin-signaling pathway in F9 cells to induce PrE, but like WNT6 [13], neither one of these transcription factors permits cells to differentiate into PE. Furthermore, GATA6 appears to have a dual role, inducing the expression of Wnt6 as well as Foxa2.
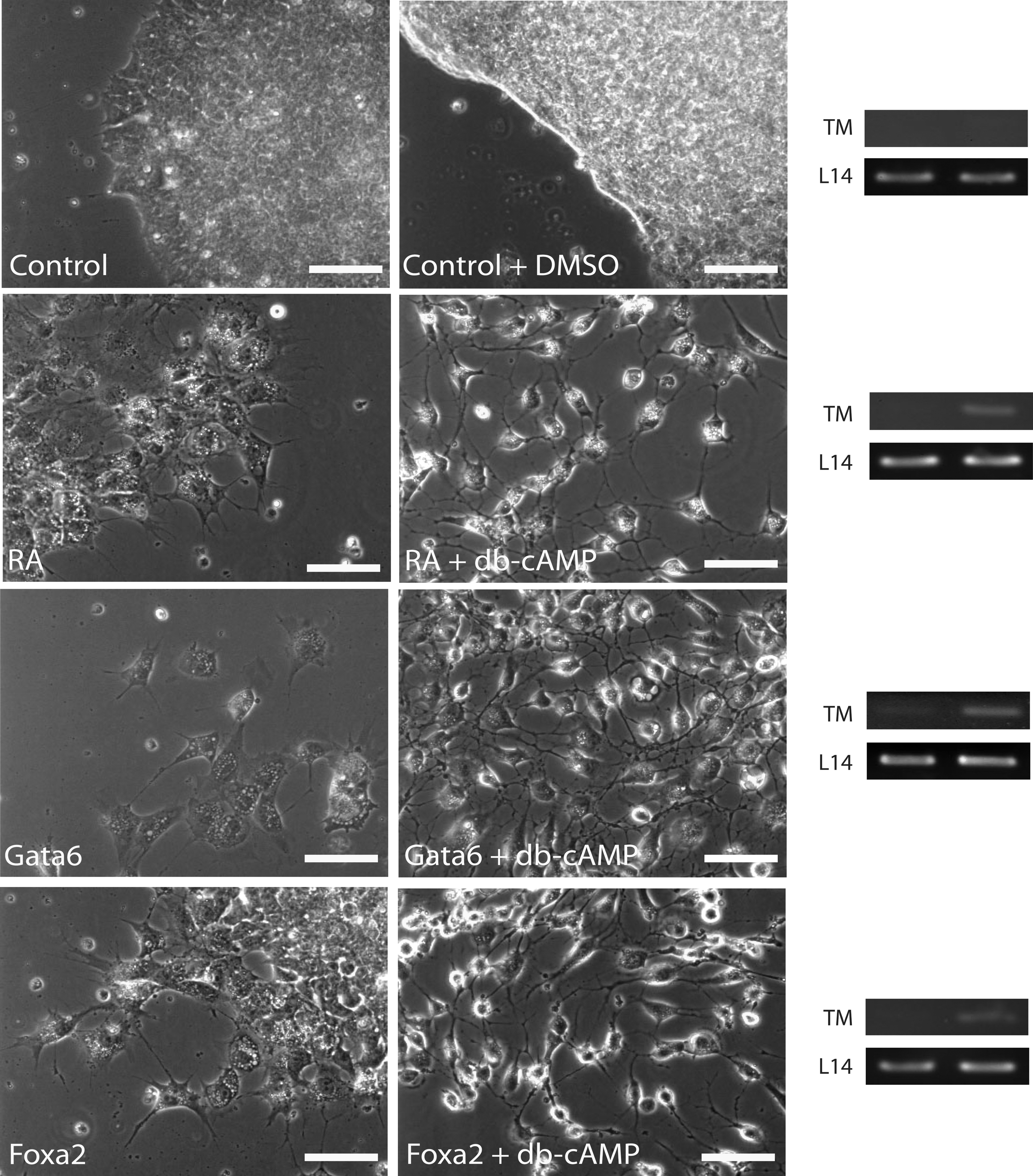
GATA6- or FOXA2-induced PrE is competent to complete the epithelial-to-mesenchymal transition and form PE. Untreated or DMSO-treated cells showed similar morphology. Likewise, cells treated with RA to induce primitive endoderm were morphologically similar to those transfected with Gata6 or Foxa2. In each case, cells migrated from the compact bodies, characteristic of the undifferentiated cells, and adopted a stellate shape with numerous filopodia. Cells transfected with Gata6 or Foxa2 and then treated with db-cAMP, showed morphological similarities to RA and db-cAMP-treated cells. Under these conditions, the cells were more spherical in shape, were highly refractile, and possessed relatively long filopodia. Total RNA from cells treated with DMSO, RA, or RA and db-cAMP and cells transfected with Gata6 or Foxa2, or transfected with Gata6 or Foxa2 and then treated with db-cAMP, were collected and reverse transcribed into first-strand cDNA for PCR. Oligodeoxynucleotide primers for PCR were used to detect Thrombomodulin (TM) expression, indicative of parietal endoderm, or L14, a constitutively expressed ribosomal gene. TM expression is only seen in cells treated with RA and db-cAMP and in those transfected with Gata6 or Foxa2 and then treated with db-cAMP. The presence of the L14 amplicon indicated that cDNAs were present under all conditions. The Gata6- or Foxa2-transfected cells were selected with G418 for 7 days or selected with G418 for 5 days and then treated with db-cAMP for 4 days under continual G418 selection. Scale bars: 20 μm.
Discussion
In the mouse embryo, several transcription factors, including those of the SOX, GATA, and FOX (HNF) families, are expressed shortly after fertilization [58]. GATA6 is expressed initially in some cells of the ICM at the time of implantation, whereas FOXA2 expression is only apparent in cells of the ExE [2,36,42,47,59]. Our study with the F9 teratocarcinoma cell line, which differentiates into PrE and PE after treatment with RA and RA and db-cAMP, respectively, confirmed that RA-induced differentiation into ExE was accompanied by the upregulation of the Gata6 and Foxa2 genes (Fig. 1). Furthermore, results indicated that the overexpression of Gata6 or Foxa2 alone was sufficient to induce biochemical and morphological markers of ExE, specifically PrE (Figs. 3, 4 and 7). TROMA-1 serves as a useful marker of ExE [50], but it does not discriminate between PrE and PE. To distinguish between the 2, a PCR-based assay was employed to detect changes in the expression of Nanog, Sox2, Utf-1, and other candidates. Unfortunately, we were unable to find a marker unique to PrE (Ren, Caraher, and Kelly, unpublished). In contrast, TM expression is used to distinguish between PrE and PE [57], and its presence together with the refractile appearance of the cells (this study) and changes to their morphology [7] indicated that cells ectopically expressing Gata-6 and treated with db-cAMP had differentiated into PE (Fig. 7). That these changes were not seen in cells ectopically expressing Gata-6 alone would indicate that PE differentiation is also dependent on other factors (Fig. 7). Not to undermine its importance, GATA6 is a master regulator of ExE differentiation. Furthermore, it is one of the first ExE-specific transcription factors to be expressed in vivo, and its loss of function results in the absence of ExE and Foxa2 expression and the death of the developing embryo [2,36,37,60]. Compelling evidence indicates that Gata6 expression is indicative of and required for the formation of the primitive endoderm lineage that precedes the parietal and/or visceral endoderm lineages [3,34,37,61,62]. The ability of GATA6 to induce the expression of Foxa2 (Fig. 1) might explain the absence of the latter reported for the Gata6 nulls. That FOXA2 is not required for early embryogenesis and visceral endoderm forms in its absence [40,42,63,64] are also indicative of the placement of GATA6 in the hierarchy of coordinated signaling required during ExE patterning and formation. Irrespective of this placement and despite the numerous in vivo and in vitro assays showing the necessity for GATA6 and FOXA2 in the proper development of ExE, many questions remain as to what genes are regulated by these transcription factors during differentiation, and what subsequent impact the proteins they encode do have on embryonic patterning. Our previous study found that one of these genes is Wnt6, which is upregulated during PrE differentiation and downregulated during PE differentiation [13]. Furthermore, we also reported that the overexpression of Wnt6 leads to the activation of the canonical WNT-β-catenin pathway, allowing for the accumulation and translocation of β-catenin into the nucleus, where it can interact with TCF/LEF to regulate the genes necessary for PrE specification. The present study is the first to report that the overexpression of Gata6 or Foxa2 can regulate the expression of the mouse Wnt6 gene (Fig. 2), and the ChIP analysis provided evidence to indicate that this regulation is directly the result of GATA6 or FOXA2 binding to the Wnt6 promoter (Fig. 6). Interestingly, this regulation may be mammalian-specific, as in Xenopus, Wnt6 appears to regulate GATA4 and 6 during heart development [65]. Despite this difference between model systems, we provide evidence that in a feed-forward manner, GATA6 and FOXA2 activate the WNT-β-catenin pathway, which leads to ExE differentiation. Inactivation of GSK-3β by phosphorylation is one of the hallmark indicators that this pathway has been activated [18]. Likewise, the presence of nuclear β-catenin is highly suggestive of active WNT signaling, but this localization does not necessarily equate to transcriptional activation [66]. To clarify this distinction in the case of ExE, we provide direct evidence for TCF/LEF-dependent transcriptional activity resulting from ectopic expression of either Gata6 or Foxa2 (Fig. 5). Thus, we are confident that the expression of these transcription factors is responsible for elevating the expression of a Wnt gene involved in canonical β-catenin signaling. Although differentiation is temporarily halted at the PrE stage, as evident by the fact that these cells do not express TM, a marker of PE, and they appear similar morphologically to PrE, these cells nevertheless remain competent to form PE when PKA activity is increased (Fig. 7). Again, this evidence corroborates our earlier work that Wnt6 expression is only sufficient to induce cells into the PrE lineage [12,13] and puts into context the limitations in inductive capabilities of GATA6 and FOXA2. Finally, that the Wnt6 reporter construct is activated by Gata6 overexpression (Fig. 6B) puts Wnt6 in the ever-growing list of genes, including Dab2, laminin, Afp, HNF4, and others, regulated by GATA6 during endoderm formation [36,67,68]. That Foxa2 overexpression also has the ability to induce the Wnt6 reporter (Fig. 6B) points to a complex signaling hierarchy involved in a transcriptional network controlling the specification of PrE in the very early development of the mouse (Fig. 8). This complexity, as evident from the fact that embryos carrying targeted deletions in either Foxa2 [40,45] or Gata6 [3,36] die from ExE defects, underscores the importance of each protein in regulating the expression of a number of genes and gene families, including those encoding WNT proteins, required for embryogenesis.
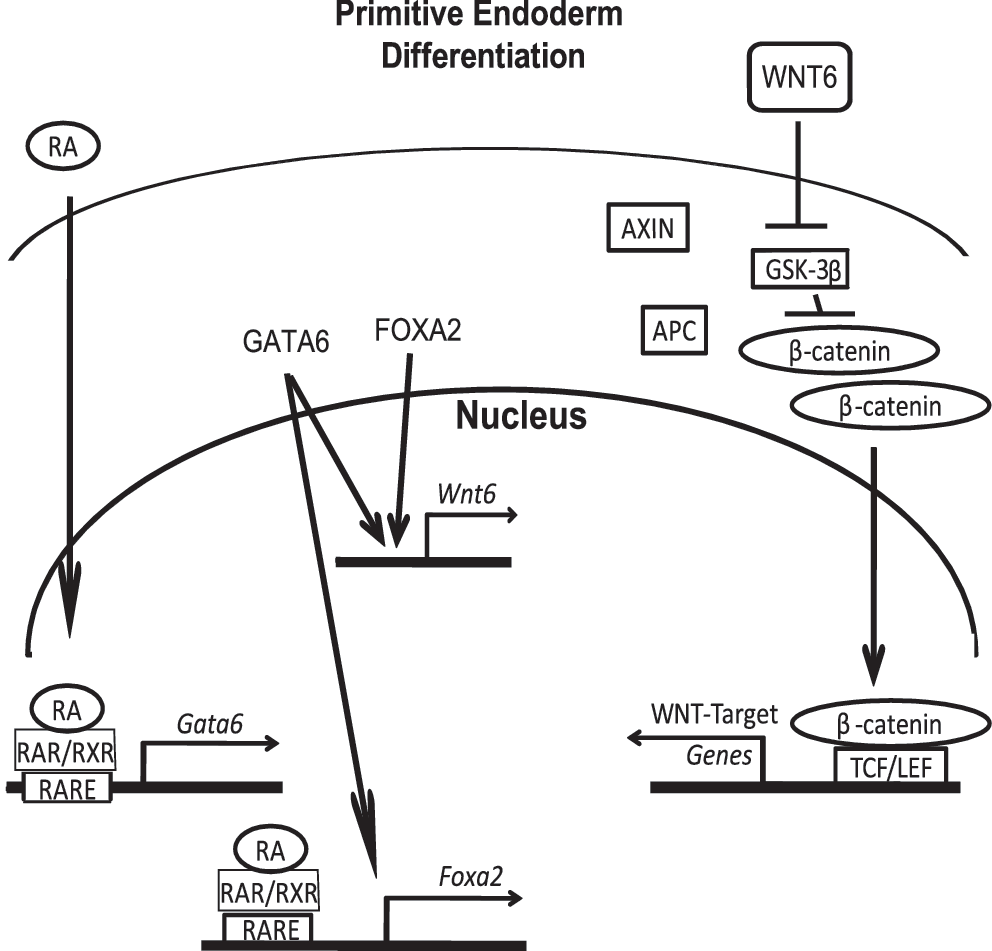
Model of the signaling hierarchy during primitive endoderm specification in F9 cells. RA induces the expression of Gata6, and GATA6, in turn, induces the expression of Foxa2, 2 transcription factors that directly upregulate the Wnt6 gene. WNT6 signals to neighboring cells by destabilizing the GSK-3β degradation complex, which allows cytoplasmic β-catenin levels to increase and eventually translocate to the nucleus, where with TCF/LEF transcription factors, activates/represses the genes required for primitive endoderm differentiation. TCF, T-cell factor; LEF, lymphoid enhances factor.
The question pertaining to the WNT family is whether or not WNT6 is the key member involved in establishing the ExE lineage in vivo. Wnt6 mRNA is expressed at the correct time [19,69], but studies have also revealed that Wnt1, 2b, 3, 3a, 4, 5a, 7a, 7b, 10b, and 11 are also expressed at early stages of mouse development [70,71], and many as protein ligands activate the canonical WNT-β-catenin pathway [72 –76]. Based on the readout of their overexpression in F9 cells, care must be exercised when extrapolating how these WNTs function in vivo. For instance, WNT3a will induce F9 cells ectopically expressing rat FZD1 to form ExE [33]; however, in RA-induced wild-type F9 cells, WNT3a treatment blocks differentiation [77]. It is also interesting to note that Wnt6 is dispensable for embryonic development [78], which means that another WNT is likely to act in a functionally redundant manner to ensure embryo viability. Identifying the WNTs compensating for the loss of WNT6 during ExE formation will require a systematic in vitro approach using si/shRNAs, but to begin, it may be more advantageous to continue focusing on how these genes are regulated at the transcriptional level. For instance, GATA6 and FOXA2 are also known to bind to and activate the Wnt7b promoter [79], a WNT that also signals through the canonical pathway [80]. While we are currently using the F9 model to test the likely possibility that more than 1 WNT is involved in ExE formation, a concerted effort to create mice with double gene knockouts will be necessary to provide evidence for this in vivo.
Footnotes
Acknowledgments
G.M.K. would like to thank the Natural Sciences and Engineering Research Council of Canada for funding. The authors also acknowledge the Ontario Graduate Scholarship Program and the Faculty of Graduate and Postgraduate Studies, Western University for scholarship support to J.T.K.H. Finally, we would like to thank Jason Wen for helpful comments during the course of the study, and we appreciate the critical comments from 2 anonymous reviewers.
Author Disclosure Statement
J.T.K.H. and G.M.K. have no commercial association or competing financial interests.