
Abstract
Endothelial cells line blood vessels and coordinate many aspects of vascular biology. More recent work has shown that endothelial cells provide a key niche in vivo for neural stem cells. In vitro, endothelial cells secrete a factor that expands neural stem cells while inhibiting their differentiation. Here, we show that a transformed mouse endothelial cell line (bEnd.3) maintains human pluripotent stem cells in an undifferentiated state. bEnd.3 cells have a practical advantage over mouse embryonic fibroblasts for pluripotent stem cell maintenance since they can be expanded in vitro and engineered to express genes of interest. We demonstrate this capability by producing fluorescent and drug-resistant feeder cells. Further, we show that bEnd.3 secretes an activity that maintains human embryonic stem cells without direct contact.
Introduction
E
Previous work on neural progenitors and endothelial cells was performed using mouse cells. We wondered whether endothelial factors also expanded human embryonic stem cell (hESC)–derived neural progenitors. However, ESCs must first be directed to neural cells before we could test their responsiveness to the endothelial factor. Previous work showed that many cells contained a so-called stromal derived induction activity (SDIA) and therefore could cause ESCs to adopt a neural cell fate [7,8]. While bone marrow stromal cells cause neural induction, neural progenitors rapidly differentiate into neurons; stromal cells do not prevent their further differentiation to neurons and glia.
Here, we tested the hypothesis that endothelial cells have an stromal derived induction activity (SDIA) activity. If true, then coculture of hESCs with endothelial cells might provide an ideal system to convert ESCs to neural progenitors without continued neuronal differentiation since endothelial cells secrete factors to inhibit neuronal differentiation and enhance neural progenitor self-renewal. This could increase the synchrony and purity of ESC-derived neural cultures, at least in principle.
In contrast to our original hypothesis, we found that the endothelial cell line bEnd.3 robustly maintained human pluripotent cells in the undifferentiated state for at least 20 passages, the longest time tested. Coculture with bEnd.3 maintained the hESC lines H1 and H9, and human induced pluripotent stem cells (hiPSCs). Direct coculture was not necessary to maintain PSCs, suggesting that bEnd.3 secretes an activity maintaining pluripotency. One important practical application of using these cells is that bEnd.3 could be passaged at least 10 times without losing the ability to maintain pluripotent cells; in contrast, primary mouse embryonic fibroblasts (PMEFs) are typically used at passage 3 since they lose the ability to maintain pluripotent cells over passage. This allows the genetic engineering of feeder lines in vitro instead of the production of a transgenic mouse. Such versatility makes these cells of practical interest for those engineering hPSCs and provides some biological insight into endothelial cells.
Materials and Methods
PSC culture
H9 (WA09) and H1 (WA01) hESCs and hiPSCs from MRC-5 fibroblasts (i202) were routinely maintained on PMEFs plated at a density of 11,500–13,500 cells/cm2, depending on the lot used (GlobalStem, Inc.). PSCs were fed daily with hPSC media [composed of Dulbecco's modified Eagle medium (DMEM)/F12 (11330-032; Gibco), 20% knockout serum replacement (10828-028; Gibco), 3.5 mM glutamine (25030-081; Life Technologies), 0.1 mM MEM NEAA (11140-050; Gibco), 55 μM 2-mercaptoethanol (21985-023; Life Technologies), and 6 ng/mL fibroblast growth factor 2 (FGF2) (R&D Systems)]. A working protocol for hPSC culture is available at
bEnd.3 culture
bEnd.3 cells (ATCC No. CRL-2299) were maintained in DMEM+10% fetal bovine serum (FBS). Cells were split with trypsin before 80% confluence at a ratio between 1:6 and 1:12, usually every 3–4 days. To prepare cells for use as feeders, cells were exposed to 103 Gy of radiation before freezing as aliquots. bEnd.3 cells were plated at a density of 13,000 cells/cm2 before coculture with PSCs.
Engineering bEnd.3 and H9 hESCs
To genetically engineer bEnd.3 and H9 hESCs, 4–5 million cells were nucleofected with 10 μg of the appropriate DNA construct using program B-16 and solution V (Amaxa Nucleofector; Lonza). Cells were allowed to recover for 2 days before starting selection. Selection was continued until the control nucleofection without DNA lacked viable cells. For most experiments, the drug-resistant population was then passaged together as an engineered line. The mCherry-expressing bEnd.3 cells were sorted 3 times to enrich for mCherry-positive cells.
Conditioned media production
MEFs or bEnd.3 cells were plated at 50,000 cells/cm2 in DMEM with 10% FBS overnight. The next day, these media were removed and the cells were washed once with phosphate-buffered saline (PBS). Then hPSC media were placed on the cells overnight for conditioning. The media-only control was added to a tissue culture dish for overnight incubation. The next day, the media were removed from each condition and FGF2 was added to 10 ng/mL before use. Confluent monolayers were used for up to 3 weeks.
Neural differentiation
H9 colonies were dissociated into smaller clusters with dispase (5 mg/mL; Stem Cell Technologies) and washed once in hESC media before a 1:30 split and cocultured with MS5 [9], MEFs, and bEnd.3 mouse brain endothelial cells (ATCC No. 2299) in serum-replacement medium [8] supplemented with recombinant Noggin (250 ng/mL); 1 μM SB431542 was changed completely on days 3, 6, and 9 to suppress SMAD activity and increase neural induction. Cultures were fixed for immunofluorescence or harvested for gene expression analysis on day 10. Quantitative polymerase chain reaction (qPCR) results are from 6 to 9 technical replicates of 3 independent biological experiments for each sample.
Directed differentiation
H9 hESCs were fed on days 1 and 2 after passage with RPMI with 10 ng/mL bone morphogenetic protien 4 (BMP4) (R&D 314-BP), 1 μM SB431542, glutamine, and 0.5% HyClone FBS. Cells were assayed on day 3 for mesoderm and trophectoderm differentiation. For endoderm, H9 hESCs were fed on days 1 and 2 with Roswell Park Memorial Institute Medium (RPMI) with 100 ng/mL Activin (R&D 338-AC), glutamine, and 0.5% HyClone FBS. Cells were assayed on day 3 for endoderm differentiation. Neural induction was performed using the dual SMAD inhibition protocol [10]. Briefly, cells were dissociated into a single-celled suspension with Accutase before plating on Matrigel in the presence of conditioned media (either MEF or bEnd.3) supplemented with 10 ng/mL FGF2 and 10 μM Y-27632. For qPCR analysis, the Chambers et al. protocol was followed verbatim until harvesting on day 15. qPCR was performed as described previously. For immunofluorescence, confluent monolayers on neural precursors were passaged using mechanical dissociation (STEMPRO EZ Passage; Invitrogen) on day 10 onto Matrigel-coated dishes in N2 media supplemented with 20 ng/mL brain derived neurotropic factor (BDNF), 200 μM ascorbic acid, 50 ng/mL Shh (C25II), and 100 ng/mL FGF8. qPCR results are from 6 to 9 technical replicates of 3 independent biological experiments for each sample.
Embryoid bodies and teratomas
Embryoid bodies were made by dispase treating hESCs and replating large fragments in hPSC media without FGF2 in a low adherence plate. Clusters were fed every 3 days. Teratomas were made by dispase treating cells before creating a near single-cell suspension through pipetting. Cells were washed and resuspended in DMEM+10% FBS. For each implant, 3 million cells were resuspended in a volume of 200 μL DMEM+10% FBS with 20 μM Y-27632 and 30% Matrigel.
Quantitative PCR
Messenger RNA levels were determined by isolating mRNA with the Qiagen RNeasy kit before cDNA production with the Qiagen Quantitect RT kit. Taqman probes were obtained from Applied Biosystems: pax6 (Hs01088108_m1), cdx2 (Hs00230919_m1), brachyury (Hs00610080_m1), sox17 (Hs00751752_s1), and the internal standard hprt (4326321E). qPCR was performed on an Eppendorf Realplex2 with the following cycling conditions: 50°C for 2 min, 95°C for 10 min, and 40 cycles of 95°C for 15 s and 60°C for 1 min. All qPCR results are from 6 to 9 technical replicates of 3 independent biological experiments for each sample.
Immunostaining
Immunostaining was performed by fixing cells in 4% paraformaldehyde for 15 min. Primary antibodies used in this study are pax6 (Covance PRB-278P; 1:500), brachyury (R&D AF2085 at 1:40), cdx2 (BioGenex CDX2-88 ready to use), sox17 (R&D AF1924 at 1:200), Oct3/4 (Santa Cruz Biotechnologies, sc-5279; 1:100), tra1-60 (Chemicon MAB4360; 1:100), and Nanog (BD Pharmingen 560482; 1:200); secondary antibodies conjugated to Alexa488, 568, or 647 (1:1,000) were used to visualize primary antibody binding, and Hoechst 33342 (Molecular Probes H-3570; 1:1,000) was added to stain nuclei. For surface markers, the staining was performed in PBS containing 1% PBS. Triton X-100 (0.1%) was added to the staining buffer for intracellular antigens.
FACS analysis
Antibodies against SSEA-3 (conjugated to Alexa 647; BD Biosciences, No. 561145) and SSEA-4 (conjugated to Alexa 647; BD Biosciences, No. 560796) were used to quantitate pluripotency markers. In brief, cells were dissociated with Accutase for 40 min, washed with PBS, and then counted before a second centrifugation. One million cells were resuspended in PBS with 0.1% bovine serum albumin (BSA) in a volume of 100 μL containing the recommended amount of antibody (5 μL for SSEA-3, 20 μL for SSEA-4 per test). Cells were incubated in the dark on ice for 1 h before 3 PBS washes. Washed cells were resuspended a final time in PBS with 0.1% BSA before analysis on a FACSAria (BD Biosciences). All FACS analysis data were derived from 3 independent experiments.
hESC marker expression on bEnd.3 feeder layers
H9 GFP.2 hESCs were passaged onto MEFs and bEnd.3 cells that had been plated at equal densities (∼12,500 cells/cm2) and FACS analysis was used to quantify stem cell marker expression on the GFP+ population. As above, FACS analysis data were derived from 3 independent experiments. Primers used for endpoint PCR are listed in Supplementary Table 1 (Supplementary Data are available online at
Results
Neural induction
We initially tested the hypothesis that coculture of hESCs with endothelial cells would convert hESCs to neural cells; that is, endothelial cells have a “stromal-cell derived inducing activity” [7]. To test this hypothesis, we cocultured H9 hESCs with PMEFs as a negative control, MS-5 bone marrow stromal cells as a positive control, and bEnd.3 endothelial cells as the experimental condition (Fig. 1). Each feeder line was irradiated to prevent overgrowth during coculture. hESC cultures were split with dispase before seeding on feeder monolayers at a relatively low density as previously described ([9]; see Materials and Methods section). Twelve days after differentiation, hESCs in the MS-5 condition had turned predominantly into circular arrangements of neural progenitors called neural rosettes (Fig. 1A) [9,11,12]. Colony morphology looked relatively flat and epithelial in the other 2 conditions. Pax6 immunostaining revealed robust neural induction in the MS-5–positive control plate (Fig. 1A). In contrast, both the endothelial cells and the MEFs had only small patches of Pax6 (Fig. 1A) and few neural rosettes. qPCR (Fig. 1B) provided further evidence for a lack of neural induction in endothelial conditions. Pax6 expression was low in the bEnd.3 and MEF coculture relative to the MS-5 coculture (n=3 independent experiments). Taken together, these data suggested that endothelial cells are inefficient in neuralizing hESCs.

Human embryonic stem cells (hESCs) are not efficiently converted to neural cells when cocultured with bEnd.3 feeder cells. hESCs were cocultured with MS-5 (as a positive control), mouse embryonic fibroblasts (MEFs) (as a negative control), or bEnd.3 cells to determine the ability of each feeder layer to cause neural induction.
hESC maintenance
We explored the use of endothelial cell coculture for hESC maintenance because the hESCs continued to appear morphologically undifferentiated after coculture with bEnd.3 cells during the neural induction experiment described previously. In preliminary experiments, hESCs (H1- and GFP-expressing H9) and an iPSC line (i202) were maintained on MEFs as a control (Fig. 2A–C) and in parallel on bEnd.3 cells (Fig. 2A′–C′). The colony morphology for all 3 lines was stable over passage and retained sharp borders as would be expected if bEnd.3 cells maintained PSCs in the undifferentiated state. To examine this more closely, we maintained H9 hESCs for over 20 passages on bEnd.3 feeder cells. Indirect immunofluorescence analysis for Oct4, Nanog, and Tra1-60 showed similar staining between the 2 different conditions (Fig. 2D, E and D′, E′). Karyotype analysis was normal for bEnd.3- and MEF-maintained H9 hESCs (Supplementary Fig. S1).
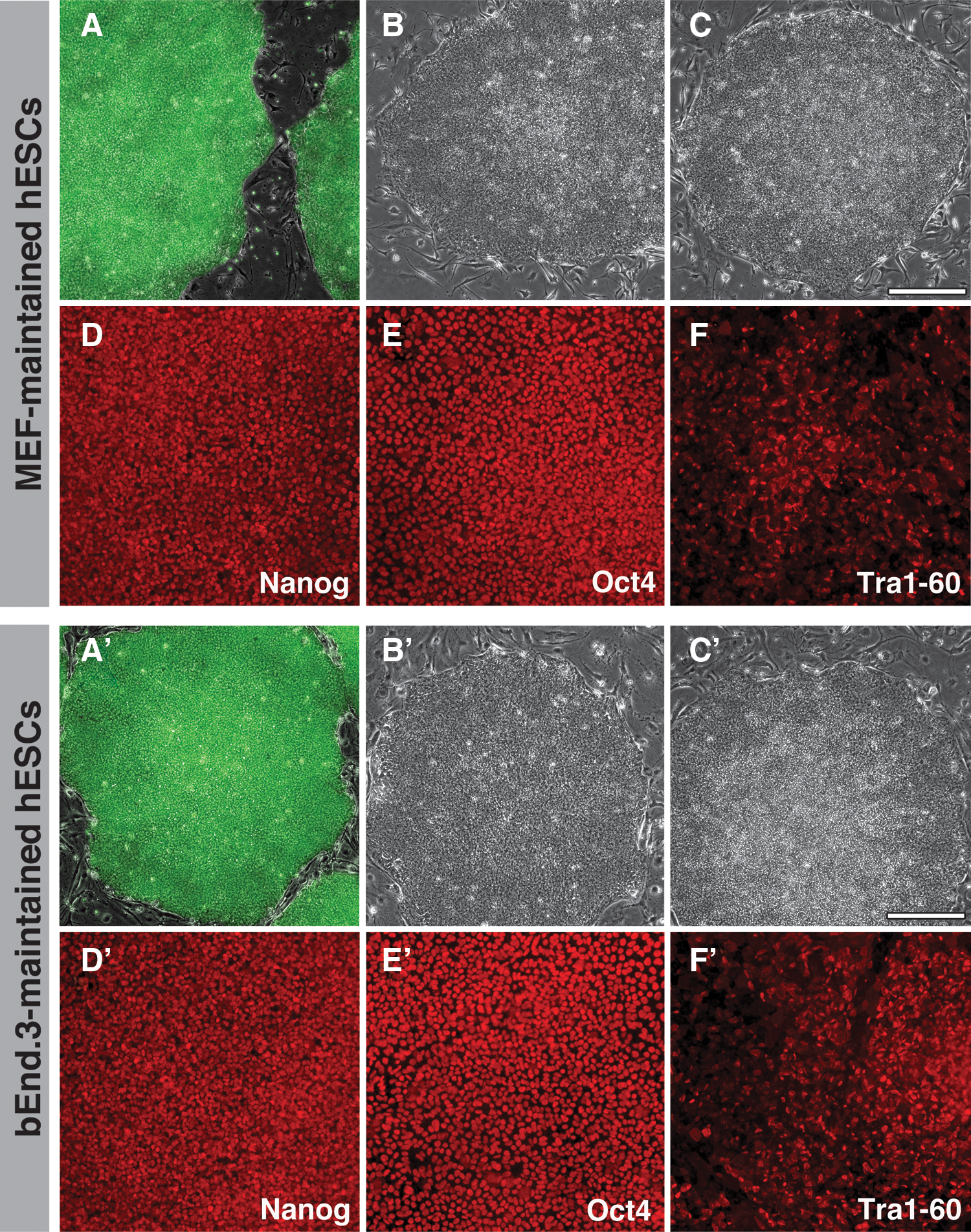
Human pluripotent stem cells (hPSCs) maintained on MEFs
H9 hESCs maintained on endothelial cells retained pluripotency since they could be directed to derivatives of all 3 germ layers and extraembryonic cell types (Fig. 3A). hESCs were cultured in RPMI with BMP4, low serum, and the TGF beta/Activin inhibitor SB431542 to create mesoderm (Brachyury+) and trophectoderm (Cdx2+). H9 hESCs that are cultured in Activin and low serum created endoderm (Sox17+), while the neural induction used dual SMAD inhibition (Pax6+) ([10]; see Materials and Methods section for details). qPCR showed similar levels of each germ layer marker after directed differentiation from both MEF- and bEnd.3-maintained hESCs (top of Fig. 3; n=3 independent experiments). Similarly, indirect immunofluorescence with antibodies against Pax6, Brachyury, Sox17, and Cdx2 showed comparable levels of staining with each marker after differentiation (Fig. 3B).
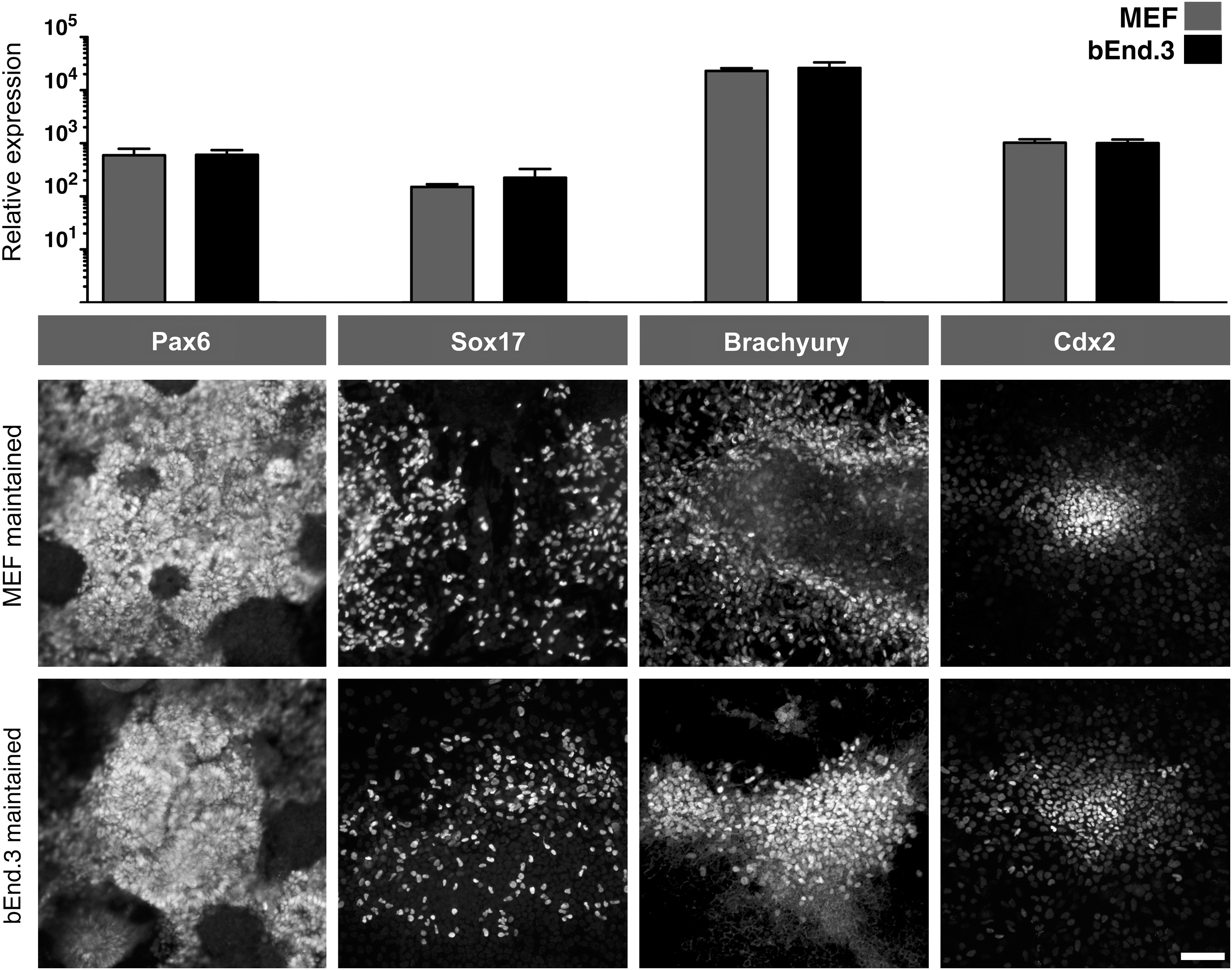
H9 hESCs maintained on bEnd.3 cells can be directed to all 3 different germ layers and trophoblasts. For differentiation details, see Materials and Methods section. (Top) After differentiation, the gene expression levels of markers characteristic of different germ layers were compared by qPCR: Pax6 for neural cells, Sox17 for endoderm, Brachyury for mesoderm, and Cdx2 for trophoblasts. (Bottom) Indirect immunofluorescence with the same markers at the protein level to verify the qPCR results. Scale bar=100 μM.
To confirm these results, we also made embryoid bodies and teratomas with H9 hESCs that had been maintained on bEnd.3 for 20 passages. Embryoid bodies derived from MEF- or bEnd.3-maintained cells had similar growth patterns and morphologies over the 21-day differentiation (Fig. 4A). Genes characteristic of the 3 germ layers were present in EBs cultured on both feeders (Fig. 4B), and bEnd.3-maintained H9 hESCs also formed teratomas in vivo (Fig. 4C). Taken together with the in vitro data described previously, these results confirmed the pluripotent nature of the bEnd.3-maintained hESCs.

H9 hESCs maintained on bEnd.3 cells can make embryoid bodies and teratomas.
bEnd.3 cell expansion
One factor that dramatically increases the cost and complexity associated with using PMEFs is that they cannot be passaged without losing the ability to maintain ESCs: most labs use MEFs at passage 3. The bEnd.3 mouse brain endothelial cell line was transformed with the polyoma middle-sized T antigen [13]. Therefore, to see whether bEnd.3 retained the ability to maintain ESCs after expansion, we performed experiments with passage 0, 5, and 10 bEnd.3 cells. A portion of each passage was irradiated before freezing down while the rest was used for further expansion. We found that passage-10 bEnd.3 cells were as effective as earlier passage cells (Fig. 5). The morphology of hESC colonies maintained the same tightly packed phenotype in each condition (Fig. 4A). Immunofluorescence analysis showed no obvious differences in the expression levels of Oct4 and Tra1-60 in each condition (Fig. 5A). To check our subjective observations, we cocultured GFP-expressing H9 cells on passage 0, 5, and 10 cells before performing FACS analysis to quantitate the surface staining of SSEA-3 and −4. Figure 5B shows the percentage of SSEA-3 and −4 on the GFP+ fraction of cells to eliminate feeders from the analysis. No differences were observed in the levels of stem cell surface markers between early and late-passage bEnd.3-maintained hESCs (n=3 independent experiments). This data suggested that bEnd.3 cells could be expanded extensively in vitro before using them to maintain hPSCs.

Passage of bEnd.3 cells does not hinder their ability to maintain hESCs. bEnd.3 cells were passaged 5 or 10 times before irradiation and used as feeders in hESC culture.
Engineered feeders
As more complicated genetic schemes are employed in hPSC research, feeders containing fluorescent markers or multiple drug resistance types are desired. The production of drug-resistant MEFs is expensive and time consuming since new transgenic mice must first be made. Since bEnd.3 is a transformed cell line, we attempted to create transgenic feeder cells in vitro. We initially engineered bEnd.3 cells to express mCherry to visualize and physically separate feeders from hPSCs using flow cytometry. In a proof-of-concept experiment, we cocultured GFP-expressing H9 hESCs on mCherry-expressing bEnd.3 cells (Fig. 6A). Flow cytometry could separate the feeders from the hESCs (Fig. 6B, 6C, 6A′–C′).
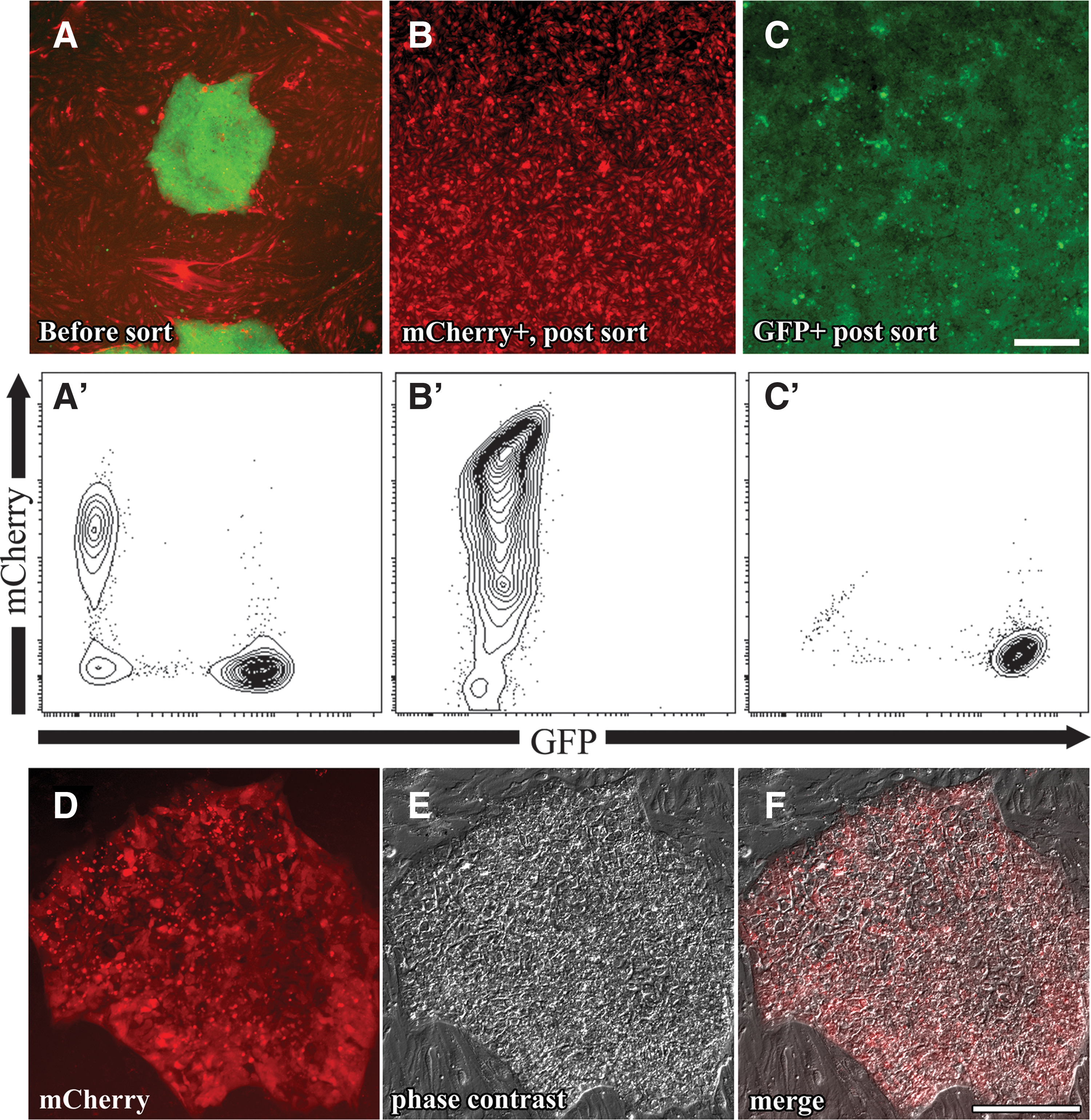
Genetically engineering bEnd.3 feeders. Fluorescent image
Another important application is the creation of drug-resistant feeders to engineer hPSCs. To this end, we nucleofected a plasmid containing a Puromycin resistance cassette into bEnd.3 cells and selected Puro-resistant lines (bEnd.3-Puro). Such Puromycin-resistant feeders were capable of maintaining hESCs. As a proof of concept, we co-nucleofected an mCherry-expressing plasmid and a Puromycin-resistance plasmid into H9 hESCs before selection on bEnd.3-Puro cells. H9 hESCs expressing mCherry were readily obtained and expanded on bEnd.3-Puro cells (Fig. 6D–F). bEnd.3 cells are also Neomycin resistant, a remnant from the original transformation [13]. Such feeders will be useful for genetic targeting or transgenesis in hPSCs and can be engineered to express any drug-resistant cassette desired.
bEnd.3 secretes an activity that maintains hESCs
Endothelial cells, including bEnd.3, secrete an activity that expands mouse neural progenitors while inhibiting their differentiation in vitro [5]. We therefore tested whether the bEnd.3 activity that maintains undifferentiated hESCs is also secreted. H9 hESCs were passaged onto Matrigel with unconditioned media or media conditioned by MEFs or bEnd.3 cells. After expansion in conditioned media for 7 days, the morphology of hESCs was noted before FACS analysis. A fraction of the cells were further expanded in the same conditions to assess further feeder-free expansion in the presence or absence of conditioned media. Three sequential rounds of feeder-free passage of H1 and i202 were examined for morphology and SSEA-4 expression through FACS analysis (Fig. 7). Both pluripotent lines expanded in hPSC media without conditioning drastically changed morphology (Fig. 7, top), lost SSEA-4 expression (Fig. 7, bottom), and nearly stopped dividing. MEF- and bEnd.3-conditioned media expanded cells with the correct morphology (Fig. 7, top) and maintained rapid cell division and SSEA-4 expression (Fig. 7, bottom). Taken together, our data show that bEnd.3-conditioned media are competent to expand hPSCs, suggesting that bEnd.3 also secretes a factor(s) that maintains pluripotency.
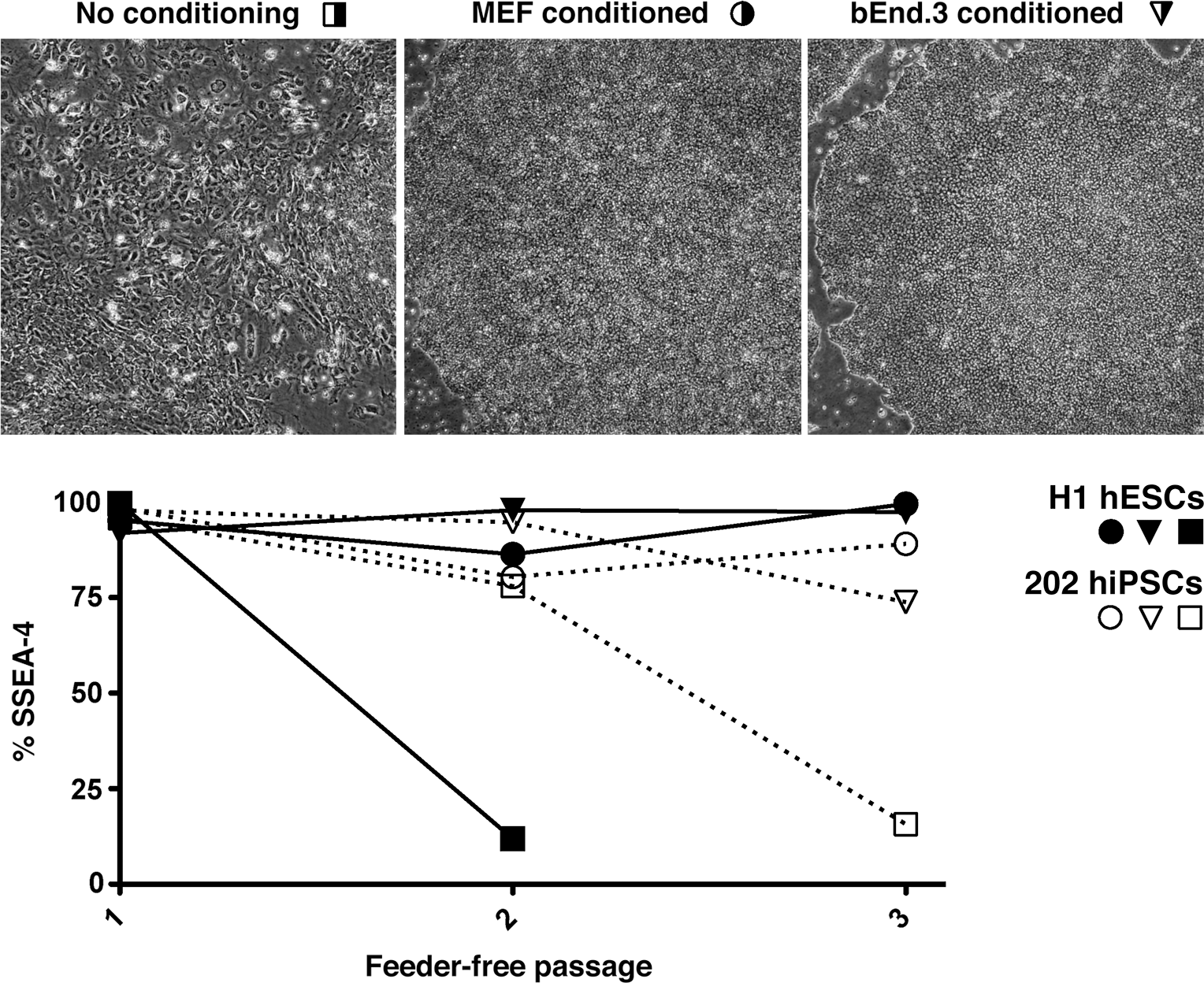
bEnd.3-conditioned media maintain PSCs without feeders. (Top) Morphology of H9 hESCs expanded on Matrigel-coated dishes with no conditioning, MEF- or bEnd.3-conditioned hESC media. (Bottom) SSEA-4 expression with H1 hESCs (black) and i202 hiPSCs (white) over 3 passages without feeders in media without conditioning (square) or conditioned by MEF (circle) or bEnd.3 conditioning (inverted triangle).
Discussion
We serendipitously discovered that hPSCs cocultured with the transformed brain endothelial cell line bEnd.3 are maintained in their undifferentiated state as well as PMEFs. This discovery is important for a number of reasons: (1) variability—each batch of MEFs can be different and therefore must be quality controlled. The use of one suboptimal preparation can differentiate all hPSCs in a lab, costing time and money. (2) Cost—it is expensive to purchase or make PMEFs. (3) Reduction of animal use—it is preferable to reduce the number of animals used in research. (4) Transgenic feeders—it is easier to engineer bEnd.3 feeder cells since they can be expanded without losing their ability to support PSCs.
While some labs have moved to feeder-free culture, many labs (including ours) feel that feeder-based hPSC culture is more robust than feeder-free culture (eg, see Ref. [14]). This is likely the reason that many influential stem cell labs continue to use mouse feeder layers (for some examples, see Refs. [15
–19]). Current feeder-free systems are also expensive for labs performing extensive cell culture. One reason that labs switch to feeder-free systems is convenience. To make feeders, animals must be bred or ordered and sacrificed on a particular day. Then, after a dissection, the primary culture must be briefly expanded since it is thought that MEFs lose the ability to maintain stem cells as they are passaged. Expanded MEFs undergo mitotic inactivation and aliquotting before quality control to verify that each lot of feeders can adequately maintain hESCs. Because of the effort required to make and quality control MEFs, many laboratories choose to purchase MEFs. Commercially purchased MEFs can still lead to variability and is a major cost associated with hPSC culture. The use of bEnd.3 cells reduces the inconvenience associated with feeder production; further, any interested laboratory can order them from the nonprofit repository ATCC (
The use of primary, early passage MEFs also restricts the genetic engineering of feeders since selection in vitro requires cell division beyond what is considered acceptable for hPSC maintenance. As such, engineered feeders are usually accomplished by the production of a transgenic mouse colony, but the time and cost associated with this can be prohibitive. We demonstrate here that bEnd.3 can be engineered to be fluorescent so that feeders can be visualized, quantitated, or physically separated by flow cytometry. Alternatively, drug-resistant feeders were made and used here to select transgenic hPSCs. Future work could be aimed at engineering bEnd.3 feeders to express FGF2 or other proteins that help support or expand hPSCs. Such a system has already been described for transformed human fibroblasts [20]. Engineered feeders secreting a constant source of growth factors could be more affordable and effective than is possible with feeder-free culture systems.
Another reason cited for switching to human- or feeder-free systems is that hPSCs will be used for clinical applications. While clinical applications will likely become increasingly important for PSCs, most studies focus on human development or disease modeling where mouse cell coculture is less of a concern. This is likely the reason that many hPSC labs continue to use mouse feeders as noted previously [13 –18]. We suggest that bEnd.3 cells are a suitable replacement for MEFs in basic science applications of hPSCs: clinical applications will require human- or feeder-free systems. Ideally, a robust, simple, defined, animal- and feeder-free culture paradigm that is cost effective would be best for all applications; in our opinion, such a system has yet to emerge.
For clinical applications, others groups have pursued many different types of human feeders including transformed lines [20], hESC-derived fibroblast-like cells [21 –24], and some from human fetuses, foreskin, or adult cells [25 –31]. We have not tried these other feeder-based systems so we cannot comment on their efficacy. Most of these studies are qualitative and do not include MEF-maintained cultures as a reference for comparison. In our opinion, this complicates their evaluation since many groups use different qualitative criteria as being optimal. Furthermore, many of the primary cells cannot be passaged extensively limiting their utility. One of the few quantitative comparisons found that MEF-maintained hESCs were more undifferentiated than human foreskin–maintained hESCs, as judged by SSEA-3 expression [32]. Further work is needed to rigorously compare different feeder systems.
Practicalities aside, the observation that bEnd.3 endothelial cells maintain hPSCs raises interesting biological questions. One possibility is that the same factor(s) secreted from bEnd.3 that maintains fetal neural progenitors is also responsible for maintaining PSCs. Alternatively, endothelial cells might secrete different activities that act on different cell populations. Future work aimed at identifying these secreted factor(s) will be interesting from a biological perspective and could have a major impact on in vitro stem cell culture.
Footnotes
Acknowledgments
This work was funded by a generous grant from the Starr Foundation and from NYSTEM (C024175-01). The authors would like to thank Margaret Leversha and Kalyani Chadalavada for karyotyping (Molecular Cytogenetics Core Lab at MSKCC), and Elisa De Stanchina and Xiaodong Huang (Antitumor Assessment Core Facility at MSKCC) and Jerrold Ward (Global VetPathology, Montgomery Village, MD/HistoServ, Inc.) for help with teratoma formation, cutting, staining, and pathology.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.