
Abstract
Although mesenchymal stem cells (MSCs) of distinct tissue origin have a large number of similarities and differences, it has not been determined so far whether tissue-resident MSCs are the progenies of one ancestor cell lineage or the results of parallel cell developmental events. Here we compared the expression levels of 177 genes in murine MSCs derived from adult and juvenile bone marrow and adult adipose tissue, as well as juvenile spleen, thymus, and aorta wall by quantitative real-time polymerase chain reaction and the results were partially validated at protein level. All MSC lines uniformly expressed a large set of genes including well-known mesenchymal markers, such as α-smooth muscle actin, collagen type I α-chain, GATA6, Mohawk, and vimentin. In contrast, pluripotency genes and the early mesodermal marker T-gene were not expressed. On the other hand, different MSC lines consistently expressed distinct patterns of Hox genes determining the positional identity of a given cell population. Moreover, MSCs of different origin expressed a few other transcription factors also reflecting their topological identity and so the body segment or organ to which they normally contributed in vivo: (1) thymus-derived cells specifically expressed Tbx5 and Pitx2; (2) spleen-derived MSCs were characterized with Tlx1 and Nkx2.5; (3) Pitx1 designated femoral bone marrow cells and (4) En2 appeared in aorta wall-derived MSCs. Thus, MSCs exhibited topographic identity and memory even after long-term cultivation in vitro. On the basis of these results, we suggest that postnatal MSCs isolated from different anatomical sites descend from precursor cells developing in the postsegmentation mesoderm.
Introduction
M
In this study the expression levels of 177 genes were examined, and compared in MSC lines established from adult adipose tissue (AAD) and adult bone marrow (ABM), as well as from juvenile bone marrow (JBM), juvenile spleen (JSpl), juvenile aorta wall (JAo), and juvenile thymus (JThy). We found that a set of genes characterizing all MSC lines did not include pluripotency genes such as Pou5f1/Oct-4, Rex-1/Zfp-42, or Nanog and the early mesodermal marker Brachyury. However, MSCs from any source uniformly expressed genes of mesenchymal cells, such as α-smooth muscle actin (α-SMA), collagen type I α-chain, GATA6, Mohawk (Mkx), and vimentin (Vim). More importantly, differential gene expression was detected regarding the patterns of Hox genes that were sufficient to identify the anatomical origin of the MSCs. Furthermore, the expression of certain transcription factors was also applicable to determine the tissue origin and hence the body segment or organ to which a particular MSC population contributed in vivo. Several examples are listed here: (1) thymus-derived cells specifically expressed Tbx5 and Pitx2; (2) spleen-derived MSCs were characterized with Tlx1 and Nkx2.5; (3) Pitx1 designated femoral BM stromal cells; and (4) En2 appeared in aorta wall-derived MSCs.
Materials and Methods
Animals
Juvenile (14 days of age) and adult (10–12 weeks of age) C57Bl/6 (National Institute of Oncology, Budapest, Hungary) mice were used for all experiments under the guidelines of the Animal Care and Use Committee of the National Blood Service (Budapest, Hungary).
Isolation and culture of MSCs
Isolation of MSCs was carried out according to Peister et al. [17] with some modifications as previously described [18]. Briefly animals were sacrificed by cervical dislocation. Femurs and adipose tissue were removed from adult C57Bl/6 mice and femurs, spleen, thymus, and aorta from juvenile C57Bl/6 mice. The femurs were flushed with complete medium (CM) containing DMEM/Ham's F-12 medium (Invitrogen), 10% fetal bovine serum, 5% horse serum (Invitrogen), 50 U mL−1 penicillin, 50 μg mL−1 streptomycin (Sigma-Aldrich), and 2 mM L-glutamine (Invitrogen) supplemented with heparin at a final concentration of 5 U mL−1. Cells were obtained from spleen, thymus, and aorta wall by mechanical disruption. They were then filtered through a 60-μm nylon mesh to remove debris and washed twice in Hanks' balanced salt solution, plated in a 25-cm2 flask (BD Falcon) at a density of 2–4×106 cells cm−2 in CM, and incubated in a humidified 5% CO2 incubator at 37°C for 72 h. Nonadherent cells were removed by rinsing and replacing the media after 72 h and the medium was replaced every 3 days. Confluent primary cultures were washed with PBS and the cells were dissociated in 0.25% Trypsin-EDTA solution (Gibco). After a subsequent wash, cells were replated in a 75-cm2 culture flasks at a dilution of 1:5.
Abdominal and inguinal fat were removed; after washing with PBS, fat was digested with 0.1% collagenase (Sigma-Aldrich) in PBS for 30 min on a 37°C shaker at 250 rpm. Mature adipocytes and connective tissues were removed by centrifugation at 600 rpm for 8 min. The pellet (SVF-stromal vascular fraction) was resuspended and washed again with PBS at 1,200 rpm for 10 min. Resuspended cells were seeded and cultured under the same conditions as the other stromal cells. All MSC cultures were used after 10–15 passages when they were free from hematopoietic contamination.
Polymerase chain reaction array analysis
Each MSC samples were established independently from 3 groups of animals to ensure true biological parallels of the polymerase chain reaction (PCR) array analysis. The PCR arrays were separately carried out 3 times involving 1 of the 3 independently established cell cultures of each cell types. Lysis of the cells was performed using TRI REAGENT™ (Sigma-Aldrich) when the 10th to 15th passage cultures became confluent. Total RNA was extracted using RT2 qPCR-Grade RNA Isolation Kit (SA Biosciences) following the manufacturer's instructions. RNA quality was assessed on Nano Drop 2000 (Thermo Scientific). About 1.5 μg of total RNA from each MSC samples was reverse transcribed to cDNA using the RT2 First Strand Kit (SA Biosciences) and then subjected to SYBR® Green–based real-time PCR using RT2 SYBR Green qPCR Master Mix (SA Biosciences) on Roche Light Cycler 480 instrument (95°C for 10 min followed by 40 cycles of 95°C for 15 s and 60°C for 1 min). 168 genes were analyzed using RT2 Profiler PCR Array System, Mouse MSC PCR Array and Mouse Homeobox Genes PCR Array (SA Biosciences) (Supplementary Tables S1 and S2; Supplementary Data are available online at
Data analysis
The fold change of mRNA in experimental and control cells was determined using the ΔΔCt method normalized to the Hprt1 housekeeping gene. It was performed with the aid of an analysis template provided by the SA Biosciences website (
Quantitative real-time PCR
To isolate the total RNA from MSCs, cells washed with 1×PBS were lysed directly in the culture dish by adding TRI REAGENT™ (Sigma-Aldrich). RNA was extracted following the manufacturer's instructions. cDNA was reverse transcribed from 0.5 μg of total RNA using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) according to the manufacturer's recommendations. Real-time PCR was performed on Eppendorf Mastercycler ep realplex4. Cycling conditions were as follows: enzyme activation at 95°C for 10 min, followed by 40 cycles of 95°C for 20 s, and 60°C for 65 s. Expression of Nanog, Pou5f1, Zfp42, Brachyury, Krüppel-like factor 4 (Klf4), Acta2, Gata4, Gata6, and Nkx2.5 of MSCs was normalized to GAPDH (TaqMan Gene Expression Assays, Applied Biosystems). The catalog numbers of gene-specific primers are listed in Supplementary Table S3.
Flow cytometry analysis
To confirm the gene expressions at protein level, 5×105 cells were labeled with PE-conjugated monoclonal antibodies against mouse CD31, CD34, CD44, CD73, and CD90.2 or with biotin-conjugated anti-CD45R/B220 (BD Pharmingen) for 30 min at 4°C. Biotinylated antibodies were incubated with PE-conjugated streptavidin (Sigma-Aldrich). Stained cells were washed with cold PBS and analyzed on a FACScan flow cytometer using CellQuest software (Becton Dickinson).
Immunfluorescent staining
Cells were fixed with 4% paraformaldehide in PBS and permeabilized with 0.5% Triton X-100 in PBS before blocking with 1% BSA in PBS. The goat polyclonal anti-Mkx, anti-Pitx1, anti-Tbx5, anti-En2, and rabbit anti-Hox11/Tlx1 (Santa Cruz Biotechnology) were applied at 1/50 at 4°C and detected by NL557 fluorochrome-labeled donkey anti-goat IgG (R&D Systems) or Alexa Flour 488–conjugated goat anti-rabbit IgG (Invitrogen) for 1 h in 1/200 dilution. The negative control was obtained by omission of the primary antibody (data not shown). Images were taken with Olympus IX51 fluorescent microscope (Olympus), SPOT RT3 camera (Burroughs), and SPOT 4.6 software.
Western blotting
MSCs were trypsinized, washed with ice-cold PSB, and resuspended in a lysis buffer with nonionic detergent (25 mM HEPES, 1% Triton X-100, 5 mM KCl, 0.5 mM MgCl2, 1 mM DTT, and 1 mM PMSF) at 2×107 cells/mL. The cytoplasmic proteins and the intact nuclei were separated by centrifugation at 13,000× g for 15 min, and then the nuclear pellet was dissolved in reducing loading buffer and boiled. Nuclear proteins of 8×105 cells were loaded onto a 7.5%–12% SDS-PAGE, and then transferred to nitrocellulose membrane, which was subsequently blocked with nonfat dry milk in TBS-Tween 20. The membrane was reacted with goat anti-TBX5 (Santa Cruz Biotechnology) followed by anti-goat IgG-HRP (Santa Cruz Biotechnology), and the signal was developed with Amersham ECL Plus (GE Healthcare). For testing the equal loading, the membrane was stripped with Re-Blot Plus Solution (Millipore) and probed with an HRP-conjugated antibody against ubiquitous nuclear protein, PCNA (Santa Cruz Biotechnology).
Plasticity of the adherent stromal cells
Osteogenic and adipogenic differentiation was performed as previously described [18]. Briefly, osteogenic differentiation was induced by DMEM supplemented with 10% FCS, 10 mM β-glycerophosphate (Sigma-Aldrich), 50 μg mL−1 ascorbic acid (Sigma-Aldrich), and 0.1 μM hydrocortisone (Sigma-Aldrich). To observe calcium deposition, cultures were stained with Alizarin Red S (Sigma-Aldrich). Adipogenic differentiation was carried out using DMEM supplemented with 10% FCS, 0.5 mM 3-isobutyl-1-methylxanthine (IBMX; Sigma-Aldrich), and 10−7 M dexamethasone (Sigma-Aldrich). After 14 days, cells were fixed and stained with Oil Red O (Sigma-Aldrich) to visualize lipid droplets for microscopic analysis. Photomicrographs were taken with an inverted microscope (Olympus CK2) and a digital camera (Nikon Coolpix 4500).
Cytokine expression analysis
Mouse Inflammatory Cytokines Multi-Analyte ELISArray Kit (SABiosciences) was used to evaluate various cytokines (IL-1A, IL-1B, IL-2, IL-4, IL-6, IL-10, IL-12, IL-17A, IFN-γ, TNF-α, G-CSF, and GM-CSF) in supernatants of the 6 confluent MSC cultures. Absorbance values were read at 450 and 570 nm after stopping the reaction. Cytokine changes were determined after absorbance correction to the absorbance of the negative control.
Dexter-type culture
Aorta wall-derived adherent cells were infected with a third-generation lentiviral vector (RRL-PPT.SF91) expressing green-fluorescent protein (GFP) (kindly provided by Dr. M. Grez, Frakfurt, Germany). Approximately 80%–90% of the infected population expressed GFP in the cytoplasm. GFP+ aorta-derived cells were cultured with a 5-10-fold excess of freshly prepared ABM cells in Dexter-like conditions as described by Dexter and Testa [19]. After 14 days, cultures were analyzed for co-expression of GFP and leukocyte markers (CD45R, CD11b, and Gr1) by FACS analysis (see above).
Differentiation toward osteoclasts
Aorta wall-derived MSCs were treated with 10−8 M 1.25 (OH)2 D3 vitamin (Supelco) for 6 days and the differentiation of JAo-MSC into osteoclast cells were examined. To detect tartrate-resistant acid phosphatase (TRAP)-positive osteoclast cells, Acid Phosphatase Leuokocyte Kit (No. 386) (Sigma-Aldrich) was applied according to the manufacturer's instructions. Freshly prepared BM cells were used as a positive control.
In vivo hematopoietic repopulation ability
Transplantation assay was used to evaluate the in vivo repopulating ability of aorta-derived cells. Recipients (adult C57Bl/6 mice) were irradiated with a single dose of 9 Gy (80 cGy/min) from a 137Cs γ-ray source and then injected intravenously with 2×106 aorta-derived cells or 106 freshly prepared BM cells as a positive control or 50 μL PBS as a negative control.
Results
Characterization of MSCs of different tissue origin
In this study pre-established and characterized ABM-MSC [20], JBM-MSC, JAo-MSC, JThy-MSC, and JSpl-MSC [18] lines were used with the exception of AAD-MSCs. Since AAD-MSCs have been used first in the present article, characterization of these cells is shown in Fig. 1. Similarly to the MSC lines isolated from other tissues [21], AAD-MSCs expressed the MSC-specific markers (Fig. 1A) and differentiated toward osteogenic, adipogenic (Fig. 1B), and chondrogenic (data not shown) lineages. The expression of mRNA levels well correlated with the cytofluorimetry results (data not shown). All MSCs were uniformly Sca-1 positive, whereas CD45 (data not shown), CD34, and CD31 (Supplementary Fig. S1) were negative based on cytofluorimetry [18,20]. Genes involved in osteogenic and adipogenic differentiation of stromal cells, Runx2 and Pparg, were similarly expressed in all types of MSCs (Fig. 1C). Notably, osteocalcin (Bglap1) transcript was more abundant in JBM-MSC than in any other MSCs tested (P=0.011).
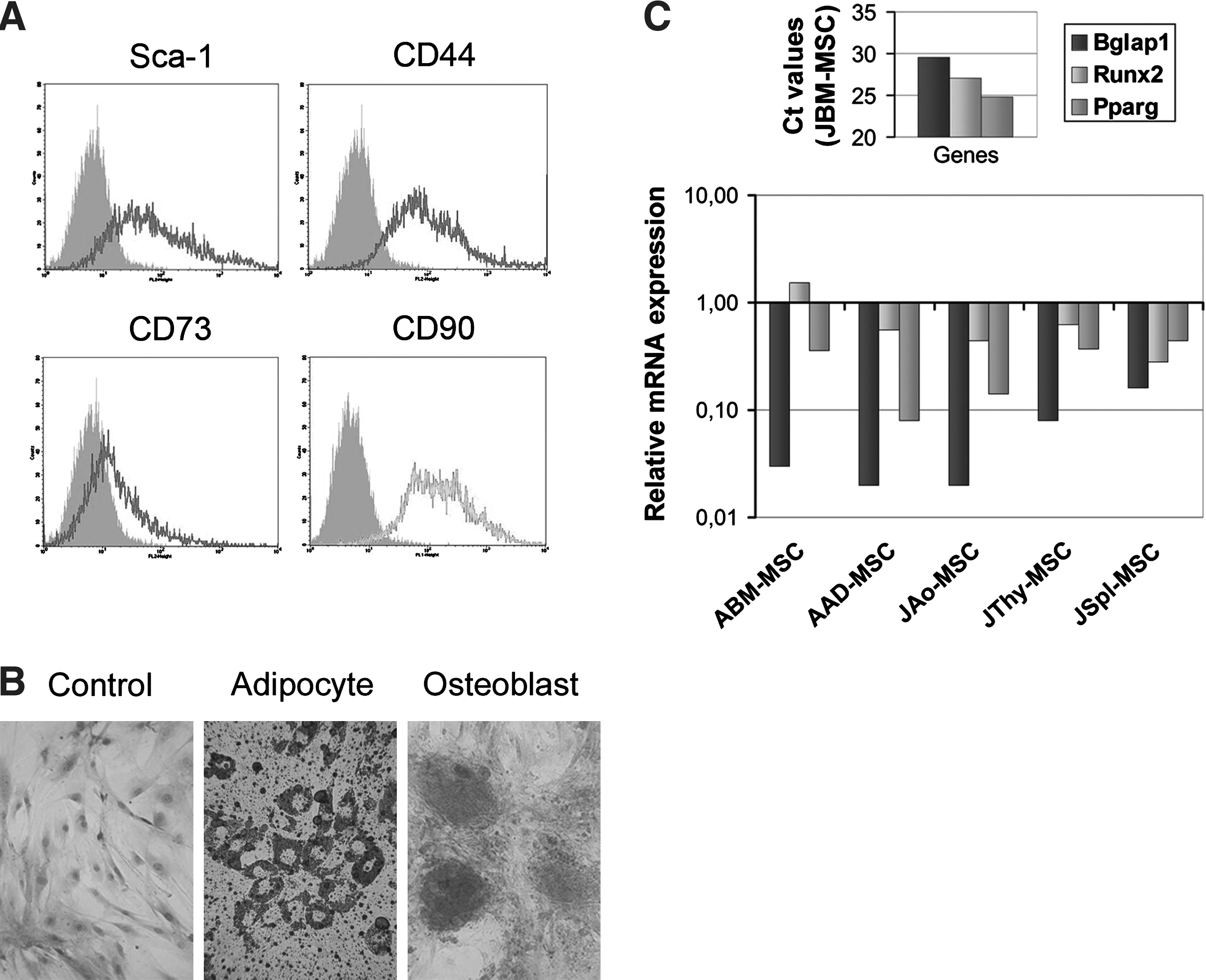
MSC markers expressed by stromal cells isolated from different organs.
The expression levels of particular genes analyzed in one type of MSC population were stable between passages 10 and 15 and independent of the length of the culture period as well as of the process of starting the culture from cryopreserved samples (data not shown).
Comparison of gene expression patterns of cytokines, growth factors, bone morphogenetic proteins, and adhesion molecules in MSCs derived from different sources
The expression levels of mRNAs encoding for different bioactive factors (cytokines, growth factors, and bone morphogenetic proteins) in the MSC lines relative to those in MSCs isolated from the BM of 14-day-old animals (JBM-MSC) were compared using the Mouse MSC PCR Array (Supplementary Fig. S2A). The original Ct values were presented in Supplementary Table S1.
Results obtained investigating the cytokine expressions are shown in Fig. 2A. Specific transcripts of IL-6 and GM-CSF (Csf2) were expressed in all MSC samples. Using the Mouse Inflammatory Cytokines Multi-Analyte ELISArray Kit, IL-6 and GM-CSF were detected in the supernatants of the different MSC cultures (data not shown). In general, all MSC lines produced higher amount of Csf3-specific (P=0.034) and IL10-specific (P=0.026) mRNA than JBM-MSCs did. In terms of expression of cytokine-specific mRNAs, the ABM-MSCs showed special features since CSF3, IL-1β (P=0.007), IFN-γ (P=0.013), and TNF-α (P=0.007) mRNA species were expressed abundantly compared to those in MSCs from any other source. However, the protein products encoded by these genes were not detected in ABM-MSC-conditioned medium (data not shown).

Cytokine and growth factor messages in MSCs derived from different anatomical location.
Expression of the analyzed growth factors (Fig. 2B) showed slight cell-type-specific differences. The most striking divergence was found in the expression of insulin II (Ins2) since this gene was highly transcribed only in JAo-MSCs (P=0.009) and slightly in JSpl-MSCs; however, other MSC populations did not express this hormone. Juvenile BM-MSCs expressed mRNAs for hepatocyte growth factor (HGF), insulin-like growth factor 1 (IGF-1), vascular endothelial growth factor A (VEGF-A), brain-derived neutrophic factor (BDNF), fibroblast growth factor 2 (FGF-2), epidermal growth factor (EGF), and fibroblast growth factor 10 (FGF-10). Adipocyte-derived MSCs failed to express HGF and IGF-1; aorta- or spleen-derived MSCs expressed much lower amount of mRNA encoding for IGF-1 (JAo-MSCs, P=0.017) and VEGF-A (JAo- and JSpl-MSCs, P=0.013) compared to those of JBM-MSCs.
Differences between the expression levels of bone morphogenetic protein (BMP) family members were observed in the various MSC groups. BMP-4 and transforming growth factor beta 1 and 3 (TGFβ1, TGFβ3) coding genes exhibited similar expression levels in all MSCs, whereas growth differentiation factor 15 (Gdf15) mRNA level decreased markedly in the aorta wall-derived (P=0.017) and, in a lesser extent, in adipose tissue-derived cells as compared to JBM-MSCs. In turn, JAo-MSCs and AAD-MSCs expressed higher levels of GDF-6 (or BMP-13) (P=0.036) and BMP-6 (P=0.033) than the other MSC types (Fig. 2C). It should be noted that BMP-13 has been shown to strongly inhibit osteogenic differentiation, especially matrix mineralization [21]; therefore, its high expression in the aorta wall and adipose tissue is not surprising.
The expression of different adhesion molecules and integrins was also analyzed. The genes encoding for CD44, β-catenin (Ctnnb1), type I collagen α-chain (Col1a1), and endoglin (CD105) (Eng) (Fig. 3A) were similarly expressed on all types of MSCs. The expression of intracellular adhesion molecule 1 (CD54) (Icam1) mRNA could not be detected in JBM-MSCs and JThy-MSCs; however, it was present in decreasing amount in the following cells: AAD-MSCs>JAo-MSCs>ABM-MSCs>JSpl-MSCs (Fig. 3A). Activated leukocyte adhesion molecule (CD166) (Alcam) mRNA was present in all MSC types but with high quantitative differences since ABM-MSC, JAo-MSCs, AAD-MSCs, and JSpl-MSC expressed this mRNA species most abundantly (P=0.010) (Fig. 3A). Melanoma cell adhesion molecule (CD146) (Mcam) and vascular cell adhesion molecule 1 (CD106) (Vcam1) mRNA were similarly expressed in all types of cells, except that Mcam showed higher expression in AAD-MSCs and JSpl-MSCs (P=0.043) and Vcam1 showed a lower expression in JSpl-MSCs. Expression of adhesion molecules belonging to the integrin family and involved in cell–extracellular matrix interaction varied in the different MSCs as shown in Fig. 3B. Integrin αx (CD11c) (Itgax) and integrin αv (CD51) (Itgav) mRNA levels were similar in all MSC lines. Integrin β1 (CD29) (Itgb1) mRNA showed similar level in JAo-MSCs, JThy-MSCs, and JBM-MSCs and slightly higher level in ABM-MSC, AAD-MSCs, and JSpl-MSCs. Analysis of the level of integrin α6 (CD49f) (Itga6) mRNA showed higher but not significant expression in AAD- and JAo-MSCs compared to the other cell types.
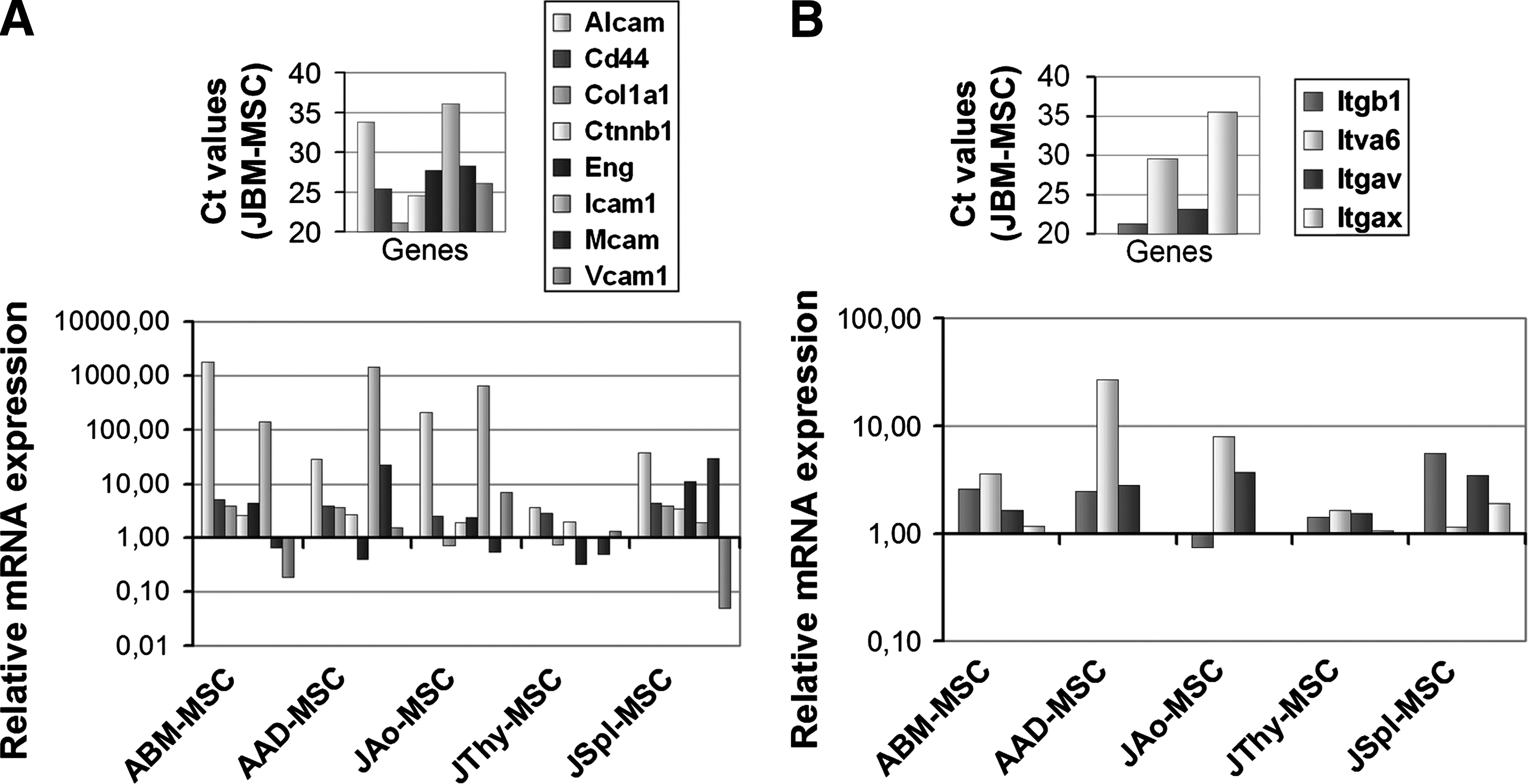
Expression of genes coding proteins involved in cell–cell and cell–matrix interactions in MSCs.
MSCs do not express pluripotency-associated genes
Recent studies have indicated that somatic stem cells, including MSCs [22,23], express pluripotency-associated genes, Pou domain containing Pou5f1/Oct-4, zinc finger family member Rex-1/Zfp-42, and/or Nanog transcription factors regulating the maintenance of undifferentiated state of embryonic stem cells. However, the expression of these transcription factors was undetectable using Mouse MSC PCR Array in the different MSC lines analyzed. To further confirm the failure of expression of the above transcription factors, single set quantitative real-time PCR (qRT-PCR) was carried out on mRNA prepared from the different MSC lines and a murine ES cell line (R1/E) as control. As shown in Fig. 4A, Pou5f1/Oct-4 (P=0.003), Nanog (P=0.002), and Rex-1/Zfp-42 (P=0.003) transcript levels were several orders of magnitude lower than in the embryonic R1/E cells. In contrast, consistent expression of Klf-4 was revealed in MSCs at a similar level to that of ES cells. This finding was not surprising considering the multifunctional role of Klf-4, participating in development, cell proliferation, and differentiation [24].

None of the MSC cultures contain pluripotency marker-expressing cells.
Genes involved in self-renewal and determination of latter cell fates of multipotent MSCs were examined. Low level of telomerase reverse transcriptase (Tert) and leukemia inhibitory factor (Lif) gene expression was detected in the various MSC samples except JAo-MSCs in which Tert and LIF-specific mRNA levels were 9- and 15-fold higher, respectively, than in the other MSC samples (data not shown). Moreover, JAo-MSCs formed unique cell clusters in overconfluent cultures that gradually organized into compact spheroids (Fig. 4B) resembling a less mature mesoangioblast-like phenotype. The name “mesoangioblast” introduced by Minasi et al. [25] was chosen to denote a common progenitor for vascular and extravascular mesodermal derivatives. To confirm or disapprove the possible hematopoietic potential of aorta-derived MSCs, GFP-labeled JAo-MSCs were co-cultured with a 5-10-fold excess of freshly prepared ABM cells in a Dexter-type culture. None of the GFP-positive cells expressed CD45R, CD11b, or Gr-1 after a 14-day incubation period (Supplementary Fig. S3A). Furthermore, when treated with D3 vitamin in vitro for 6 days, they did not express the osteoclast marker TRAP enzyme, did not acquire a multinucleated osteoclast-like morphology (Supplementary Fig. S3B), and did not form hematopoietic colonies in semi-solid media [18]. Finally, aorta-derived adherent cells were unable to repopulate the marrow of lethally irradiated (9 Gy) syngeneic (C57Bl/6) mice (Supplementary Fig. S3C). Thus, JAo-MSCs were not able to differentiate into hematopoietic cells in vivo or in vitro.
Expression of a transcription factor, Brachyury, regulating the specification and differentiation of posterior mesoderm during gastrulation [26], was hardly detectable in JBM-MSCs (CT value 37.7) similarly to that of MSCs from other organs (Fig. 4C). This result suggested that Brachyury was dispensable in controlling the MSC's fate and function.
Expression of mesodermal marker genes in MSCs
Mesodermal origin of the various MSC lines was analyzed by detecting expression of connective tissue proteins, Col1a1, a major component of collagen I, and Vim, a member of the intermediate filament family, as well as α-SMA (Acta2), an actin isoform typical of smooth muscle cells. JBM-MSCs expressed high levels of Col1a1, Vim, and Acta2. MSCs from other tissue sources showed similar expression levels of Col1a1 and Vim as JBM-MSCs; however, α-SMA expression showed remarkable differences. While adipose- and spleen-derived MSCs expressed more abundant levels (10-fold) of Acta2 mRNA compared to JBM-MSC (P=0.024) (Fig. 5A), aorta-derived MSCs expressed lower amount of this message. The analysis of α-SMA protein, however, showed a uniform low expression in 100% of JAo-MSCs. In contrast, 40%–60% of MSCs from other sources expressed α-SMA protein detected by immunostaining [18].
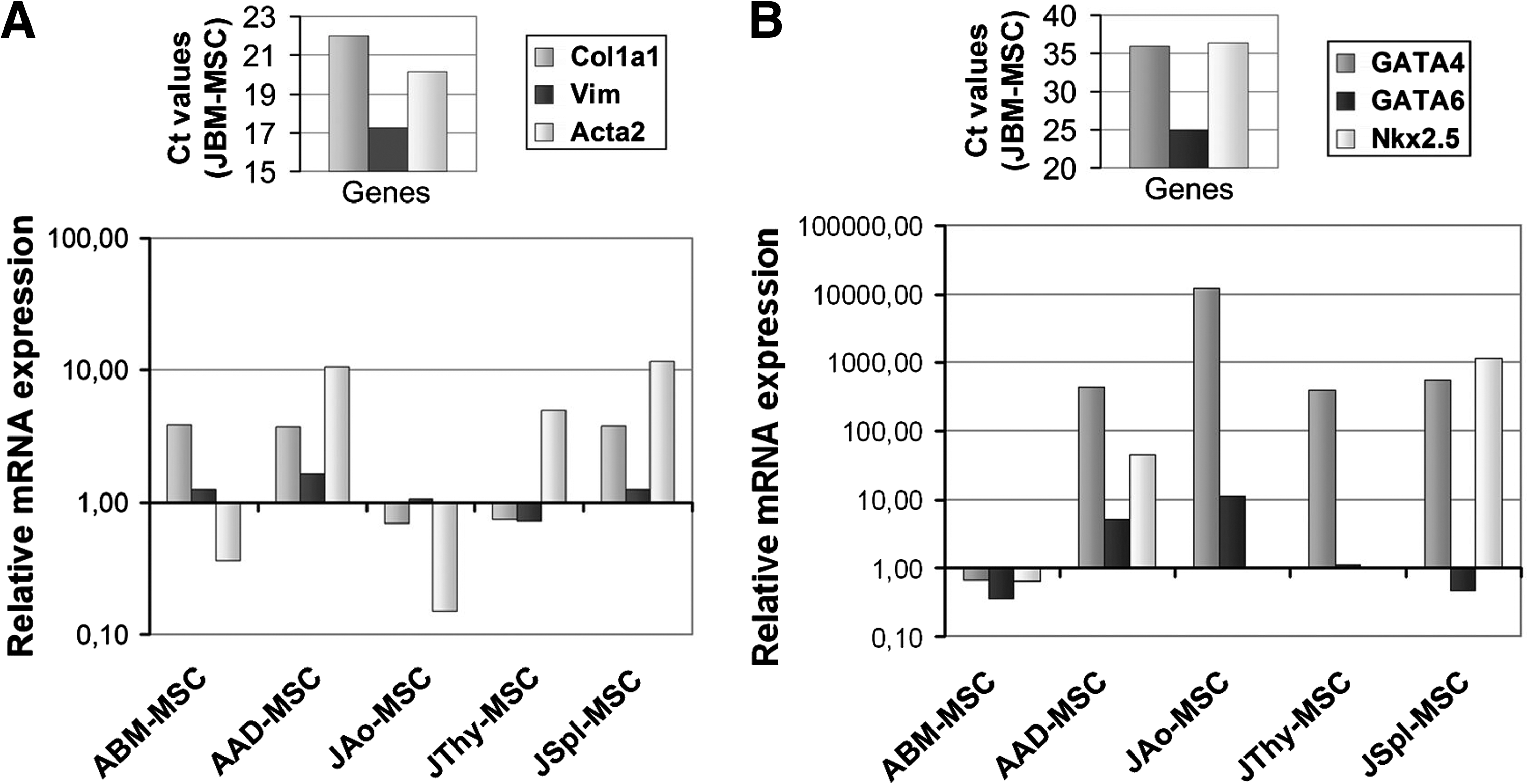
All of the MSCs express mesenchymal and mesodermal markers.
Next, the expression of 3 transcription factor (GATA4, GATA6, and Nkx2.5) coding genes, also involved in the specification and differentiation of cells with mesodermal origin, was evaluated by quantitative RT-PCR. Among these, the Gata6 gene exhibited similarly high expression levels in all MSC samples (Fig. 5B). The other 2 factors, GATA4 and Nkx-2.5 mRNAs, were detected at low levels in JBM-MSC and ABM-MSC (Ct values >35). Moreover, Nkx-2.5, but not GATA4 mRNA, level was also equally low in JAo- and JThy-MSCs. In contrast, GATA4 mRNA level was ∼800- and ∼10,000-fold higher in AAD-, JThy-, JSpl-MSCs and JAo-MSC (P=0.011), respectively. High expression of Nkx2.5 was found in AAD-MSCs and JSpl-MSCs, ∼80- and ∼1,000-fold as in JBM-MSCs (P=0.049), respectively. These data imply that all MSC populations can be classified as mesenchymal cells, likely deriving from the mesoderm.
Positional memory of MSCs
Homeotic selector (Hox) genes encode a family of evolutionary-conserved transcription factors that specify embryonic positional identity along the anterior-posterior and secondary axes in animals, and their expression in adult cells constitutes a form of positional memory [27]. Thus, to assess a possible role for Hox genes in tissue-specific differences between MSCs from distinct sources, Hox expression profiles were generated from each MSC samples. The primary basis of these experiments was the Mouse Homeobox (HOX) Genes PCR Array, including in total 24 Hox-specific primer pairs (Supplementary Fig. S2B). The original Ct values are presented in the Supplementary Table S2.
Among these, consistent expression of 5 Hox genes (Hoxa1,7, Hoxb3,4, and Hoxd9) and no expression of 6 other transcripts (Hoxb1, 9, Hoxd1,3, 12,13) were detected in all MSC lines (Fig. 6A–D). Thirteen Hox genes (Hoxa9, Hoxb2,7,8, Hoxc6,8,9,10,11,12,13, and Hoxd4,8) were differently expressed in MSCs isolated from distinct sources. Thus, the MSCs have individual Hox code or Hox expression fingerprint that may reflect their anatomical origin.
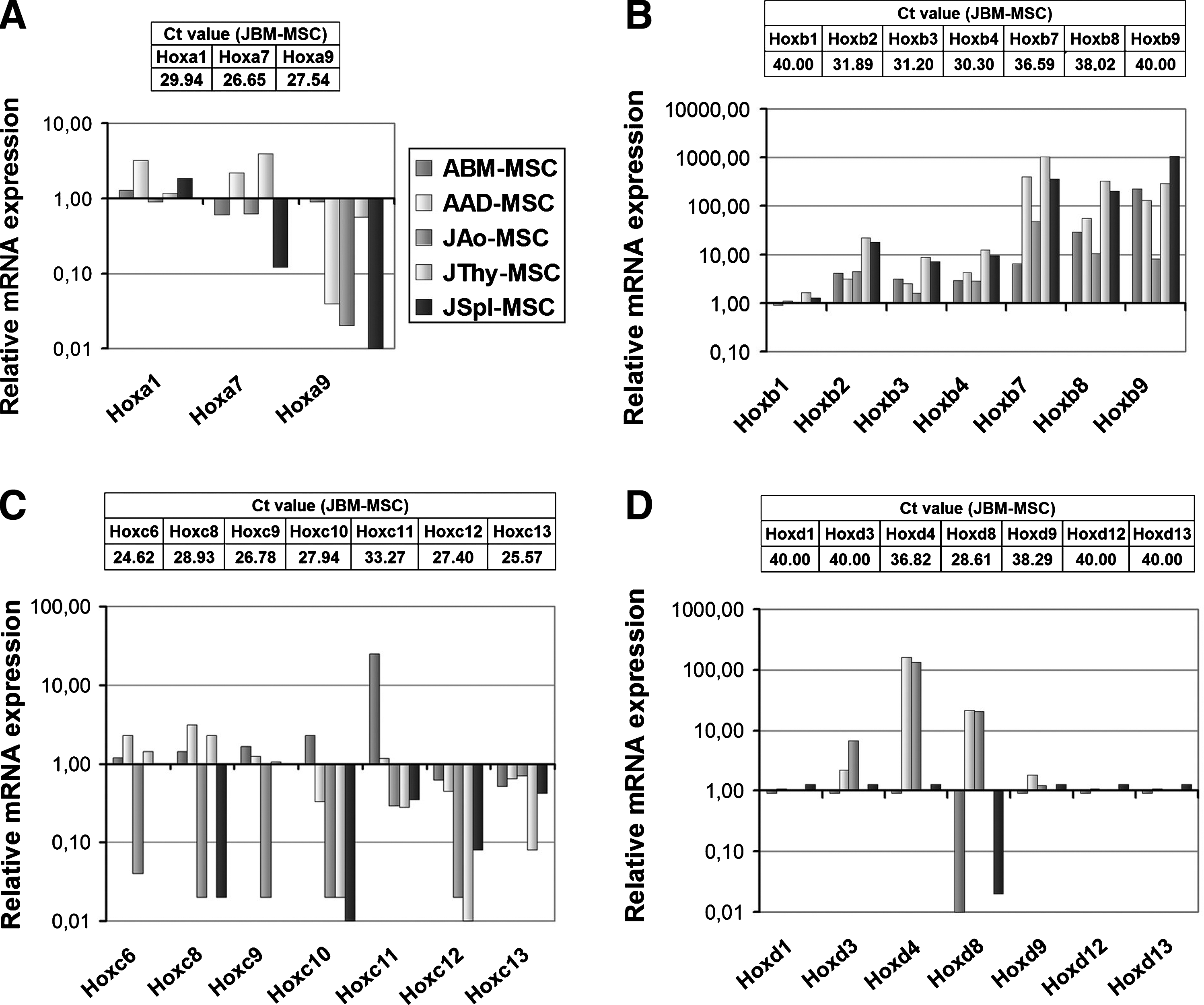
Genes of the 4 classical Hox clusters exhibit different mRNA expression patterns in MSC samples.
Twenty-one genes encoding homeodomain-containing transcription factors, such as Arx, Barx1, Cdx1,2,4, Dmbx1, Emx1, Hopx, Lbx1, Lmx1a,1b, Nkx3-1, Otx2, Pdx1, Phox2b, Pitx3, Prop1, Six3, Vax1, Vsx1, were not detected in any MSC lineages (data not shown). However, another 30 genes encoding homeobox-containing transcription factors—Alx1,3,4, Dlx2,3,4,5,6, Emx2, En1 (Supplementary Fig. S4A), Hhex, Isl1, Isl2, Lbx2, Lhx1, Meis1, Meox1, Mixl1, Msx1,2 (Supplementary Fig. S4B), Otp, Otx1, Pax3, Pitx2, Prox1, Shox2, Six1,2,6, and Vax2 (Supplementary Fig. S4C)—involved in body pattern formation, morphogenesis and other developmental processes, were expressed at variable levels in MSC populations with distinct origin. For example, Alx1, which is involved in brain, craniofacial bone, and limb—especially in chondrocyte lineage—development [28], is expressed at much higher levels in both ABM-MSCs (∼100-fold, P=0.014) and JThy-MSCs (∼1,000-fold, P=0.003) in comparison with JBM-MSCs (Supplementary Fig. S4A). On the other hand, Pax3, which is involved in development of neural crest derivatives and also detected in somite compartments [29,30], is expressed more than ∼100-fold higher in ABM-MSCs, JAo-MSCs, and JSpl-MSCs compared with JBM-MSCs (P=0.006).
Homeodomain-containing transcription factor-encoding genes—cut-like homeobox 1 (Cux1) involved in cytoskeleton remodeling, cell–cell and cell–matrix adhesion, epithelial-to-mesenchymal transition, and transcriptional regulation [31]; distal-less homeobox 1 (Dlx1) implicated in patterning and development of the brain craniofacial structures, and the axial and appendicular skeleton [32]; Mkx transcribed in cell lineages derived from the somites [33,34]; and sine oculis-related homeobox 4 (Six4) expressed in somites and limb buds [35]—shared high expression levels in all MSC samples (Fig. 7A). The expression of Mkx protein was also confirmed by immunofluorescence (Supplementary Fig. S5). Most importantly, T-box 5 (Tbx5) involved in the formation of the forelimb and heart [26,36]. Tbx5 was expressed at an especially high level in JThy-MSCs (P=0.037), Engrailed homeobox 2 (En2) in JAo-MSCs (P=0.028), T-cell leukemia homeobox 1 (Tlx1), participating in the early patterning and proliferation of the splenic primordium [37,38] in JSpl-MSCs (P=0.017), and paired-like homeodomain transcription factor 1 (Pitx1), implied in the development of hindlimb [39] in the femoral BM-derived samples (JBM-MSCs and ABM-MSCs) (P=0.024) (Fig. 7B). Validation of Tbx5, En2, Tlx1, and Pitx1 expression was carried out at the protein level by immunofluorescence using specific antibodies (Fig. 7D). Furthermore, Western blot analysis revealed that Tbx5 protein was detected in thymus-derived MSCs, but it was not present in JBM-derived cells (Fig. 7C). The results confirmed the expression of the particular gene products in the proper MSC lines, for example, Tbx5 in JThy-MSCs, Tlx1 in JSpl-MSCs, Pitx1 in JBM-MSCs, and En2 in JAo-MSCs. These data indicate that MSC populations derived from different tissues and organs could also be distinguished based on the higher expression of genes involved in regionalization and development of the mesoderm.
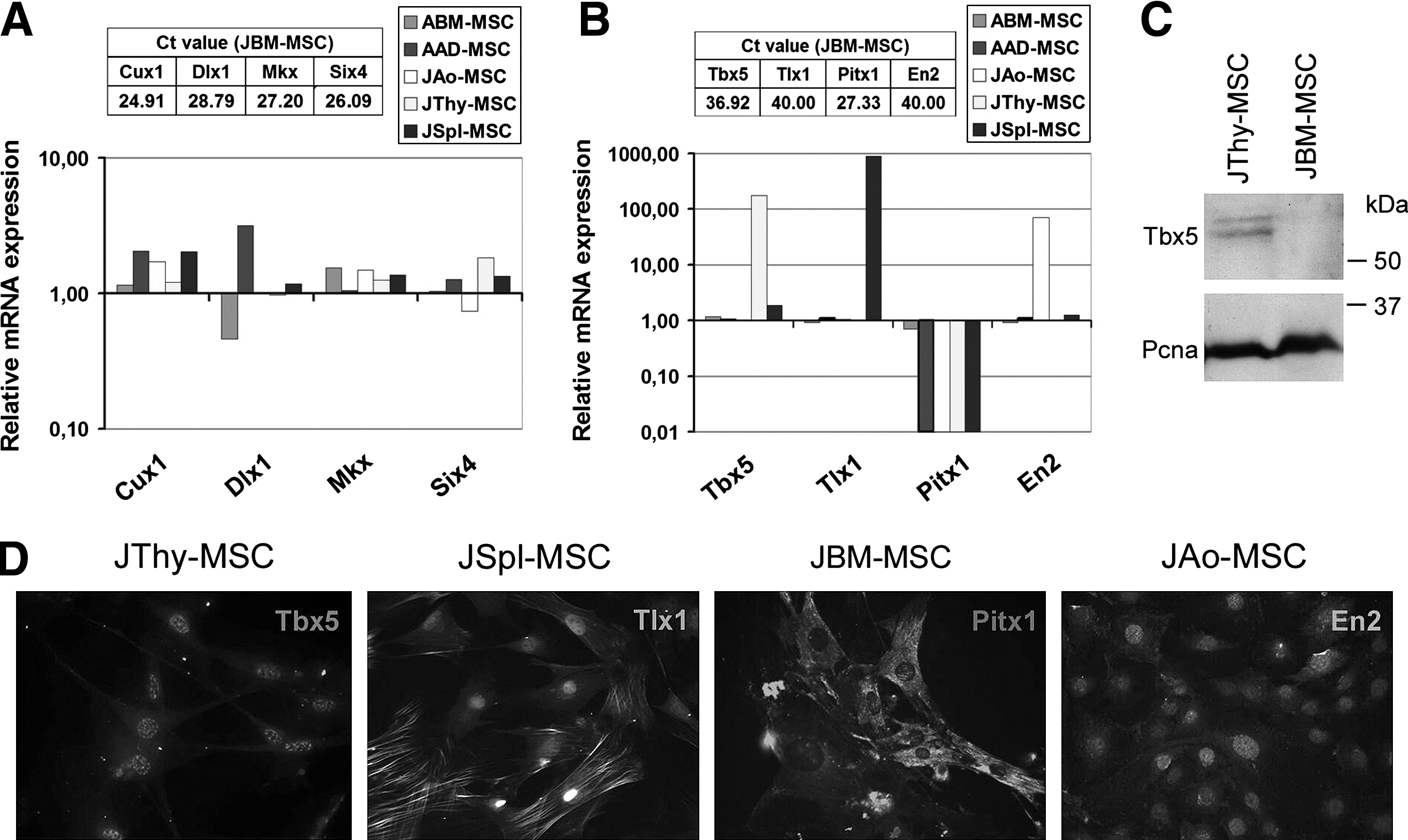
Expression of other homeodomain-containing transcription factors in the MSCs.
Discussion
One significant unrevealed question of MSC biology is whether MSCs' cell fate and tissue positioning are determined and controlled during early embryogenesis or later upon specific tissue environments. So far, the main difficulty of getting an answer comes from the failure to identify specific lineage markers defining MSCs as a specific cell type. Therefore, we analyzed MSCs of various tissue of origin by expressing gene profiles, classified into 4 groups: (1) pluripotency-associated genes, (2) genes classically characterizing MSCs, (3) mesenchymal genes, and (4) genes involved in mesoderm segmentation and somite development.
Recently, it has been reported that a small subpopulation of murine [22] and human [23] stromal cultures express pluripotency-associated genes, especially Pou5f1/Oct-4, and these pluripotent embryonic-like stem cells serve in vivo to maintain the turnover of skeletal tissues in homeostasis or tissue repair during adulthood. In contrast, Lengner et al. [40] have failed to detect the expression of pluripotency-associated genes in MSC cultures and excluded the presence of a pluripotent cell subpopulation in the MSC compartments. Our results are in accordance with the latter data since Pou5f1/Oct-4, Nanog, and Rex-1/Zfp-42 messages were absent in MSCs isolated from any tissues compared to those expressed in ES cells. Hence our data do not support the existence of embryonic remnant, that is, embryo-derived pluripotent stem cells within the postnatal MSC compartments or, alternatively, the view that pluripotency genes play role for the self-renewal of MSCs and in maintaining tissue homeostasis.
In previous studies [18,20] and in the present work, we have compared MSCs of various tissue origins analyzing the expression of accepted MSC markers and differentiation factors (Bglap1, Runx2, and Pparγ) and their ability to differentiate into adipocyte and osteoblasts under specific tissue culture conditions. As expected, all cell cultures carried MSC features although quantitative but not qualitative differences were found in the expression of the cell surface markers well-correlating with the PCR data and differentiation ability. Mesodermal origin of MSCs of any tissue origin was reinforced by commonly expressing core transcriptome and our data were in accordance with those of Menicanin et al. [41]. High expression of genes encoding for typical mesenchymal and mesodermal markers (α-smooth muscle actin, collagen I α-chain, PPARγ, Runx2, and vimentin), growth factors (BDNF, EGF, FGF2, HGF, IGF1, and VEGFα), morphogens (BMP4, TGF-β1, and TGF-β3), cytokines (IL-6 and GM-CSF), cell adhesion molecules (CD44, β-catenin, endoglin, MCAM, and VCAM1), and integrins (CD29, CD49f, CD51, and CD11c) was detected in all MSC samples (Fig. 8). However, expression of several genes encoding for secreted or cell surface molecules such as BMP6, FGF10, and HGF showed marked quantitative, but not qualitative difference. Importantly, the common core expression profile included a set of transcription factor encoding genes, such as the Cux1, Klf4, and Gata6, which were shown to play role in many fundamental biologic processes, including embryonic development, organogenesis, cell proliferation, and differentiation [31,42]. Notably, knockdown of GATA6 by small inhibitory RNA suppressed the self-renewal capacity of human MSCs [43]. The transcription factor Gata4 is expressed in all tissue- but BM-derived MSCs. Gata4 is regulated by signaling pathways that act in the early specification of endoderm and mesoderm (heart, intestine, liver, pancreas, and smooth muscle) [44], hence controlling the formation of many organs. Another transcription factor, Nkx2.5, showed higher expression in JAo-MSCs and JSpl-MSCs, up-regulating Gata4 [45] in the latter cells. Notably, Pax3 message is also found in BM, spleen, and aorta wall-derived cells. Since Pax3 is expressed in the neural crest and involved in development of neural crest derivatives [46], we cannot completely exclude the possibility that a small subpopulation of postnatal MSCs originate from the neural crest, as suggested by Morikawa et al. [46]. However, Pax3 expression is also detected in somite compartments that give rise to embryonic skeletal muscle progenitors, and in cells that give rise to skeletal muscle compartments of the developing limb buds [47]. Furthermore, expression of several genes involved in the development of brain and nervous system, such as Arx, Dmbx1, Lbx1, Otx2, Phox2b, Prop1, and Six3, is not detected in any of the analyzed MSC samples. The high similarity identified in the gene expression profiles of various MSC populations suggests that adherent stromal cells isolated from different tissues have similar phenotype and may be defined by an akin, but not identical, transcriptome. Moreover, the above results strongly support our and others assumption [15] that postnatal MSCs derive from the mesoderm. Note that MSCs located in the craniofacial region derived from the neural crest have not been analyzed in this study.
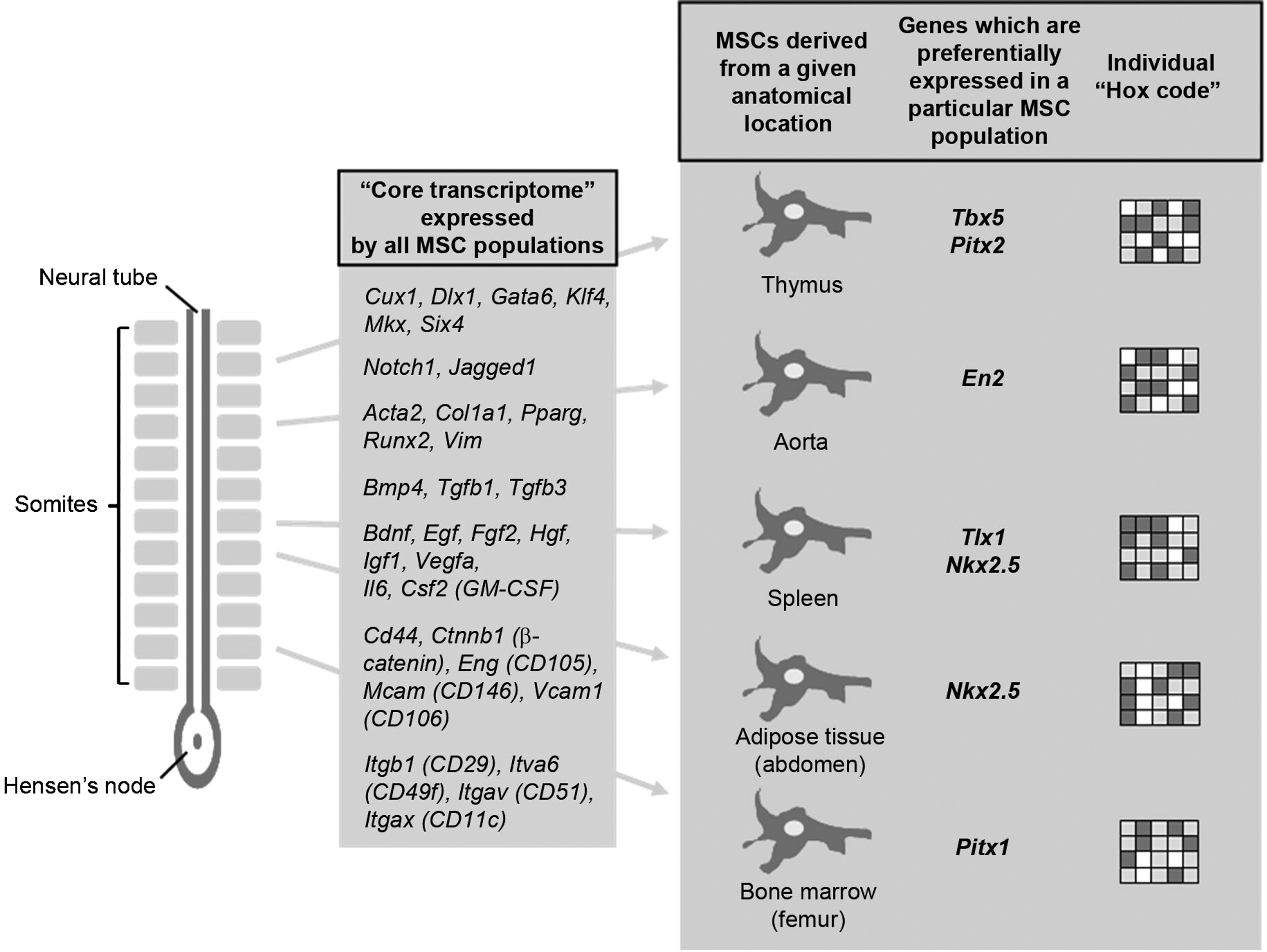
Schematic representation of gene expression profiles and developmental origin of MSCs derived from different tissues and organs.
To examine whether the fate of the MSCs is determined at the stage of development of postsegmentation mesoderm or somites, transcription of Dlx1, Six4, and Mkx genes were investigated. The Dlx genes have been implicated in patterning and development of the brain, craniofacial structures, and the axial and appendicular skeleton [32]. The Six4 gene is expressed from E8 in overlapping expression patterns in somites, limb buds, dorsal root ganglia, and branchial arches, and involved in different type of organogenesis [48]. More recent studies also indicate that Six4 protein plays critical roles in regulating several transition points of the cell cycle as well [49]. During mouse development, Mkx is transcribed in cell lineages derived from the somites. As early as embryonic day 9.0, Mkx is expressed in an anterior to posterior gradient in the dorsomedial and ventrolateral lips of the dermomyotome. As somites mature, Mkx is transcribed in the tendon-specific syndetome and the sclerotome-derived condensing mesenchyme prefiguring the proximal ribs and vertebral bodies [33]. Mkx is a strong inhibitor of myogenic differentiation, a process that is dependent on DNA-binding via the Mkx homeodomain [50]. Knockout mice have hypoplastic tendons throughout the body. Despite the reduction in tendon mass, the cell number in the tail tendon fiber bundles in Mkx −/− mice are similar to that of wild-type animals [34]. Our results show that all of these genes are highly expressed in MSCs derived from any tissues supporting the ancestry of all MSC populations from the postsegmentation mesoderm, presumable from the somites. Our findings are supported by the reports showing that mesoangioblasts, a class of multipotent mesodermal progenitor cells [51], and all smooth muscle cells in the postnatal descending aorta are derived from the somites [52]. Furthermore, Pouget et al. [53] described that all mural cells—pericytes and vascular smooth muscle cells of the trunk, limbs, and aorta—originate from the sclerotome.
Consistent with the above results, we have found that MSC populations have individual Hox code or Hox expression fingerprint that are specific for their anatomical origin. In a recent publication Ackema and Charite [54] have described that murine MSCs isolated from bones from different anatomical sites have slightly distinct developmental potentials, and this feature can be correlated with the anatomic site-specific expression of certain Hox genes. Another study by Chang et al. [55] has revealed that fibroblasts from numerous finely mapped anatomic sites across the human body consistently express distinct patterns of HOX genes, sufficiently determining the position of a given cell along the 3 developmental axes. Others have confirmed that a typical Hox code properly marks out the positional identity of smooth muscle cells [56] and adipocytes [57]. Thus, postnatal tissue stem and differentiated somatic cells—including MSCs—carry their positional information. In addition to controlling the embryonal segmentation process, positional identity also ensures the body-segment-specific development of skin, muscle, or bone [27]. The retention of positional identity in postnatal MSCs might limit their plasticity, leading to a restricted regenerative ability. Accordingly, Creuzet et al. [58] have shown that overexpression of Hoxa3 in neural crest cells prevents the formation of the skeleton derived from the first branchial arch, but allows the development of the nasal septum. By contrast, Hoxb4 overexpression hampers the formation of the nasal bud-derived skeleton, while allowing that of a proximal segment of the lower jaw. Simultaneous overexpression of the Hoxa3 and Hoxb4 genes prevents the formation of facial skeletal structures. Formation of neural derivatives of the crest is not affected by the function of these genes. Conversely, when Leucht et al. [59] grafted Hoxa11-positive MSCs from the tibia into a site of Hoxa11-negative (mandibular) injury, the transplanted cells showed a persistence of Hoxa11 expression in the Hoxa11− environment and failed to differentiate into new bone, instead formed a scar tissue. However, Hoxa11-negative MSCs from the mandible readily adopted Hoxa11 expression when transplanted into a Hoxa11+ tibial injury site. These grafted cells formed new bone, with no evidence of scar formation. Hence, it is indicated from this study that MSCs may regain a novel Hox-code within the proper environment but they are not able to loose a preprinted code. Moreover, co-operation between the stem cells occupying an injury site and the injury environment itself could be a crucial component of normal healing or regeneration.
Of particular interest in the new findings reported here is the identification of those messages that are highly and preferentially expressed in a particular MSC population derived from a given anatomical location. In this term, Tbx5 and Fgf10 mRNAs are expressed at an especially high level in thymus-derived MSCs. Functional consequence of deficiency in FGF10 has been described [60] since the thymus development was blocked at early embryonic stage. The possible reason for the thymic developmental failure is that Tbx5/FGF10 signaling pathway is essential for normal thymus development, especially for the proliferation and maturation of thymic epithelial cells [60]. Tbx5 has been shown to be essential for the formation of the forelimb in several vertebrate species, including mouse and human. In the absence of Tbx5, no forelimb bud is formed [61]. Additionally, Tbx5 directly activates the Fgf10 gene, which, in turn, maintains Tbx5 expression during limb outgrowth [62]. This is consistent with the defects seen in Holt-Oram syndrome, an inherited disorder characterized by abnormalities of the upper limbs and heart due to the haploinsufficiency of human TBX5 [63]. In addition, Pitx2 is expressed in almost all muscle anlagen from embryogenesis until adulthood during forelimb development. Its overexpression during forelimb development results in severe tendon, muscle, and bone anomalies [64]. Finally, Alx1 encodes a paired-like homeoprotein expressed selectively in chondrocyte lineage during embryonic development. Although its target gene remains unknown, gene disruption studies have revealed that Alx1 plays an important role for craniofacial bone formation as well as for limb development by cooperating with another homeoprotein, Alx4 [28,65]. Tlx1 mRNA shows specifically high expression in spleenic MSCs, a result that is in accordance with Roberts et al's data [66]. It has been previously well established that Tlx1 is required for spleen patterning, morphogenesis, and expansion. Although viable, Tlx1-null mice are asplenic, without any other detectable abnormalities. The spleen primordium of Tlx1 mutant embryos develops normally until E13.5 but fails to expand thereafter, probably as a result of reduced mesenchymal proliferation [37,38]. The putative splenic mesenchyme is closely associated with the dorsal pancreatic mesenchyme and also expresses Nkx2.5, another evolutionary conserved homeobox gene detected in JSpl-MSCs. This gene has a repressor activity on myofibroblast differentiation as well, which may be of homeostatic significance as a mean of suppressing myofibroblast differentiation in the absence of tissue injury [67]. Pitx1, a paired-type homeodomain transcription factor specifying the identity and structure of hindlimb [39], is similarly present in both juvenile- and adult femur-derived MSCs but not in other stromal cells. JAo-MSCs also express a couple of specific genes. First, En2, a gene whose expression is essential for the formation of the mesencephalon and metencephalon and for patterning the region that determines the cerebellum, and also for specifying head muscle identity, [68,69] is highly and uniquely expressed in these cells. However, the biological significance of its expression in JAo-MSCs is unclear. Second, high Gdf6 expression in MSCs isolated from the aorta wall can contribute to the maintenance of aorta stiffness and the prevention of its calcification. In fact, role of the Gdf6 gene product BMP-13 in precluding the osteogenic differentiation has been perceived in a variety of mesenchymal derivatives such as tendon and cartilage [70]. BMP-13 induces neo-tendon/ligament formation and the expression of chondrocyte markers such as proteoglycans during connective tissue healing [71]. In vivo, mutational inactivation of the BMP-13 gene is associated with Klippel-Feil syndrome, characterized by congenital fusion of the cervical spine vertebrae [72], and causes defects in joint, ligament, and cartilage formation in a knockout mouse model [73]. The expression of En2, Pitx1, Tbx5, and Tlx1 gene products analyzed by immunofluorescence confirmed that the proteins were present homogenously in the cell cultures of aorta-, femoral BM-, spleen-, and thymus-derived MSCs, respectively.
Based on the above results, we conclude that the postnatal mesenchymal stem/progenitor cell populations, resident in different tissues, develop in a parallel manner within different body segments after the regionalization of the mesoderm. This conclusion is supported by the diversity and positional memory of MSCs based on their unique Hox code and the selective expression of key transcription factors such as the Mkx (somite), Pitx1 (hindlimb), Tbx5 (thymus), and Tlx1 (spleen) involved in patterning of the mesoderm and in the somitogenesis. Stable topographic identity and memory was retained in the cells of various tissue origins even after long-term (10–15 passages) tissue culturing in vitro. These data were in accordance with previous findings presenting remarkable retention of genes determining the positional identity of diverse cell types [54,56,57]. Our findings may have crucial consequences in regenerative medicine. MSCs isolated from diverse anatomical locations are similarly able to differentiate into bone, cartilage, and adipose tissues in vitro. Nevertheless, their regenerative potential may differ in vivo since difference between the tissue-specific molecular imprint of the MSC graft and the injured tissue or organ of the recipient may be critical determinants of the successful tissue repair and regeneration. This hypothesis is supported by the finding that the origin of human MSCs determines the healing performance of these cells in cardiac regeneration [11] and their engraftment potential into dystrophic muscle [74]. Additionally, Badri et al. [75] describe that tissue-specific MSCs engraft in their organ of origin and establish a pathway of bi-directional interaction between these mesenchymal progenitors and adult somatic epithelial cells in the lung. However, it cannot be excluded either that MSCs originated from different tissues may also substitute each other since it has been shown that BM MSCs are mobilized in response to heavy injury or tumor formation [76]. Moreover, we have described that BM-, spleen-, and aorta-derived, but not thymus-derived, MSCs, in combination with hematopoietic cells, are efficient in preventing streptozotocin-induced diabetes in mice [18,20]. To clarify this contradiction, further in vivo lineage-tracing studies are required.
Footnotes
Acknowledgments
We are grateful to Dr. Anikó Szilvási (Laboratory of Molecular Diagnostics, National Blood Service) for her invaluable help with the statistical analysis of array data. This work was supported by the Hungarian Scientific Research Foundation (NKTH-OTKA CK 78007, CK 78188, and OTKA K 69047).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.