
Abstract
The therapeutic potential of bone marrow mesenchymal stem cells (MSCs) in kidney failure has been examined in some studies. However, recent findings indicate that after transplantation, these cells home to kidneys at very low levels. Interaction of stromal derived factor-1 (SDF-1) with its receptor, CXCR4, is of pivotal importance in migration and homing. Recently, CXCR7 has also been recognized as another SDF-1 receptor that interacts with CXCR4 and modulates its functions. In this study, CXCR4 and CXCR7 were separately and simultaneously overexpressed in BALB/c bone marrow MSCs by using a lentiviral vector system and the homing and renoprotective potentials of these cells were evaluated in a mouse model of cisplatin-induced acute kidney injury. Using flow cytometry, immunohistochemistry, and real-time PCR methods for detection of GFP-labeled MSCs, we found that although considerably entrapped in lungs, native MSCs home very rarely to kidneys and bone marrow and this rate cannot be significantly affected by CXCR4 and/or CXCR7 upregulation. Transplantation of neither native nor genetically engineered MSCs ameliorated kidney failure. We concluded that overexpression of CXCR4 and CXCR7 receptors in murine MSCs cannot improve the homing and therapeutic potentials of these cells and it can be due to severe chromosomal abnormalities that these cells bear during ex vivo expansion.
Introduction
R
Although several studies have examined bone-marrow-derived mesenchymal stem cells (MSCs) for acute kidney failure, the absolute potential of these cells to recognize and locate in the site of injury needs further elucidation. Initial studies reported considerable repopulation of kidney cells with bone-marrow-derived cells following injury [10]; however, it is now believed that some of these studies had suffered from technical problems [11,12] and the repopulation of injured kidney cells with bone-marrow-derived cells is currently considered to be minimal [13 –15]. Interestingly, not only by systemic transplantation, but also by direct intra-parenchymal injection of MSCs after ischemic injury, homing of transplanted cells is reported to be very low and transient [11,16]. Therefore, it seems reasonable to assume that enhancing the homing potential of MSCs to preferentially reach the kidney may increase the renoprotective properties of these cells.
Several mediators and receptors are involved in homing of cells to sites of injury. Interaction between the chemokine stromal-derived factor 1 (SDF-1), also referred to as CXCL12, and its receptor CXCR4 is of pivotal importance in this process. Indeed, the local concentration of SDF-1 increases following tissue damage that recruits stem cells to the location of injury [17 –19]. For most cell therapy experiments, in vitro expansion of cells is unavoidable, but it is shown that expression of CXCR4 declines following a few passages in the culture [20,21], which probably decreases the homing and engraftment potentials. Therefore, overexpression of CXCR4 on the surface of stem cells [22,23] or increasing the levels of SDF-1 in the injured tissue [24 –26] has been examined by some investigators as strategies to overcome this limitation. In addition to chemotaxis and migration, SDF-1/CXCR4 axis has been shown to be a potential regulator of proliferation and survival of bone marrow stromal stem cells [27]. SDF-1 is also a ligand for another chemokine receptor, CXCR7, whose exact role and interactions with SDF-1 and CXCR4 are yet to be determined. It has been shown that CXCR4 and CXCR7 can form heterodimers but reports on the role of these heterodimers are controversial [28,29]. Mazzinghi et al. investigated the role of these 2 chemokine receptors in renal progenitor cells and have stated that both receptors are crucial for homing and therapeutic potentials of these cells, with CXCR7 being more involved in cell survival and adhesion to endothelium and CXCR4 in chemotaxis [30].
In this study, we hypothesized that overexpression of CXCR4 and/or CXCR7 in bone marrow MSCs might enhance the homing of these cells to injured kidneys; therefore, the therapeutic effects of these cells could be augmented. The surface expression of these 2 receptors was separately or simultaneously upregulated in mouse bone marrow MSCs by lentiviral vectors carrying the receptor genes. Using a mouse model of cisplatin-induced nephropathy, the homing and renoprotection potentials of these cells were assessed.
Materials and Methods
MSC isolation and characterization
After humanely sacrificing the mice, the ends of tibia and femur bones were clipped to expose the marrow. The bones were inserted into adapted centrifuge tubes as described previously [31,32] and centrifuged for 1 min at 400 g. The cell pellet derived from 2 tibia and 2 femur bones was suspended in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum, penicillin G (100 U/mL), streptomycin (100 μg/mL), as well as 0.25 μg/mL amphotericin B (all from Gibco-BRL, Grand Island, NY), and cultured in a 25-cm2 culture flask. Nonadherent cells were removed after 24 h. The medium was changed regularly every 3 days and, at ∼80% confluency, the cells were detached using trypsin-EDTA (Gibco-BRL) and re-plated. The cultures were maintained at 37°C and 5% CO2.
To induce osteogenic differentiation, the cells were treated with 10 mM beta-glycerol-phosphate (Merck, Darmstadt, Germany), 50 μg/mL ascorbic acid bi-phosphate (Sigma-Aldrich, St. Louis, MO), and 100 nM dexamethasone (Sigma-Aldrich) for 3 weeks. Osteogenic differentiation was assessed with Alizarin red staining. Adipocyte differentiation was achieved in the presence of 250 nM dexamethasone and 0.5 mM 3-isobutyl-1-methylxanthine (Sigma-Aldrich), for 3 weeks. Oil Red O staining was used to determine the accumulation of oil droplets in the cytoplasm. For differentiation to chondrocytes, 2×105 cells were centrifuged to form a pelleted micromass and then treated for 3 weeks with 10 ng/mL transforming growth factor -beta, 10 ng/mL basic fibroblast growth factor (both from Peprotech, Rocky Hill, NJ), 50 μg/mL ascorbic acid bi-phosphate, and 100 nM dexamethasone. Chondrocyte differentiation was assessed with Alcian blue staining, performed on 4-μm-thick sections.
The expression of surface markers was evaluated using monoclonal antibodies against mouse CD11b, CD29, CD31, CD34, CD44, CD45, CD105, CXCR4, and CXCR7 (all from eBioscience, San Diego, CA). The cells were detached with trypsin/EDTA and incubated with the specific antibodies or isotype control antibodies in 100 μL of 3% bovine serum albumin (Sigma-Aldrich) in phosphate-buffered saline (PBS; Gibco-BRL) for 1 h at 4°C. The cells were then fixed with 1% paraformaldehyde (Sigma-Aldrich) and analyzed with a PAS flow cytometry machine (Partec, Munster, Germany).
Production of lentiviral vectors and transduction of BALB/c MSCs
For gene delivery to MSCs, a second-generation lentiviral vector system [33] was employed. pLV-IRES-GFP, a bicistronic lentiviral vector in which transgene expression is regulated by EF-1 alpha promoter, was selected as backbone (kindly provided by Dr. S. Ghazizadeh). Murine CXCR4 cDNA was generated from total RNA isolated from a mixture of mouse tissues suspected to express CXCR4 (including eye, muscle, spinal cord, thymus, lymph nodes, and gut) by RT-PCR using specific primers. The PCR product was cloned in pTZ57R using a TA-cloning kit (Fermentas, Burlington, Canada) and then excised with SmaI restriction enzyme (Fermentas) and subcloned in pLV-IRES-GFP to generate pLV-CXCR4-IRES-GFP plasmid. Similarly, murine CXCR7 was amplified from mouse genomic DNA and inserted in pLV-IRES-GFP backbone to generate pLV-CXCR7-IRES-GFP construct.
To produce viral particles, psPAX2 (lentiviral packaging vector), pMD2.G (encoding VSV-G), and each of the lentiviral plasmids described previously were transfected to 293T packaging cells by calcium phosphate precipitation method. Medium was changed 16 h after transfection, and virus-containing supernatants were harvested 24, 36, and 48 h later, pooled, and concentrated by centrifugation at 40,000 g for 2.5 h at 4°C. The titer was determined by transduction of 293T cells with serial dilutions of the vector and assessment of eGFP expression by flow cytometry after 72–96 h.
BALB/c MSCs were transduced with pLV-IRES-GFP (encoding eGFP), pLV-CXCR4-IRES-GFP (encoding CXCR4 and eGFP separated by IRES sequence), or pLV-CXCR7-IRES-GFP (encoding CXCR7 and eGFP separated by IRES sequence) lentiviral vectors at the multiplicity of infection of 30 in the presence of 5 μg/mL polybrene followed by a second transduction after 48 h. Afterward, MSCs transduced with pLV-CXCR4-IRES-GFP, pLV-CXCR7-IRES-GFP lentiviral vectors, or both vectors will be referred to as CXCR4-MSC, CXCR7-MSC, or CXCR4&7-MSC, respectively. In this study, MSCs transduced with pLV-IRES-GFP lentiviral vector (referred to as GFP-MSC) and not-transduced MSCs (native MSCs) were included as controls.
Real-time PCR for CXCR4 and CXCR7
RNA was extracted with TRIzol (Sigma-Aldrich) and treated with DNase I (Sigma-Aldrich) before cDNA synthesis. Parallel reactions were run without adding reverse transcriptase (minus RT control) to check for contaminating DNA. Random hexamer primed cDNA synthesis was carried out using revert aid first-strand cDNA synthesis kit (Fermentas). The expression of CXCR4 and CXCR7 genes was quantified with QuantiFast SYBR green PCR kit (Qiagen, Valencia, CA) and Rotor Gene 6000 Real-Time PCR Machine (Corbett, Sydney, Australia). The temperature profile consisted of an initial step of 95°C for 10 min followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. Melting curve analysis and agarose gel electrophoresis were performed after the amplification. For quantitative gene expression analysis, ΔΔCt method was applied and β-actin was used as internal control. Each sample was examined in duplicate. Primer sequences are shown in the Supplementary Table S1 (Supplementary Data are available online at
Animal model of cisplatin-induced nephropathy and cell transplantation
Normal adult male BALB/c mice, weighing 20–25 g, were obtained from Pasteur Institute of Iran (Tehran, Iran). Animal care and experiments were carried out in accordance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals (U.S. Department of Health and Human Services Public Health Services, NIH Publication No. 8523, revised 1996) and approved by the Institutional Animal Care and Use Committee.
Kidney injury was induced in 2–3-month-old male BALB/c mice by subcutaneous injection of cisplatin as described previously [34,35] with some modifications. Briefly, 8 h before cisplatin administration, food and water were withheld. Mice were given 18 mg/kg cisplatin, after which the mice again had free access to food and water. The dose of cisplatin was chosen based on preliminary experiments. Twenty-four hours later, through tail vein injections, the mice were given 5×105 of one of the following cell types suspended in 0.5 mL PBS: CXCR4-MSCs, CXCR7-MSCs, CXCR4&7-MSCs, GFP-MSCs, or native MSCs. Another group of mice received 0.5 mL PBS alone (n=8 per group). Sixteen hours after cisplatin injection, blood samples were obtained; mice were humanely sacrificed; and kidneys were removed and kept in 10% buffered formalin for histopathologic analysis, in liquid nitrogen for DNA extraction, or in cold PBS for preparation of single-cell suspension for cytofluorometric analysis.
The physical activity of mice was scored according to a previously described method [36] (1: lazy, slow movement; 2: intermediate level of activity; 3: active movement or searching).
For survival analysis, 24 h after cisplatin injection, mice were divided into 6 weight-matched groups and received either PBS or one of the stem cell types as described previously (n=10 per group). These mice were inspected every few hours for more than 10 days and mortalities were recorded.
Histopathologic analysis
Hematoxylin and eosin–stained coronal sections of kidneys were examined in a blinded manner and scored for injury, as described previously [34] with some modifications. Briefly, random cortical fields were analyzed using a 40× objective. One hundred tubules were examined for each kidney section, and a score from 0 to 3 was assigned for each tubule: 0, normal histology; 1, tubular cell swelling, brush border loss, and nuclear condensation, with loss of up to one-third of the tubule nuclei; 2, same as for score 1, but greater than one-third and less than two-thirds of the tubular profile showing nuclear loss; and 3, greater than two-thirds of the tubular profile showing nuclear loss. The total score per kidney was calculated by addition of all 100 scores, with a maximum possible injury score of 300. To quantify the mean number of hyaline casts, a total of 40 random cortical fields in 2 sections for each kidney were assessed.
Biochemical analysis
Creatinine measurement in serum samples was performed through an enzymatic method by Cobas Integra analyzer (Roche, Indianapolis, IN). Urea measurement was performed manually with diacetyl monoxime method. Briefly, 2.5 mL of solution A [6.7% (v/v) diacetyl monoxime, 6.7% (v/v) thiosemicarbazide, and 0.5% (v/v) brij detergent in distilled water], 2.5 mL of solution B [12.8% (v/v) high-concentration sulfuric acid, and 0.1% (v/v) ferric chloride in distilled water], and 20 μL of serum samples were mixed and incubated in boiling water for 10 min and then cooled down to room temperature. Optical density was measured at 520 nm and urea levels were calculated based on the standard curve.
Assessment of homing by flow cytometry
The homing potential of the cells was examined with a quantitative method, as described previously [9,37,38] with some modifications. Briefly, before transplantation the cells were incubated with 25 μM CellTracker Green (Cambrex, East Rutherford, NJ) at 2°C −8°C for 1 h and then washed with PBS. Thirty-six hours after transplantation, kidneys and lungs were removed, minced and digested with 200 IU/mL collagenase I (Gibco-BRL), treated with RBC lysis buffer (Dako, Glostrup, Denmark), washed with PBS, and then analyzed with CyFlow machine and FloMax® software (Partec) to investigate for the presence of fluorescent-labeled MSCs in single-cell suspensions made from the tissues.
Assessment of homing by GFP immunohistochemistry
Five-micrometer sections of paraffin-embedded kidneys were incubated at 70°C for 40 min followed by putting in xylene for 2×5 min and then rehydrated in an alcohol series and water. Antigen retrieval was performed by incubation with 0.05% trypsin (Gibco-BRL) in 37°C for 25 min. A 1:1,000 dilution of chicken polyclonal antibody to GFP (Abcam, Cambridge, United Kingdom) was applied to slides and kept overnight in a wet box at 4°C. After washing, endogenous peroxidase was blocked with 3% H2O2 (Merck) and a dilution of 1:500 of HRP conjugated-rabbit polyclonal antibody to chicken Ig Y (Abcam) was applied for 1 h at 37°C. The immune complexes were visualized with 3, 3′-diaminobenzidine (Sigma-Aldrich). The sections then were counterstained with hematoxylin solution (Padtan Teb, Tehran, Iran), washed and dehydrated with ascending grades of alcohol, and mounted with Entellan (Merck). To assess the homing of transplanted cells in bone marrow, 4-μm sections of paraffin-embedded femur bones were processed with a similar GFP immunostaining protocol. However, for these samples, antigen retrieval was performed by boiling in citrate buffer (pH=6) for 30 min.
Mice receiving PBS or native MSCs served as negative control in this method. As well, to show that the method is operational, kidney-positive control samples were prepared by direct intra-parenchymal injection of GFP-labeled cells and rapid fixation in 10% formalin. In addition, to provide positive controls for bone marrow immunohistochemistry, a wild-type mouse was exposed to total body irradiation (a single dose of 8.2 Gy) and after 24 h was transplanted with whole BM from syngeneic GFP-transgenic mice.
Assessment of homing by GFP real-time PCR
After humanely sacrificing the animals, kidneys were removed and kept in liquid nitrogen until analysis. TissuLyser (Qiagen) was applied to make homogenates. DNA was extracted from tissue homogenates as well as MSC cultures with QIAamp DNA mini kit (Qiagen) according to the manufacturer's instruction. The presence and level of eGFP were determined with SYBR Premix Ex Taq™ kit (Takara Bio, Shiga, Japan) and Rotor Gene 6000 Real-Time PCR Machine (Corbett). TATA binding protein (TBP) was used as internal control. The temperature profile consisted of an initial step of 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. Melting curve analysis and agarose gel electrophoresis were performed after the amplification. Each sample was examined in duplicate. Primer sequences are shown in the Supplementary Table S1.
Apoptosis assay
TACS•XL®-Blue Label In Situ Apoptosis Detection Kit (Trevigen, Gaithersburg, MD), which is based on terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL), was used to detect apoptotic cells in 7-μm-thick kidney paraffin sections, according to manufacturer's instructions.
Statistical analysis
Statistical analysis was carried out using SPSS software version 13 (SPSS, Inc., Chicago, IL). Kruskal–Wallis H and Mann–Whitney U tests were used to assess differences between groups. For survival analysis, Kaplan–Meier method was used and groups were compared using the log-rank test. Values of P≤0.05 were considered statistically significant. Data are expressed as mean±standard error of mean.
Results
Isolated cells from bone marrow samples displayed the properties of MSCs
Bone marrow contents from BALB/c mice were collected, cultured, and after 24 h nonadherent population was removed. The remaining adherent cells were small and round in the first days (Fig. 1a) but later became larger and multiangular. The cells were expanded in normal culture conditions and after a few passages the cells had a uniform morphology (Fig. 1b). To confirm the identity of the isolated cells as MSCs, differentiation potential and surface markers were assessed. The cells could be differentiated to osteocyte lineage when cultured in favorable conditions as shown by Alizarin Red S (Fig. 1c) staining. In addition, the expanded cells had adipogenic and chondrogenic differentiation potentials as shown by Oil Red O (Fig. 1e) and Alcian blue (Fig. 1g) staining, respectively. Undifferentiated cells were used as negative controls for Alizarin Red S (Fig. 1d), Oil Red O (Fig. 1f), and Alcian blue staining (Fig. 1h). To determine the immunophenotype of the cells, the expression of some surface markers was examined by flow cytometry. The cells expressed CD29, CD44, and CD105 but were negative for CD45, CD11b, VEGFR2, CD31, and CD34 (Fig. 1i). This pattern of surface markers is compatible with previous studies on mouse MSCs [32,39,40]. Therefore, based on published guidelines [41], the isolated cells fulfill the criteria of MSCs.
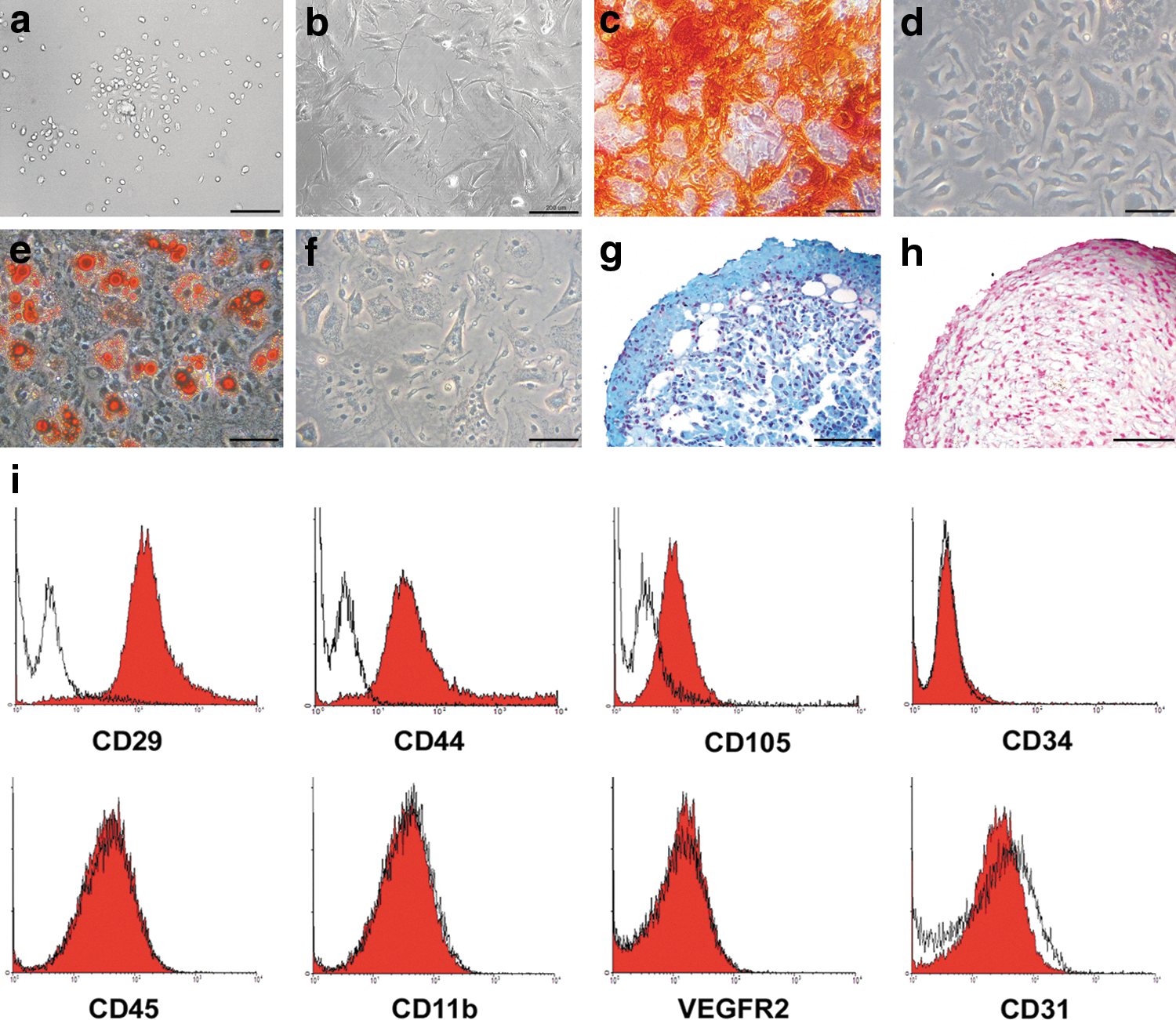
Isolation and characterization of BALB/c bone marrow MSCs. The isolated MSCs were round and small at 3 days after isolation
CXCR4 and CXCR7 were overexpressed by lentiviral transduction of MSCs
pLV-IRES-GFP (encoding IRES-eGFP), pLV-CXCR4-IRES-GFP (encoding CXCR4-IRES-eGFP), and pLV-CXCR7-IRES-GFP (encoding CXCR7-IRES-eGFP) plasmids (Fig. 2a) were used to produce lentiviral particles. BALB/c MSCs were transduced with pLV-IRES-GFP (GFP-MSCs), pLV-CXCR4-IRES-GFP (CXCR4-MSCs), pLV-CXCR7-IRES-GFP (CXCR7-MSCs), or both pLV-CXCR4-IRES-GFP and pLV-CXCR7-IRES-GFP lentiviral vectors (CXCR4&7-MSCs). In addition, a population of cells not genetically manipulated (native MSCs) was used as control. Real-time PCR revealed that the expression of CXCR4 mRNA was increased in CXCR4-MSC and CXCR4&7-MSC populations. Similarly, CXCR7 mRNA was overexpressed in CXCR7-MSC and CXCR4&7-MSC populations (Fig. 2b). As all vectors encoded eGFP, we looked at the expression of the transgenes by inspecting cells with a fluorescent microscope. As expected, native MSCs did not express eGFP, whereas the other populations did. GFP signal was brighter in GFP-MSCs compared with CXCR4-MSCs and CXCR7-MSCs (Fig. 2c). To confirm that the receptors are expressed at the surface of the cells, the expression of CXCR4 and CXCR7 was examined by flow cytometry in the 5 MSC populations. Cytofluorometric data for eGFP expression were in accordance to microscopic observations. CXCR4 was not expressed in native MSCs but its expression was significantly increased in CXCR4-MSC and CXCR4&7-MSC populations. In addition, native MSCs had a baseline expression of CXCR7 and this rate increased after transduction with pLV-CXCR7-IRES-GFP lentiviral particles in CXCR7-MSC and CXCR4&7-MSC populations (Fig. 2d).

CXCR4 and CXCR7 were upregulated in MSCs by lentiviral vectors. pLV-IRES-GFP—a lentiviral vector with eGFP gene, IRES sequence, and EF-1 alpha promoter—was used as backbone to generate pLV-CXCR4-IRES-GFP and pLV-CXCR7-IRES-GFP constructs
Both transduced and native MSCs had similar differentiation potentials and surface marker expression (data not shown), indicating that viral transduction did not alter the characteristic features of the cells.
Native and genetically manipulated MSC populations home to the injured kidney at very low levels, if at all
For in vivo studies, a mouse model of cisplatin-induced nephropathy was developed. To validate the method and make sure that kidney injury is reproducibly induced, biochemical and histopathologic parameters were measured at 60 h after cisplatin injection. The means of serum urea, serum creatinine, pathology score, hyaline casts, and weight loss were higher in the cisplatin-treated mice than in the control animals. As well, activity score was less in the cisplatin-treated animals (P<0.05 for all parameters; Fig. 3).
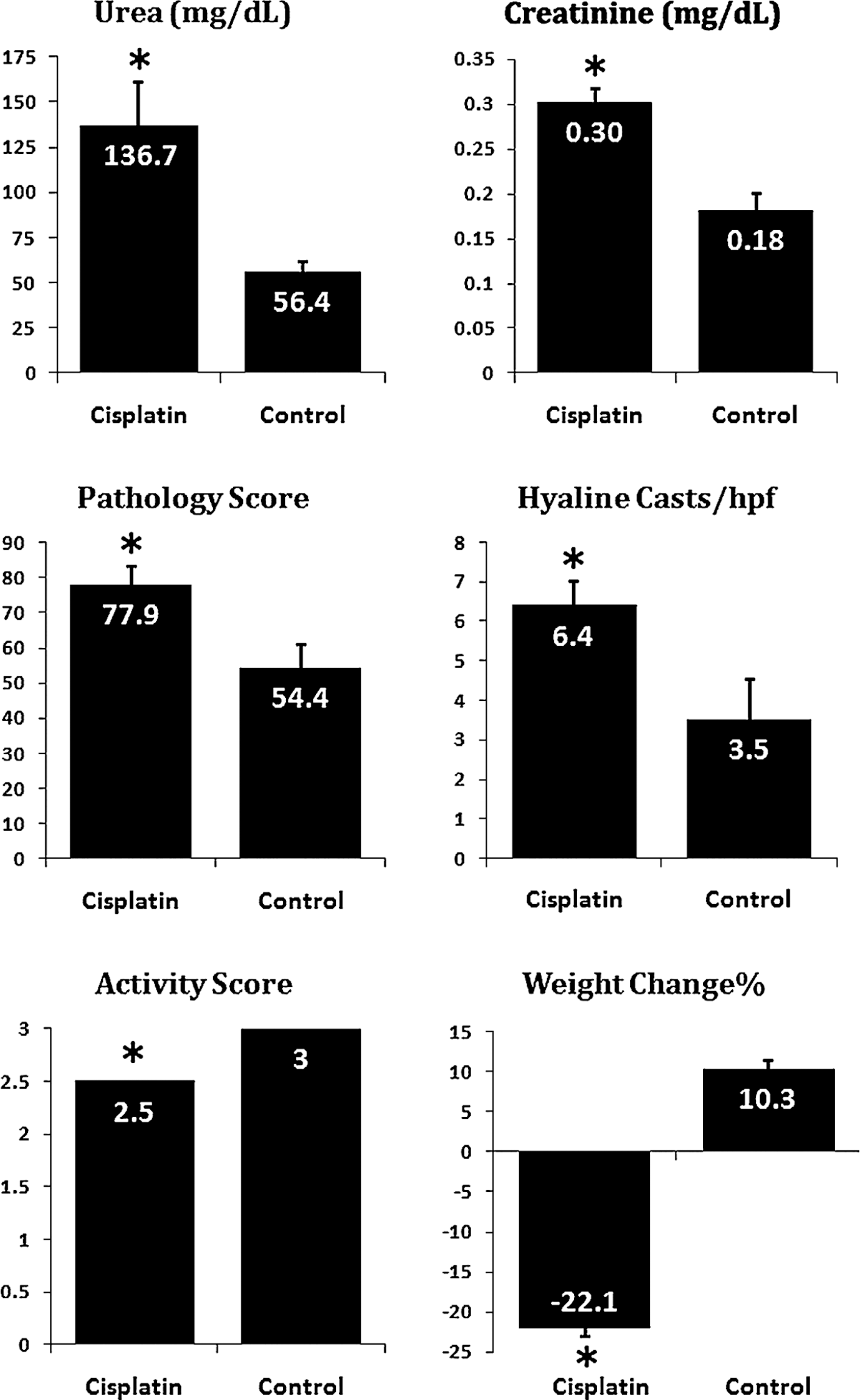
Validation of cisplatin-induced acute kidney injury mouse model. Mice were fasted for 8 h and then subcutaneously received 18 mg/kg cisplatin. After 60 h, serum urea and creatinine, kidney pathology score and cast formation, animal activity score, and weight loss were assessed. *In all of the parameters, cisplatin-treated mice had statistically significant differences (P<0.05) with normal controls (n=6 per group). Data are mean±SEM. SEM, standard error of mean.
To compare the homing potential of different MSC types, 24 h after cisplatin administration, mice were randomly divided into 6 experimental weight-matched groups and received either one of the MSC populations, or 0.5 mL PBS alone through tail vein injections. Thirty-six hours later, tissue samples were harvested and the engraftment rate of transplanted cells was assessed by a cytofluorometric quantitative method (Fig. 4a). Previous studies indicate that following systemic transplantation of stem cells, considerable amount of cells entrap in lungs [37,38,42]; therefore, we examined not only the homing potential of cells in the kidney as the site of injury, but also the entrapment of cells in the lung. As native MSCs did not show fluorescent signal and considering that the strength of GFP expression in genetically manipulated cells was not identical, all cell types were labeled with CellTracker Green, a fluorescent dye, just prior to transplantation. After harvesting kidneys and lungs from humanely sacrificed animals, they were digested with collagenase I and single-cell suspensions were analyzed for fluorescently labeled cells by flow cytometry. This assay could detect the presence of labeled cells in lungs, but we did not detect the homing of any MSC population in kidneys (Fig. 4b). To exclude the possibility of transient homing of the cells in kidneys, we repeated the experiment by performing the assessments only 10–12 h after cell transplantation. In this second experimental design, the number of transplanted cells was also increased to 1.4×106 per animal, delivered in 2 episodes (0.7×106 each) with a 2 h-interval (Fig. 4c). Even with this protocol, we could not detect the labeled cells in kidneys but they could again be detected in lungs (data not shown). Therefore, homing of the cells to kidneys is below the detection limit of this assay.
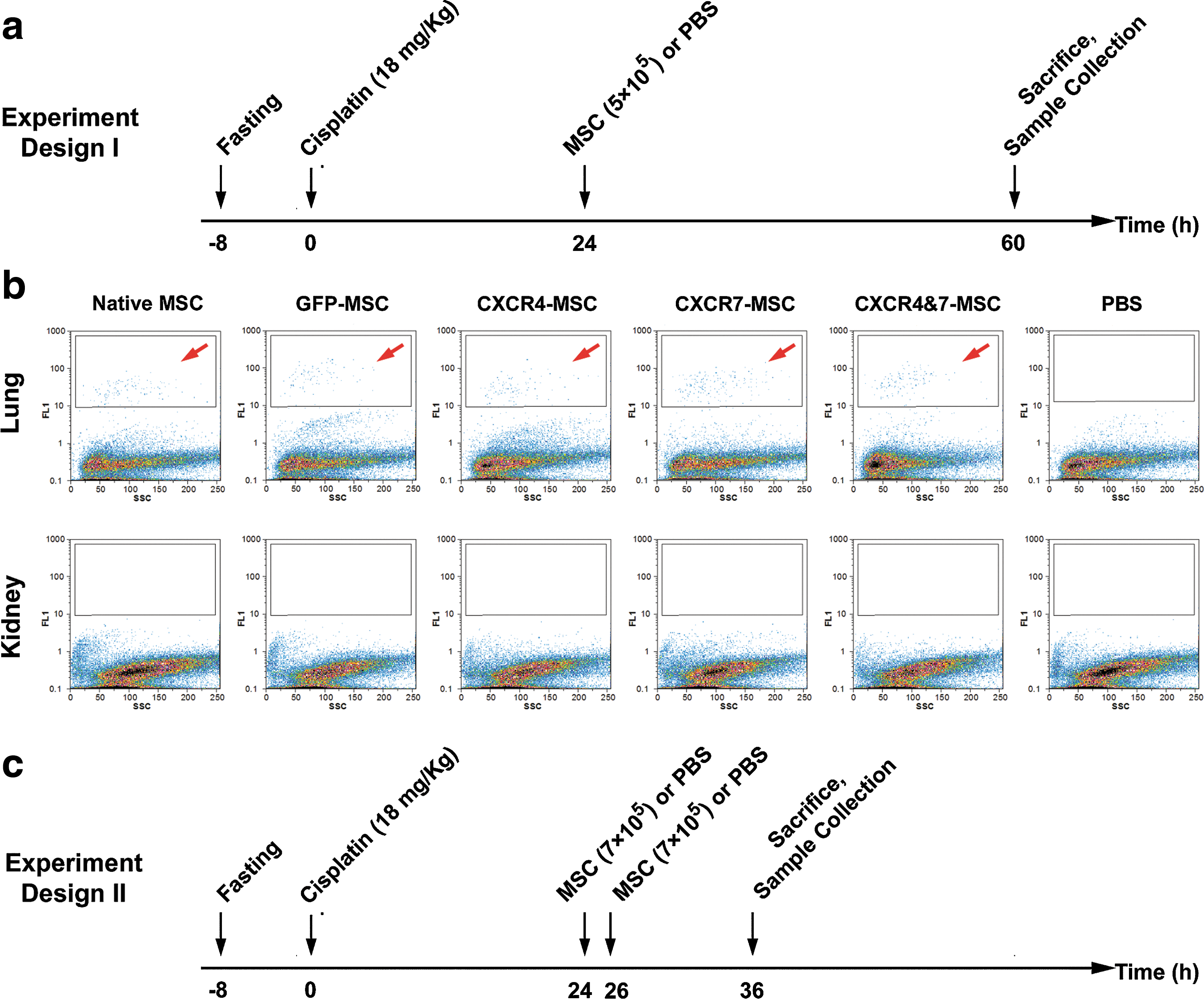
Assessment of homing by flow cytometry. Nephropathy was induced by injection of 18 mg/kg cisplatin after 8 h of fasting. In the next day, various MSC types were transplanted at the dose of 5×105 cells per mouse. Thirty-six hours after cell transplantation, kidneys and lungs were harvested. Single-cell suspensions made from these tissues were assessed by flow cytometry for the presence of fluorescent-labeled cells
As flow cytometry could not detect the homing of transplanted cells, we tried to perform this assessment by GFP immunohistochemistry that is more commonly used for this purpose. To check the validity of this assay, a kidney tissue with direct intra-parenchymal injection of GFP-MSCs was examined in parallel with other samples. The presence of GFP-labeled cells could clearly be detected in this positive control sample (Fig. 5a). In addition, kidneys of mice that had received PBS or native MSCs were completely negative (Fig. 5b, c). After validation of the method with these positive and negative controls, we assessed the test slides. To increase the sensitivity, we processed about 12 coronal sections for each tissue sample and all microscopic fields were carefully inspected by 2 investigators. However, we could not detect even 1 positive cell in any of the sections (Fig. 5d–g).
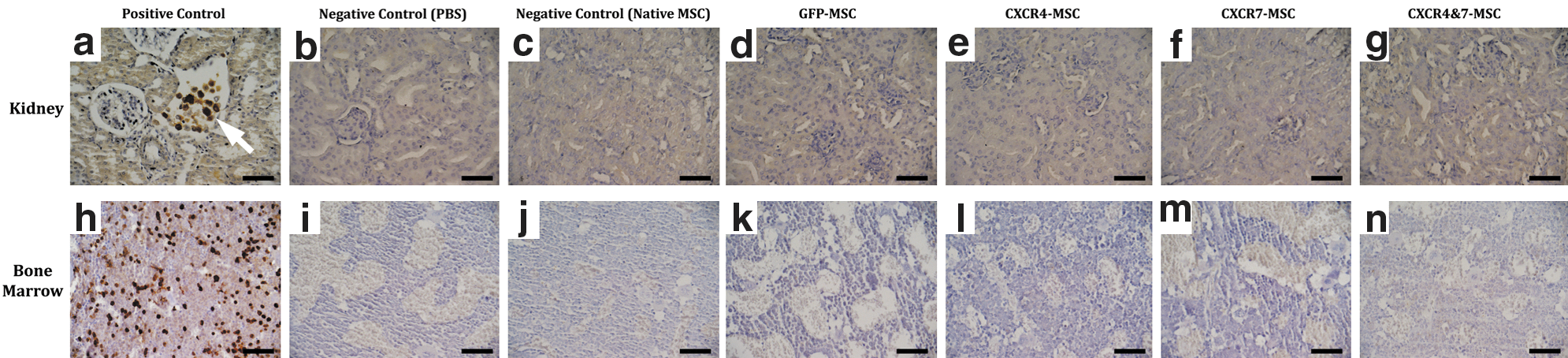
Assessment of homing by GFP immunohistochemistry. To investigate the homing of transplanted cells to kidneys and bone marrow, tissues were harvested 36 h after cell transplantation and GFP immunohistochemistry was performed. A kidney tissue with direct intra-parenchymal injection of GFP-labeled MSCs served as positive control in this assay. The presence of GFP-positive cells (arrow) was clearly detectable in this sample
To verify flow cytometry and IHC results and to increase the sensitivity, we looked for any trace of transplanted cells in kidneys by real-time PCR. DNA was extracted from kidney tissues as well as from the various MSC types and real-time PCR was performed for detection of eGFP. TBP that was selected as house-keeping gene was positive in all samples but eGFP could not be detected in DNA extracted from any tissue sample. The only exceptions were 1 kidney sample from mice receiving CXCR4-MSCs and 1 sample from CXCR4&7-MSC group that showed a very weak positive signal for eGFP (threshold cycle: 38). The specificity of these 2 positive signals was confirmed by melting curve analysis and visualization on agarose gel (Supplementary Fig. S1).
As we found that the transplanted cells did not home to kidneys, we were interested to see whether they instead homed to bone marrow. Therefore, femur bones were processed for GFP immunostaining. The femur of a mouse that had been irradiated and transplanted with the bone marrow of syngeneic GFP-transgenic mice served as a positive control sample in this assay. The presence of GFP-expressing cells in this sample was demonstrated (Fig. 5h) and as expected, PBS and native MSC group samples were negative (Fig. 5i, j). GFP-labeled cells were undetectable in the samples of GFP-MSC, CXCR4-MSC, CXCR7-MSC, and CXCR4&7-MSC groups (Fig. 5k–n).
Transplantation of the native and genetically modified MSCs fails to ameliorate renal failure
To assess the therapeutic effects of native and genetically engineered stem cell populations, blood and tissue samples were harvested 36 h after cell transplantation and biochemical and histopathologic analyses were performed (n=8 per group). Multiple group comparison of means of serum urea, serum creatinine, pathology score, and hyaline casts showed that for none of these parameters, the difference between the 6 experimental groups reached a statistically significant level (Fig. 6). Actually, assessment of kidney function and structure showed that the transplantation of different stem cell types did not have any therapeutic effect above the administration of PBS alone.

Transplantation of neither native nor CXCR4/CXCR7 overexpressing cells was renoprotective. Blood and kidney tissues were harvested at 36 h after cell transplantation. Serum urea, serum creatinine, pathology score, and hyaline casts/hpf were similar in all the experimental groups and the differences did not reach a statistically significant level, indicating that cell transplantations were not beneficial. Data are mean±SEM (n=8 per group).
To examine the effect of our interventions on mice survival, a group of mice with cisplatin-induced nephropathy was inspected for mortality for several days after stem cell or PBS administration (n=10 per group). Most animals died between 80 and 160 h after cisplatin injection. However, in different groups, 0%–20% of mice spontaneously improved after a period of renal failure and survived (Fig. 7). In agreement with biochemical and histopathologic examination, the log-rank test showed that the difference of survival in the 6 groups did not reach a statistically significant level.
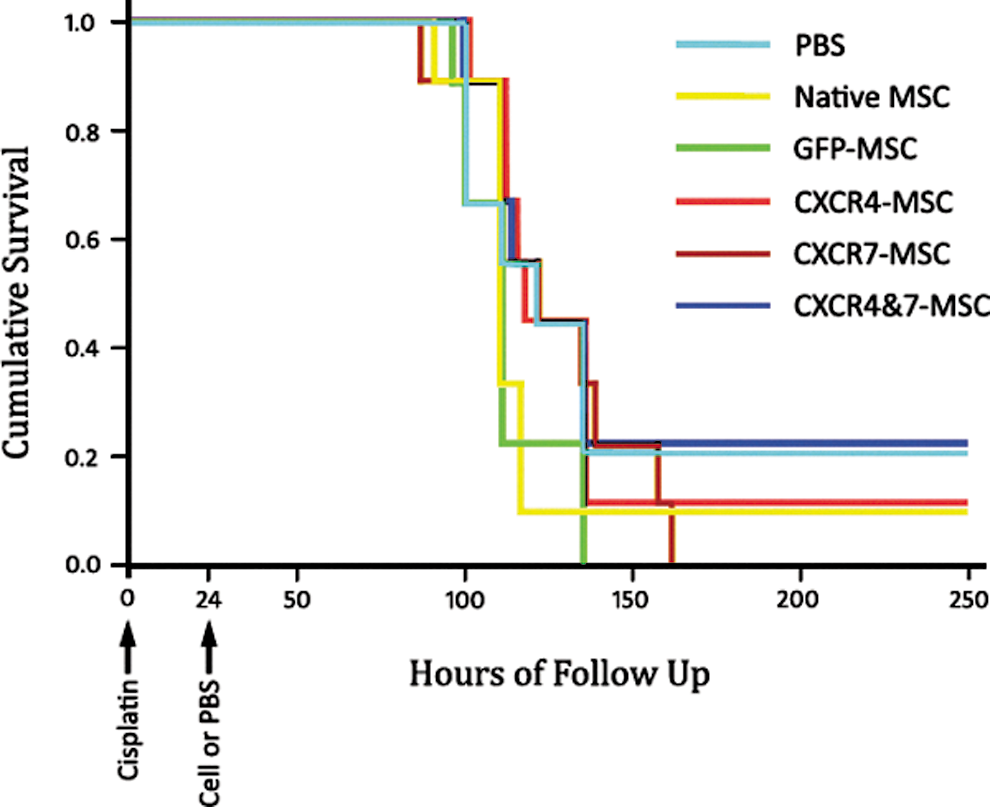
Transplantation of different stem cell types had no effect on animal survival. Twenty-four hours after cisplatin injection, mice were allocated in 6 weight-matched groups and received PBS or one of the MSC types. The survival probability is shown in Kaplan–Meier curves. The survival probability of mice in the 6 groups was not statistically different. Color images available online at
To investigate the mechanism of kidney injury, we used a TUNEL-based kit that detects DNA breaks as markers of apoptotic cells. According to the manufacturer's instruction, a sample treated with nuclease to produce artificial DNA nicks was used as positive control in this assay. Most nuclei of this sample showed blue stain deposit showing sites of DNA breaks (Fig. 8a, b). However, apoptotic cells were not detectable in kidney sections from any of the experimental groups (Fig. 8c–h).
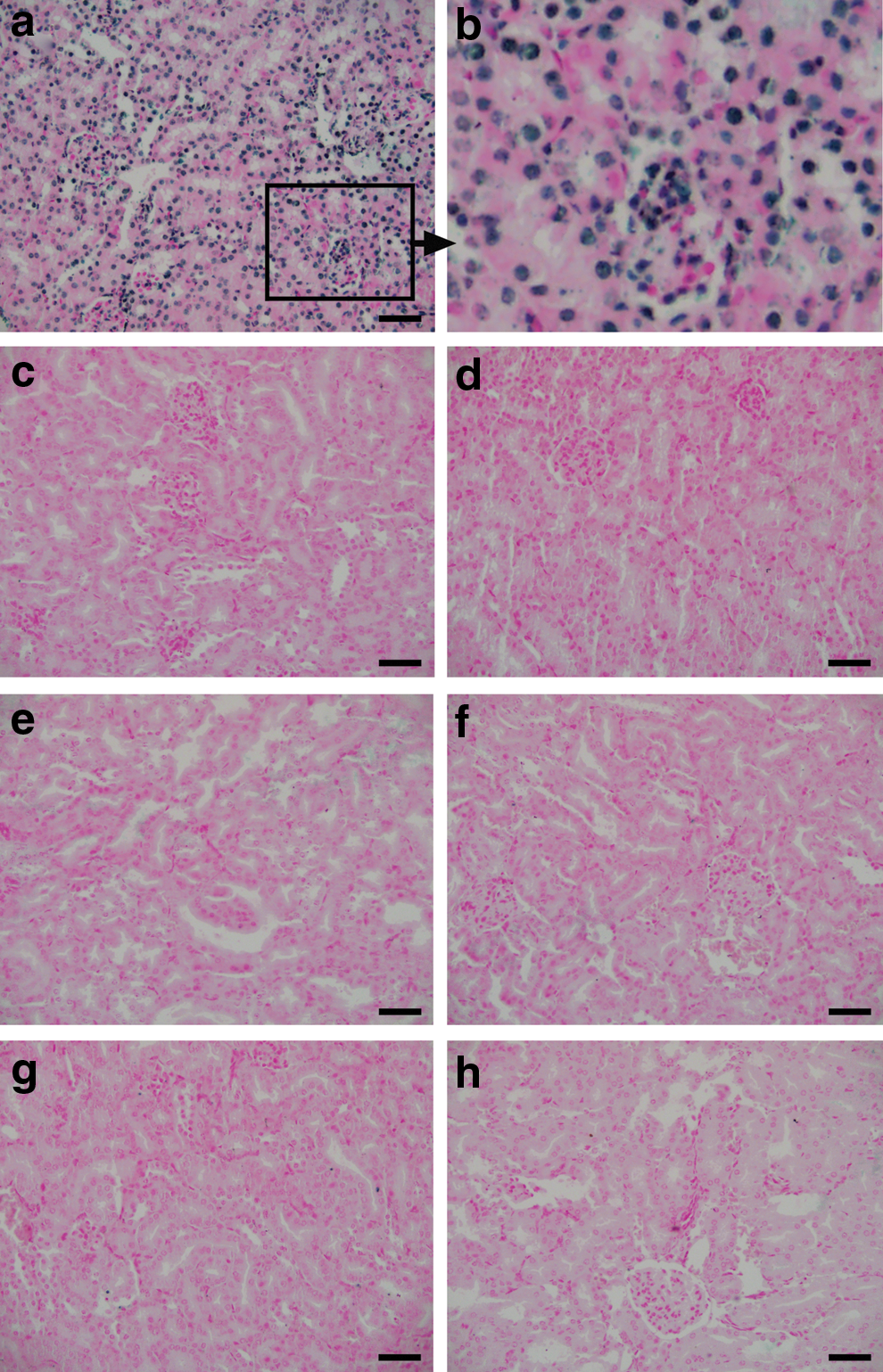
Apoptosis was not responsible for tissue damage in our animal model of kidney injury. Apoptosis was assessed by a TUNEL method. The nuclei in positive control sample showed DNA nicks
Discussion
In the current study, CXCR4 and CXCR7, the chemokine receptors responding to SDF-1, were overexpressed in mouse bone marrow MSCs and homing potential and renoprotective features of these cells were assessed in a mouse model of cisplatin-induced kidney injury. Our data indicate that MSCs very rarely home to kidneys and overexpression of neither CXCR4 nor CXCR7 alters this low engraftment rate. We also found that the transplantation of neither native nor genetically modified MSCs improved kidney performance, tissue injury, and the survival of mice with renal failure.
Selection of lentiviral vector system for gene transfer to MSCs was based on our previous study indicating that MSCs are resistant to common nonviral gene delivery methods [43]. The efficiency of lentiviral vectors for overexpression of CXCR4 and CXCR7 at mRNA and protein levels was confirmed by real-time PCR and flow cytometry techniques. Transduction efficiency was further confirmed by assessment of eGFP expression. Notably, the expression of this protein in CXCR4-MSCs and CXCR7-MSCs was less than in GFP-MSCs, consistent with a previous report indicating that in bicistronic systems that IRES segment locates between 2 open-reading frames, the expression of the second protein is less than the first one [44]. Taken together, based on flow cytometry and quantitative PCR data as well as fluorescent microscopy, acceptable expression level of the transgenes was achieved. In addition, similar to a previous report [23], we found that viral transduction does not affect the surface markers and plasticity of the cells.
Using robust assessments, Togel et al. [13] and Duffield et al. [11] in 2 independent studies showed that homing of bone-marrow-derived cells in kidneys following injury occurs at very low levels. The low homing rate of transplanted cells in kidneys is also reported by other investigators [14,16]. In agreement with these reports, by using 3 validated methods, we found that native MSCs home to the injured kidneys at almost undetectable levels. Additionally, at least in the examined time point, these cells even do not home to their “mother land,” the bone marrow space. Actually, our data, in line with some other studies, indicate that they become mainly entrapped in lungs after systemic intravenous injection [37,38,42,45]. Surprisingly, we found that neither separate nor simultaneous overexpression of CXCR4 and CXCR7 significantly improves the homing potential of MSCs to kidneys and bone marrow. This finding is in accordance with 2 previous reports indicating that migration of intravenously injected hematopoietic stem cells to kidneys with ischemic injury could not be altered by manipulation of SDF-1/CXCR4 axis [46,47]. In sharp contrast, some other researchers have shown that bone marrow cells migrate to kidneys with ischemic injury and this migration is SDF-1/CXCR4 dependent [48]. The findings of Bonig et al. [49] shed light on these discrepant findings. Their results indicate that cooperative and compensatory mediators are involved in migration and homing that can flexibly be substituted. Indeed, they have shown that VLA-4/VCAM-1 pathway can compensate for SDF-1/CXCR4 axis and homing of hematopoietic cells can occur even in the absence of SDF-1-induced signaling. They suggested that cytokine milieu and probably source of cells determine the main operating pathway in cell migration.
To investigate the renoprotective properties of different MSC populations, biochemical and histopathologic analyses were performed at 36 h after cell transplantation. We found that transplantation of native MSCs, MSCs transduced with lentiviral vector backbone, and MSCs with separate or simultaneous overexpression of CXCR4 and CXCR7 failed to ameliorate the kidney injury. Likewise, stem cell transplantation did not increase the survival of mice suffering from renal failure. TUNEL assay for kidney sections showed that in all experiment groups apoptotic cells were undetectable, indicating that the kidney injury in our animal model was not mediated by apoptosis and probably necrosis is the main mechanism of tissue damage. This could be due to the relatively high dose of cisplatin in our study as previous studies have shown that cisplatin at low concentrations induces apoptotic cell death of kidney-cultured cells while at higher concentration necrosis proceeds [50,51]. Our finding that bone marrow MSC transplantation does not improve renal failure is in line with the data of some other investigators who examined the efficacy of autologous MSC transplantation for kidney protection in a large animal model of acute renal failure [52]. However, it is in sharp contrast with several previous studies that have shown that transplantation of MSCs is renoprotective [4,13,42,53 –55]. These controversies can be explained by differences in experimental design, such as the protocol of cell transplantation, the animal strain, and the type and severity of kidney injury.
In conclusion, the results of this study indicate that the engraftment rate of murine bone marrow MSCs in injured kidneys following systemic infusion is very low, if at all, and it cannot considerably be influenced with separate or simultaneous overexpression of CXCR4 and CXCR7. In addition, we found that the transplantation of neither native nor genetically engineered MSCs improves renal failure in terms of survival and biochemical and histopathologic measures. Previous studies by our group and other investigators have shown that after a period of ex vivo expansion, murine bone marrow MSCs bear severe chromosomal abnormalities, such as balanced and unbalanced translocations, deletions and amplifications, as well as other morphological and functional alterations [56 –59]. With this huge amount of genomic instability, it would not be surprising that re-stabilization of 2 chemokine receptors cannot restore complex cell functions, such as migration, homing, and repair, that are dependent on the coordinated actions of numerous cellular elements. This study evokes several questions to be answered on stem cell homing as a crucial obstacle in clinical stem cell transplantation and suggests stimulation of resident stem cells as a more feasible approach instead of transplantation of ex-vivo-expanded stem cells for kidney repair.
Footnotes
Acknowledgments
This study was supported by grants from Pasteur Institute of Iran and Stem Cell Technology Research Center, Tehran, Iran. The authors are grateful to Dr. Soosan Ghazizadeh (Stony Brook University, Stony Brook, NY) for technical advice during generation of lentiviral vectors and to Dr. Sima Rafati (Pasteur Institute of Iran) for wise comments on experiment design. We also appreciate the assistance of Dr. Minoo Saeidi in this study.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.