
Abstract
Inducing a stable and predictable program of neural cell fate in pluripotent cells in vitro is an important goal for utilizing these cells for modeling human disease mechanisms. However, the extent to which in vitro neural specification recapitulates in vivo neural specification remains to be fully established. We previously demonstrated that in the mouse embryo, activation of fibroblast growth factor (FGF) signalling promotes definitive neural stem cell (NSC) development through the upregulation of the transcription factor Zfhx1b. Here, we asked whether Zfhx1b is similarly required during neural lineage development of embryonic stem (ES) cells. Zfhx1b gene expression is rapidly upregulated in mouse ES cells cultured in a permissive neural-inducing environment, compared to ES cells in a standard pluripotency maintenance environment, and is potentiated by FGF signalling. However, overexpression of Zfhx1b in ES cells in maintenance conditions, containing serum and leukemia inhibitory factor (LIF), is sufficient to induce Sox1 expression, a marker found in neural precursors and to promote definitive NSC colony formation. Knockdown of Zfhx1b in ES cells using siRNA did not affect the initial transition of ES cells to a neural cell fate, but did diminish the ability of these neural cells to develop further into definitive NSCs. Thus, our findings using ES cells are congruent with evidence from mouse embryos and support a model, whereby intercellular FGF signaling induces Zfhx1b, which promotes the development of definitive NSCs subsequent to an initial neural specification event that is independent of this pathway.
Introduction
M
Previous mouse studies have highlighted the presence of another type of NSC, that is, the primitive neural stem cell (pNSC), which shows characteristics of an intermediate state between ES cell and the more committed (or definitive) NSC [4]. It has been shown that the initial specification of the pNSC was dependent on BMP inhibition and the mitogenic/survival effects of leukemia inhibitory factor (LIF), but not on fibroblast growth factor (FGF) signaling or Notch signaling [4,7]. The addition of exogenous LIF is sufficient to promote pNSC-derived clonal colony formation [4]. In contrast, definitive NSCs are dependent on both Notch for their maintenance and FGF for clonal colony formation, but LIF signaling is not required [4,7]. This differential growth factor requirement for pNSC-versus-NSC colony formation provides a convenient assay for studying these 2 stem cell populations during NSC lineage ontogeny [8].
Neural lineage commitment is a multistep process requiring complex regulation of gene expression as cells transit from an ES cell identity to a tissue-specific NSC identity [8 –10]. It is generally accepted that any method of directed differentiation in vitro should recapitulate developmental programs of lineage specification that would normally occur in vivo, yet this goal often yields to the equally important objective of generating large numbers of neural cells for application in medicine. Nonetheless, in vitro differentiation of ES cells into stem/progenitor cells of the appropriate tissue in a robust, predictable, and stable manner could be achieved by drawing on the developmental biology literature.
Evidence from vertebrate neural-induction studies in chick [11], frog [12 –15], fish [16], and human [17,18] suggests that FGF signaling, and likely its downstream effector, Zfhx1b, is required for the specification of neural tissues. The mouse Zfhx1b encodes a bipartite zinc-finger homeobox transcription factor and is one of the 2 members of the vertebrate δEF1/ZFH protein family [19 –21]. Its protein product, also referred to as Smad-Interacting Protein 1 (SIP1), was originally identified as a Smad1 interactor through a yeast 2-hybrid screen [22] and has since been implicated to downregulated the BMP-signaling pathway through its interaction with SMAD1 in the Xenopus embryo [12], thereby functioning as an essential component of early neural induction. In the chick, Zfhx1b has been shown to be required for neural plate formation, as well as to suppress mesoderm formation through its downregulation of Brachyury [11]. Expression of mouse Zfhx1b is initiated at 7.5 dpc, and a knockout mutation in this gene results in a lack of neural tube closure, shortened somites, failure to turn, and death by 9.5 dpc [20]. Results from these notable studies suggest a potential role for Zfhx1b in the neural lineage specification. We have previously reported that activation of FGF signaling promotes NSC development within the mouse ectoderm, and described a novel role for the transcription factor Zfhx1b, which we have shown to act downstream of FGF signaling [23]. Together, these studies suggest that definitive NSC fate specification, but not pNSC fate specification, in the mouse anterior neurectoderm is facilitated by FGF-dependent activation of Zfhx1b [23].
Wnt [24 –26] and/or BMP inhibition [17,27,28] in ES cell cultures results in a clear enhancement of neural specification and neuronal differentiation, but there are conflicting views over whether FGF acts a positive inducing signal during neural cell fate specification [17,29 –31]. Recent findings in mouse ES cells suggest that FGF-induced Erk1/2 signaling controls the transition from a FGF5+ neurectoderm identity (perhaps comparable to a pNSC identity) to a definitive neural identity, and in its absence causes the maintenance or expansion of this neurectoderm state [32], consistent with the results from our previous ex vivo studies [23]. Here, we wanted to expand on this previous work to address 2 related questions: (1) does FGF signaling regulate the expression of Zfhx1b during neural specification of ES cells? and (2) what is the functional role of Zfhx1b during differentiation from a pluripotent ES cell to a definitive NSC?
Materials and Methods
ES cell maintenance and neural differentiation
Mouse ES cell lines R1 (a gift from Dr. Andras Nagy) and Sox1:GFP (46C) (a gift from Dr. Austen Smith) were used in this study and were maintained on mitomycin-C-treated mouse embryonic fibroblasts in a standard ES cell medium [Dulbecco's modified Eagle's medium (DMEM) high-glucose, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, 100 mM beta-mercaptoethanol, 15% fetal bovine serum, 50 μg/mL penicillin and 50 μg/mL streptomycin (all Gibco, Invitrogen), and 1,000 U LIF/mL (Chemicon)]. Before differentiation, cells were dissociated into a single-cell suspension using 0.05% Trypsin-EDTA (Invitrogen), feeder-depleted for 30 min on uncoated plates, and plated at 10 cells/μL (low density) or 50 cells/μL (high density) on laminin/fibronectin (BD Biosciences)-coated culture slides (Nunc) in an ES cell medium for 24 h and then washed in Dulbecco's phosphate buffered saline (DPBS) (Invitrogen) and changed into a neural medium (NM) consisting of DMEM/F12 (1:1), 0.6% D-glucose, 5 mM HEPES, 3 mM NaHCO3, 2 mM glutamine, 25 μg/mL insulin, 100 μg/mL transferrin, 20 nM progesterone (all Invitrogen), 60 μM putrescine (Sigma Aldrich), and 30 nM sodium selenite (Sigma Aldrich). Cells were fixed for immunohistochemistry after 24 h or 4 days, analyzed by fluorescence-activated cell sorting (FACS) using anti-GFP or anti-Oct4 antibodies, or collected for reverse transcription (RT)-polymerase chain reaction (PCR) analysis after 4 or 24 h in NM. Treatment conditions included FGF8 (20 ng/mL), SU5402 (5 μM in 0.05% DMSO; Calbiochem), WNT3A (100 ng/mL), DKK1 (100 ng/mL), BMP4 (10 ng/mL), SB431542 (5 μg/mL in 0.05% DMSO; Invivogen), or Chordin (100 ng/mL). WNT3A and DKK1 were also used at 200 ng/mL each in separate experiments. All recombinant proteins were obtained from R&D Systems. About 1× DPBS (Invitrogen) as well as 0.05% DMSO (Sigma Aldrich) were used as controls.
NSC colony assay
NSC colony assay performed as previously described [4]. ES cells as a single-cell suspension were plated onto uncoated culture slides (Nunc) or 96-well plates (Corning) at 10 cells/μL in NM with the addition of 1,000 U/mL LIF (Chemicon), or FGF2 (10 ng/mL; R&D Systems) and heparin (2 μg/mL; Sigma Aldrich). The medium was partially replenished every 2 days. Colonies were quantified at 7 days after plating with at least 3 replicates per experiment, and n=3 separate experiments were performed. Means and standard deviations were calculated for the control and experimental groups. Statistical analyses between groups were performed using the Student's t-test. Differences were regarded as significant for P<0.05.
Overexpression
The full-length mouse Zfhx1b construct was obtained through Origene (NM_015753), and the DsRed overexpression construct was from Invitrogen. Transfections were performed using Lipofectamine 2000 (Invitrogen) using a 1 μg/well DNA concentration in 96-well plates or culture slides at time of plating. Cells were washed after 24 h, and the medium was replenished with the indicated growth factors or inhibitors. For overexpression experiments in ES cell maintenance cultures, the number of cells positive for the fluorescent reporter gene (DsRed) was estimated by counting live cells in 2 randomly selected counting arenas per well after 24 h after transfection. A total of n=6 experiments were performed. GFP+ cells were either counted in live cultures (as a percentage of DsRed+ cells) in the control of Zfhx1b transfection groups (n=3 experiments with 2 replicates each) and with a transfection efficiency of ∼50%, or cells were fixed in 4% PFA and immunostained with anti-Oct4 (1:500), anti-GFP (1:1,000), and Hoechst (n=3 experiments with 3 replicates each) and with a transfection efficiency of ∼15%. Here, GFP+ cells were quantified as a percentage of Hoechst+ cells in 2 randomly selected counting arenas per well using confocal imaging (Leica SP5). For colony-forming experiments, GFP+ colonies were visualized and counted at 7 days after plating using a fluorescent compound microscope (Leica 4500B).
In situ hybridization
Plasmids containing Zfhx1b (a gift from Dr. Pzemko Tylzanowski) were used to generate antisense RNA probes using digoxigenin-11-uridine 5’-triphosphate (Roche) following a standard protocol. Cells were fixed in 4% paraformaldehyde after 4 days culture in NM, FGF8, or SU5402, and in situ hybridization was performed. Digoxigenin-labeled probes were detected with an anti-DIG alkaline phosphatase-coupled Fab fragment (1:2,000, Roche). The Alkaline Phosphatase reaction was performed using BM Purple (Roche).
Reverse transcription-quantitative polymerase chain reaction
Cells were collected by trypsinization, and total RNA was extracted with the mini-to-midi RNA extraction kit (Invitrogen). First strand cDNAs were reverse transcribed from oligo(dT)12–18-primed total RNA (DNase treated, 0.5 μg; New England Biolabs) using Superscript III (Invitrogen). Semiquantitative and quantitative RT-PCRs were performed as described [23]. Annealing temperatures and cycle number for each primer pair were determined using gradient PCR (MJ Research). Products were resolved by electrophoresis on a 1% agarose gel with ethidium bromide (Sigma Aldrich). Semiquantitative RT-PCR was quantified using the gel analyzer plug in for ImageJ (
Immunohistochemistry
Cells were fixed in 4% paraformaldehyde (Sigma Aldrich) and permeabilized with 0.3% Triton X-100 (Sigma Aldrich). About 5% normal goat serum (Invitrogen) was used as blocking agent. Primary antibodies were used included anti-Oct4 mouse monoclonal (1:500, gift from Dr. W. Stanford) and anti-GFP-Alexa 488 rabbit polyclonal (1:1,000; Invitrogen). Secondary antibodies were used at 1:200, and all the secondary-only controls were negative for staining. Cell nuclei were visualized with Hoechst (Sigma Aldrich).
siRNA knockdown
Transient knockdown of Zfhx1b and GAPDH was carried out using Zfhx1b siRNA (s76954), GAPDH siRNA (4390849), and Scrambled siRNA (Scr) Negative Control No. 1 (4390843) (all Ambion Silencer Select Predesigned siRNAs) by Lipofectamine 2000 (Invitrogen) transfection following the manufacturer's suggestions in Sox1:GFP ES cells. A dose–response experiment was carried out to determine the optimal concentration of siRNA for knockdown. All groups were cotransfected with 1 μg pCMV-DsRed overexpression construct, including the control to visualize the transfection efficiency. Successful transfection occurred when at least 50% of the population was transfected. RT-qPCR was used to monitor the knockdown efficiency, and the FACS analysis was performed at various time points after knockdown to quantify GFP+ cells. For assaying FGF2-dependent (definitive NSC) colony formation, pNSC colonies were generated from the Sox1:GFP ES cell line in serum-free media containing 1,000 U/mL LIF (Chemicon). GFP+ colonies were selected and dissociated into a single-cell suspension using 0.05% Trypsin-EDTA (Invitrogen) for 5 min at 37°C, triturated, and filtered using a 40-μm cell strainer (BD Biosciences). Cells were then transfected as mentioned above, and cultured in serum-free media containing FGF2 and Heparin as mentioned above. Cells were washed after 24 h. SU5402 was only added during the first 24 h after transfection. Five days after transfection, GFP+ colonies were identifiable and quantified (see NSC colony assay above).
Fluorescence-activated cell sorting
Analytical flow cytometry was performed on a FACSCalibur flow cytometer (BD Biosciences) to evaluate GFP+ cells derived from Sox1:GFP ES cells. Cells were fixed in 4% paraformaldehyde (Sigma Aldrich) and were permeabilized 0.3% Triton X-100 (Sigma Aldrich) for 10 min on ice and then washed with PBS. The cell pellets were resuspended in anti-GFP-Alexa 488 rabbit polyclonal (1:200; Invitrogen) diluted in an FACS buffer containing 1% w/v bovine serum albumin and 2 mM EDTA in PBS for 30 min on ice. The negative control for this analysis was undifferentiated ES cells. Alternatively, cells were suspended in anti-GFP rabbit polyclonal (1:200; Invitrogen) and probed with Alexa-488 anti-rabbit secondary with a secondary-only control as a negative control. For the 4-day NM experiment, anti-Oct4 mouse monoclonal was included with the primary antibody at 1:100 and probed with Cy5-anti mouse secondary (1:100). Cells were washed in PBS and analyzed. Undifferentiated Sox1:GFP ES cells were used as a negative control. The data were analyzed and plotted using Flo-Jo Software (Treestar, Inc., version 7.5). Three separate experiments (n=3) were performed with 2 replicates for each time point analyzed. Means and standard deviations were calculated for the control and experimental groups. Statistical analyses between groups were performed using the Student's t-test. Differences were regarded as significant for P<0.05.
Results
Zfhx1b expression is upregulated during neural specification of ES cells and can be potentiated by FGF and Chordin
Previous studies showed that the ES-to-neural transition in high-density serum-free cultures over the course of several days requires endogenous FGF signaling [29,32]. We asked whether the dependency of FGF to promote neural specification over this relatively long time frame correlated with an induction of Zfhx1b gene expression. First, we cultured mouse ES cells harboring a Sox1:GFP transgene reporter (line 46C) or R1 ES cells for 4 days in a serum-free medium supplemented with N2 and B27 [3,4] at a relatively high initial cell density of >50 cells/μL [29,32] in the presence or absence of exogenous FGF8 or SU5402, an FGFR inhibitor [33], to confirm whether neural cell development under these conditions required FGF signaling. We quantified cell fate changes using FACS. Consistent with previous observations, after 4 days, there was a significant decrease in the number of Oct4+ cells by ∼60% in NM cultures (Fig. 1A, F) when compared to ES cell maintenance cultures (Fig. 1D, F). In contrast, there was a dramatic increase in the percentage of GFP+ neural cells after 4 days (Fig. 1A, D, F). The change in the percentage of Oct4+ or GFP+ cells was not altered when FGF8 was added to the NM (Fig. 1B, F). On the other hand, cultures treated with SU5402 resulted in a decrease in the number of cells that were GFP+ only by ∼50%; however, a substantial percentage of ES cells retained both Oct4 expression and GFP expression (Fig. 1C, F). Cultures treated with SU5402 contained ES cell colonies with diminished growth that were well circumscribed, instead of the typical wide-spread distribution of cells found in 4-day cultures with only NM or NM+FGF8 (Supplementary Fig. S1A–C; Supplementary Data are available online at
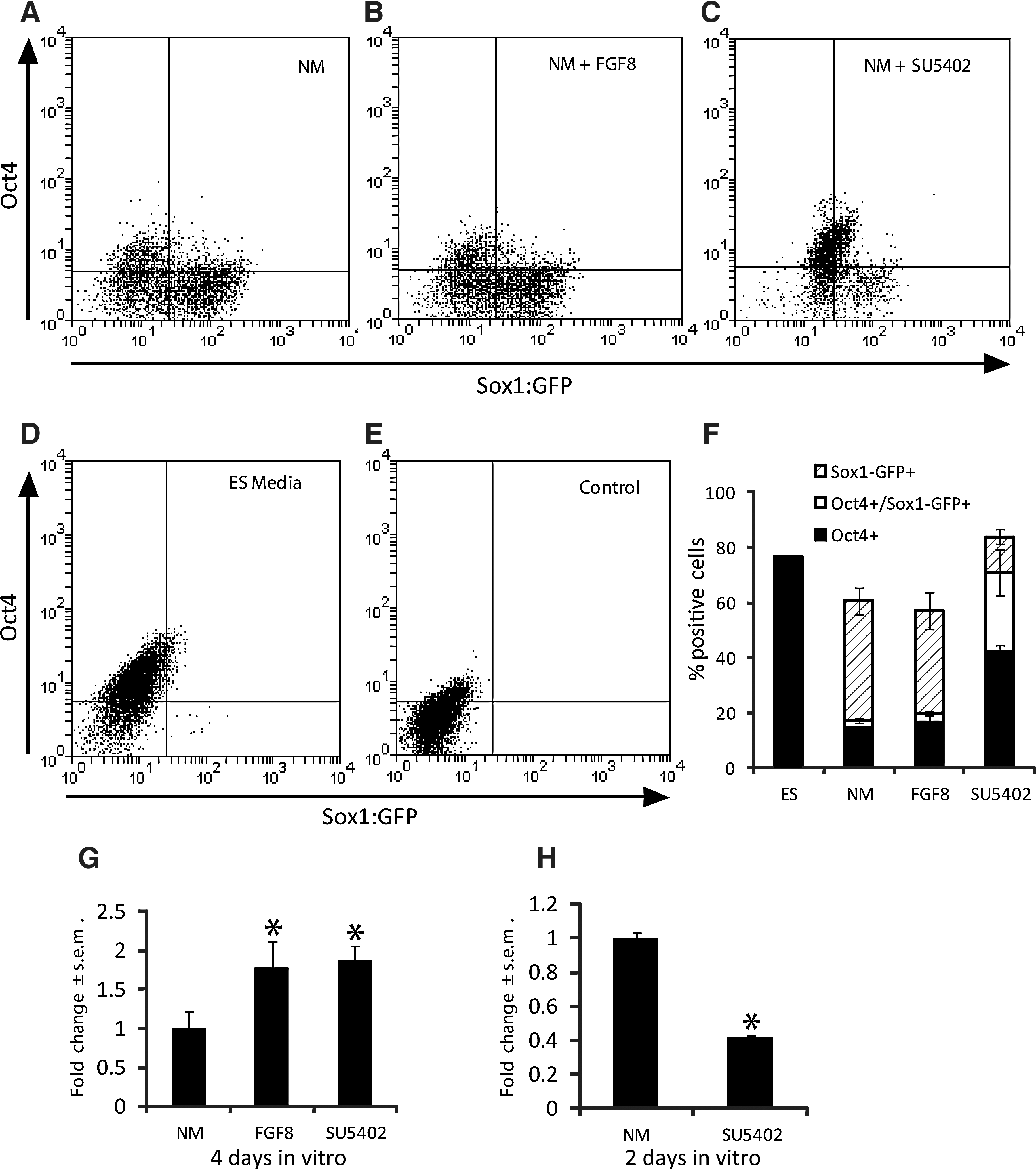
FGF signaling regulates the expression of Zfhx1b and the development of committed neural precursors in long-term cultures. Sox1:GFP embryonic stem (ES) cells cultured in a neural medium (NM) for 4 days with or without FGF8 or SU5402. The number of GFP+ cells, Oct4+ cells, or GFP+/Oct4+ cells was analyzed by FACS in
We next assayed for the expression of Zfhx1b and tested whether this expression is modulated by FGF signaling. As above, we assume that most of the effects of exogenous factors will occur within the first 1–2 days, and that assaying for gene expression after 4 days represents the cumulative effects of these events over time. Under baseline serum-free NM conditions, cells cultured after 4 days express Zfhx1b, as revealed by in situ hybridization (Supplementary Fig. S1D). The extent of expression was not uniform across the cell population and instead varied in both intensity and spatial distribution. This is expected, since at high densities, cells will develop microenvironments that will both positively and negatively regulate neural gene expression over time (in this case, 4 days). Nonetheless, despite these high cell density cultures, the presence of exogenous FGF8 resulted in a consistent upregulation of Zfhx1b gene expression (Supplementary Fig. S1E). Using RT-qPCR, we confirmed that exogenous FGF8 signaling enhanced Zfh1xb expression by ∼75% (Fig. 1G). This is the first indication that Zfhx1b gene expression is regulated by FGF signaling during neural cell development from ES cells in vitro.
We also treated 4-day, high-density cultures with the FGFR inhibitor SU5402. Since we observed a significant increase in Zfhx1b expression in the presence of FGF8 after 4 days, we anticipated that SU5402 would cause a decrease in Zfhx1b expression. Surprisingly, SU5402 resulted in an increase in Zfhx1b expression after 4 days (Fig. 1G, and Supplementary Fig. S1F). Comparing these results with the data in Fig. 1F is informative. In the presence of FGF8, the higher levels of Zfhx1b correlate with a predominate population of definitive NSCs (GFP only), but the effect of extra FGF signaling on neural specification is saturated by 4 days (ie, no difference when compared to NM only), although at earlier time points, the extra FGF signaling may have increased Zfhx1b expression above control values (see below). In contrast, in the presence of SU5402, the higher Zfhx1b levels correlate with a predominate population of GFP/Oct4 double-positive pNSCs (as well as ES cells) presumably through their selected proliferation/survival over 4 days (independent of FGF signaling). We tested the effects of SU5402 on Zfhx1b expression after only 2 days in the NM culture and found that the levels were significantly reduced (Fig. 1H). Therefore, these data suggest that the accumulation of more pNSCs by 4 days in SU5402 may have resulted in a delayed upregulation of Zfhx1b expression.
Since neural induction of ES cells occurs relatively rapidly, a 4-day culture period is not sufficient to reveal the initiating events that lead to a neural cell fate, despite the fact that this is a standard method used in the field. Indeed, cells can expand through selected proliferation of neural (or other) subpopulations, and gene expression can become more heterogeneous over time. Thus, to determine whether Zfhx1b gene expression is upregulated during neural specification of ES cells, we turned to a more sensitive 24-h culture assay. We plated dissociated Sox1:GFP ES cells at a low density of 10 cells/μL in serum-free conditions without exogenous growth factors to limit any extrinsic neural-inducing effects. Using immunocytochemistry, we observed that most ES cells can adopt a neural fate within 24 h as evidenced by the expression of GFP and the downregulation of Oct4 expression (Supplementary Fig. S2A–A″, B–B″). To test for Zfhx1b induction, we examined gene expression using RT-qPCR. We observed that Zfhx1b expression is increased 5-fold within the first 4 h and continues to increase significantly during the course of neural lineage specification (Fig. 2A). Increased Zfhx1b expression in the first 24 h parallels that of Sox1 (Fig. 2B) and coincides with the significantly reduced expression of the pluripotency marker Nanog (Fig. 2C). We also observed a modest decrease in E-cadherin expression, which occurs in vivo as cells transit from an epiblast cell fate to a neuroepithelial cell fate, although this was not statistically significant (Fig. 2D). Consistent with previous findings [4,7,34], the ES-to-neural transition occurs rapidly and does not require exogenous inducing factors. Furthermore, these data indicate that Zfhx1b induction correlates with neural specification.
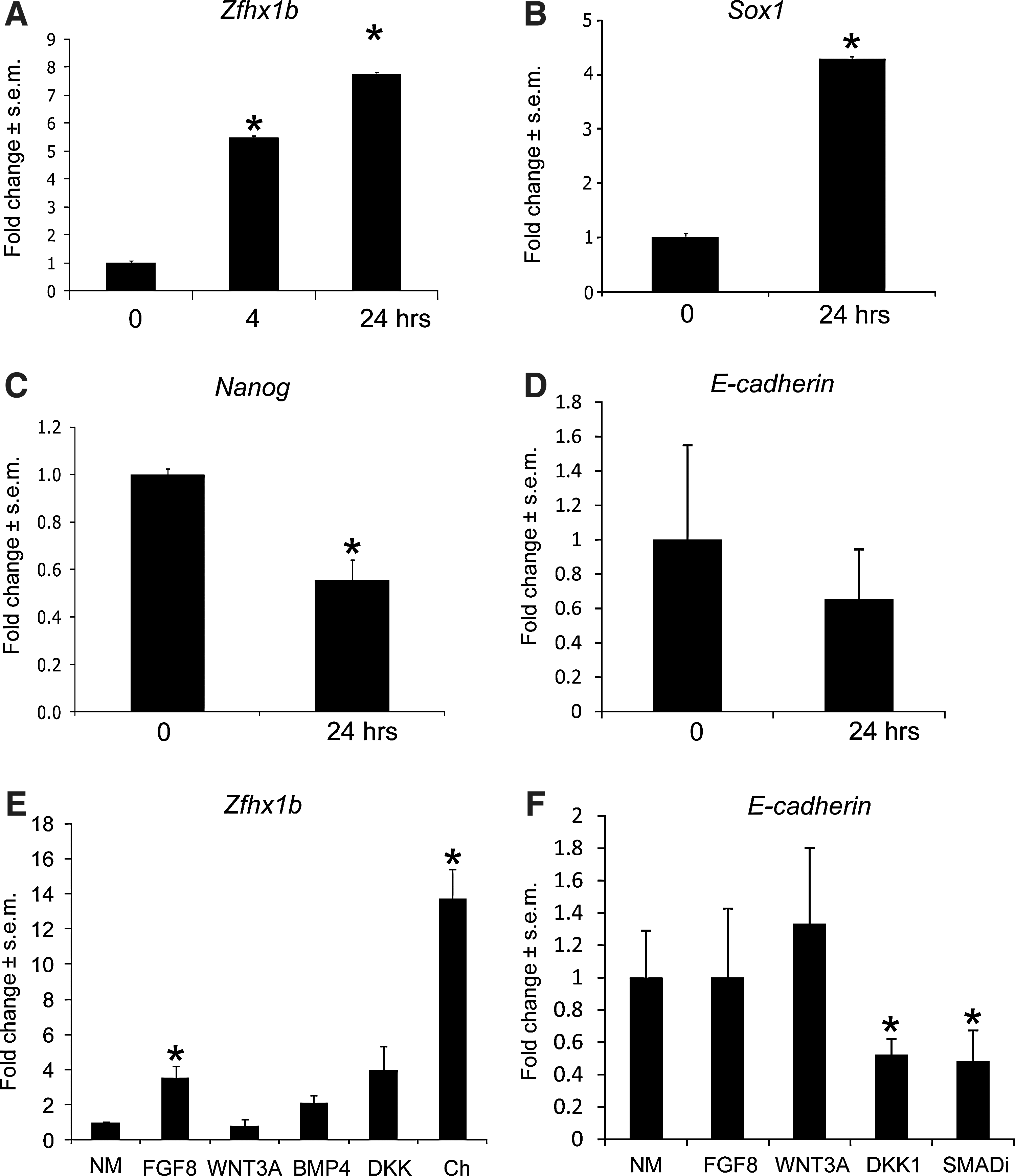
Zfhx1b gene expression is rapidly induced and modulated by FGF8 and Chordin.
Next we asked whether Zfhx1b expression can be modulated by exogenous factors during this 24-h period. Treatment with exogenous factors, such as FGF8 and Chordin, significantly increased Zfhx1b expression. Inhibition of the endogenous BMP-signaling pathway with Chordin showed a very robust ∼14-fold increase in the expression of Zfhx1b within 24 h in serum-free conditions, whereas FGF8 resulted in a ∼4-fold increase (Fig. 2E). We previously showed that modulation of the canonical Wnt-signaling pathway with WNT3A and DKK1 altered Zfhx1b expression in the mouse ectoderm [23]. In contrast, using the same WNT3A and DKK1 recombinant proteins and concentrations, we were not able to detect any changes in Zfhx1b expression in ES cells cultured in NM after 24 h (Fig. 2E). Doubling the concentration of WNT3A or DKK1 did not have any effect (data not shown). Thus, in contrast to its role in vivo, Wnt signaling does not seem to have a modulatory function in regulating Zfhx1b expression during neural specification of ES cells in vitro.
Interestingly, exogenous BMP4 had no significant effect on Zfhx1b gene expression (Fig. 2E). Judging by the robust influence of BMP inhibition by Chordin, this surprising result could indicate that other BMPs are primarily responsible for neural cell fate inhibition, or that BMP4-dependent inhibition is already saturated with respect to Zfhx1b expression. Therefore, we tested if the Smad inhibitor (SB431542) was sufficient to enhance Zfhx1b expression. We did not observe any changes in Zfhx1b expression compared to controls (data not shown). However, we did observe that the Smad inhibitor was very effective at suppressing E-cadherin expression (Fig. 2F). Thus, it is possible that changes to Zfhx1b in response to BMP inhibition by Chordin could be indirect through the activation of another signaling pathway (eg, FGF) that induces Zfhx1b as cells lose their epiblast identity. It would appear that simply downregulating E-cadherin expression is not sufficient for this effect, since DKK1 alone significantly decreased E-cadherin expression (Fig. 2F), but had no effect on Zfhx1b expression (Fig. 2E). Further experiments are required to clarify the specific role of indirect signaling pathways in this context. In summary, these data reveal that Zfhx1b gene expression is rapidly induced during neural specification of ES cells, and that this effect can be potentiated by either FGF signaling or BMP inhibition.
Zfhx1b gain of function is sufficient to promote NSC development from ES cells
Our gene expression analysis suggested that Zfhx1b might play a functional role in regulating neural cell fate specification. To determine whether Zfhx1b is sufficient to promote neural specification of ES cells, we overexpressed Zfhx1b in the Sox1:GFP transgenic cell line. First, we tested for the ability of Zfhx1b to induce a neural fate within a nondifferentiating ES cell maintenance environment, containing LIF and serum. ES cells were either transiently transfected with a control plasmid containing the DsRed reporter gene alone, or cotransfected with a Zfhx1b expression plasmid. Transfected cells were not selected and subsequently expanded as uniformly expressing populations. Instead, we aimed to simply transfect ES cells in their normal growth conditions, and after 24 h, fix and stain cultures for GFP and Oct4. Transfection efficiencies for both the control and the Zfhx1b overexpression cultures were ∼16%. Virtually all cells in cultures transfected with the control plasmid expressed Oct4, and there were few GFP+ cells that were observed (Fig. 3A, C, E). In a given counting arena with hundreds of cells (Hoechst+), ∼2% of the cells were GFP+ (Fig. 3G). The fact that there were GFP+ cells is consistent with previous findings of spontaneous neural specification in ES cell maintenance cultures [4]. In contrast, cultures transfected with Zfhx1b+DsRed showed a significant ∼3-fold increase in GFP+ cells, and most of these cells could be observed to also express Oct4 (Fig. 3B, D, F, G).
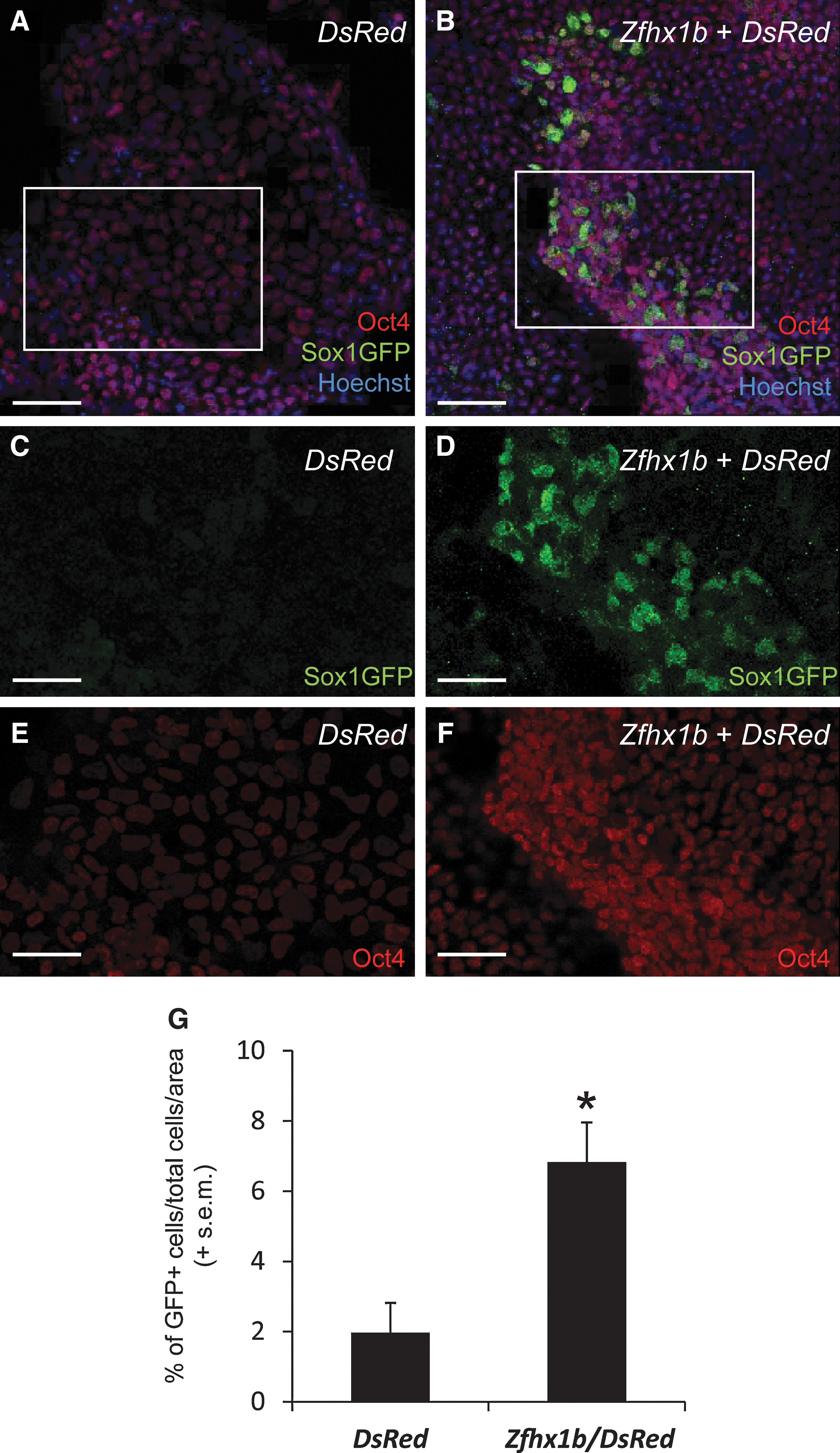
Zfhx1b overexpression is sufficient to induce Sox1:GFP expression in ES cell maintenance cultures. ES cell maintenance cultures overexpressing the DsRed reporter gene only
We corroborated this analysis (using fixed and stained cultures) in a separate transfection experiment by quantifying only live (unstained) DsRed+ cells to determine the fraction that were GFP+ in cultures that were cotransfected with Zfhx1b compared to cultures transfected with the DsRed control plasmid only. In this experiment, the transfection efficiency was ∼50% for both groups. In control cultures, we did not observe any GFP+ cells (n=35 DsRed+ cells analyzed). In contrast, transient overexpression of Zfhx1b along with the same reporter construct resulted in 29.2% GFP+ cells (n=41 DsRed+ cells analyzed) (Supplementary Fig. S3). Despite the differences in the transfection efficiency and method of analysis (fixed/stained versus live/unstained) between experiments, our data indicate that overexpression of Zfhx1b in ES cell maintenance cultures is sufficient to precociously induce the neural cell fate.
Next, we asked whether Zfhx1b overexpression was sufficient to promote a definitive NSC identity directly from ES cells, without first generating LIF-dependent pNSC colonies, by assaying instead for the formation of primary GFP+ colonies in the presence of exogenous FGF2. As expected, without Zfhx1b overexpression, we observed predominantly LIF-responsive pNSC-derived colonies, but not FGF2-responsive NSC colonies (Fig. 4A, C). However, we found that overexpression of Zfhx1b resulted in a significant increase in FGF2-responsive definitive NSC colony formation (Fig. 4B, C). Overall, our results suggest that expression of Zfhx1b is sufficient to initiate neural specification (even under nonpermissive conditions). Although many cells are probably not yet committed to a neural lineage, at least some of these induced cells precociously develop characteristics of definitive NSCs.
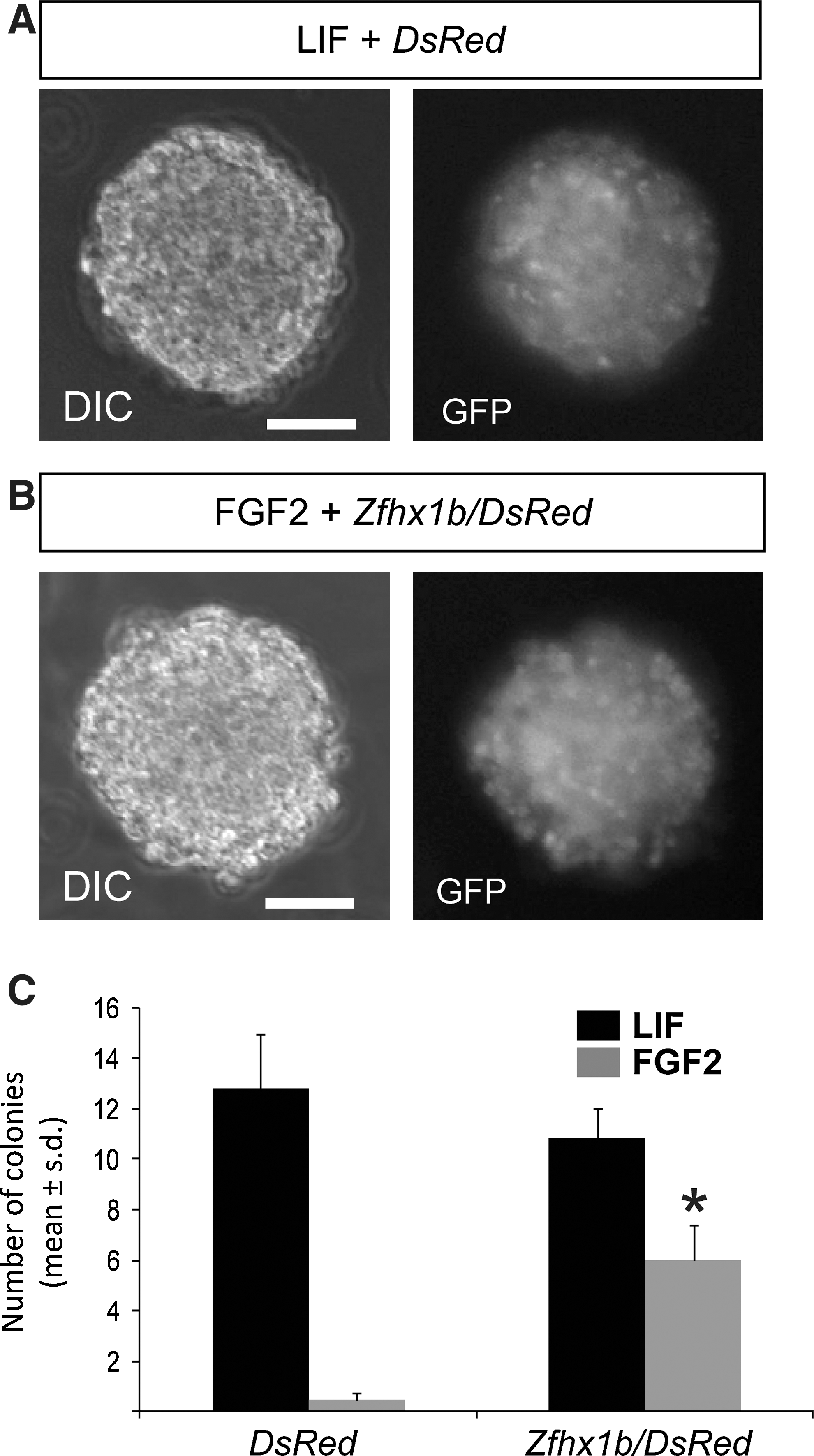
Zfhx1b overexpression is sufficient to promote precociously FGF-dependent colony formation. In serum-free, low-density cultures, Sox1:GFP ES-derived neural cells transfected with the DsRed reporter gene predominantly form LIF-dependent primitive neural stem cells (pNSC) colonies that are GFP+
Zfhx1b is not necessary for the initial neural specification of ES cells
Although Zfhx1b gene expression is induced within 4 h of low-density, serum-free culture, and overexpression of Zfhx1b is sufficient for a modest fraction of ES cells to overcome the inhibitory effects of an ES cell maintenance culture environment and initiate neural lineage specification, this does not necessarily indicate that Zfhx1b is required for this initial transition. Furthermore, the number of LIF-responsive pNSC colonies was not altered in ES cells overexpressing Zfhx1b, suggesting that the ability of Zfhx1b to influence NSC development might be independent of the initial steps of neural specification when pNSCs are first established and when Zfhx1b expression is being upregulated.
To determine if Zfhx1b is necessary for the initial transition of ES cells into neural cells, we performed a loss-of-function experiment, knocking down Zfhx1b in mouse Sox1:GFP ES cells using siRNA. ES cells were transiently transfected with the Zfhx1b siRNA, a scrambled siRNA, or GAPDH siRNA along with a DsRed reporter gene, and compared to DsRed gene transfection alone as a control. Upon successful transfection (∼50% of cells expressed the DsRed reporter after 24 h), ES cells were transferred to serum-free neural-inducing conditions and assayed for Sox1:GFP expression. First, we found that reduction of Zfhx1b gene expression in the Zfhx1b siRNA-transfected group was still evident after 48 h in neural-inducing conditions. As shown in Fig. 5A and B, expression of Zfhx1b decreased significantly in the Zfhx1b siRNA group compared to the transfection control in our standard NM conditions and compared to ES cells transfected with a scrambled (Scr) siRNA.

Zfhx1b is not required for the initial specification of neural cells. Zfhx1b siRNA, GAPDH siRNA, and scrambled siRNA (Scr siRNA) were transfected into mouse Sox1:GFP ES cells with DsRed as a reporter gene in a maintenance medium, and transferred to NM in monolayer conditions after 24 h, without the addition of any exogenous growth factors. SU5402 (SU) was added to determine the role of FGF signaling during the initial specification of the neural fate in mouse ES cells.
Second, to evaluate the extent to which reduced levels of Zfhx1b expression affected early neural specification, we quantified the percentage of transfected ES cells that expressed the Sox1:GFP transgene after either 4 h or 48 h. Our experiments showed that a total of 48 h (instead of our usual 24-h assay) is an optimal time frame to assay for changes in neural specification, as the siRNA construct would require sufficient time for effective gene knockdown. FACS analysis revealed that there is a robust shift in the percentage of GFP+ neural cells from 4 to 48 h in serum-free, low-density media (NM; Fig. 5C), from <1% of the population to ∼90% of the population (Fig. 5D, E). However, neither FGFR inhibition (cells treated with SU5402) nor knockdown of Zfhx1b by siRNA resulted in a change in the percentage of GFP+ neural cells that developed after 48 h compared to untreated controls (NM), or cells transfected with Scr siRNA (Fig. 5C–E). These results reveal that Zfhx1b is not necessary for the initial neural specification of ES cells.
Zfhx1b is required for the development of NSCs from pNSCs
Knockdown of Zfhx1b had no effect on the initial neural specification of ES cells, but the FGF-dependent regulation of Zfhx1b and the ability of this gene to enhance FGF-responsive clonal colony formation when overexpressed suggested that this gene might instead regulate downstream NSC development. To test this possibility, we generated primary LIF-responsive (pNSC) colonies using the Sox1:GFP transgenic ES cells in standard low-density serum-free cultures. Primary GFP+ colonies were then dissociated, transfected with either Zfhx1b siRNA or Scr siRNA, and cultured in similar low-density serum-free conditions, except this time in the presence of FGF2 to assay for definitive NSCs that normally developed from the pNSC-derived colonies [4]. The results showed that knockdown of Zfhx1b caused a significant decrease in the number of definitive NSCs present in the cultures, compared to either untreated controls (NM) or cells transfected with Scr siRNA (Fig. 6A). This is the first evidence to suggest that Zfhx1b is not only sufficient to promote NSC development but it is also necessary for NSC development. We also asked whether blocking FGFR signaling using SU5402 during the initial stages of plating (initial 24 h) under these conditions would similarly compromise FGF colony formation. Blocking FGF signaling with SU5402 for only 24 h at the start of the culture, followed by changing media to NM without an inhibitor, did not significantly affect definitive NSC colony formation a week later (Fig. 6A). Definitive NSC colonies derived from Zfhx1b knockdown cultures or Scr siRNA cultures were of similar average diameter to colonies generated in untreated control cultures (∼100 μm). However, treatment with SU5402, for the first 24 h, yielded colonies that were approximately half the size compared to all other groups, which does subsequently increase to ∼100 μm in diameter with prolonged culture (data not shown). The deficient growth of the passaged LIF-derived colonies in SU5402 suggests that the cells do not proliferate normally, as one might expect in this context, but it is possible that with the addition of SU5402, cells persist in a pNSC state for longer, as suggested by our finding that a larger fraction of cells treated with this inhibitor in NM expresses both Oct4 and Sox1 (Fig. 1F). Taken together, these results indicate that Zfhx1b is required for the development of definitive NSCs rather than the initial specification from ES cells. Furthermore, it seems that FGF signaling acts to facilitate this process.
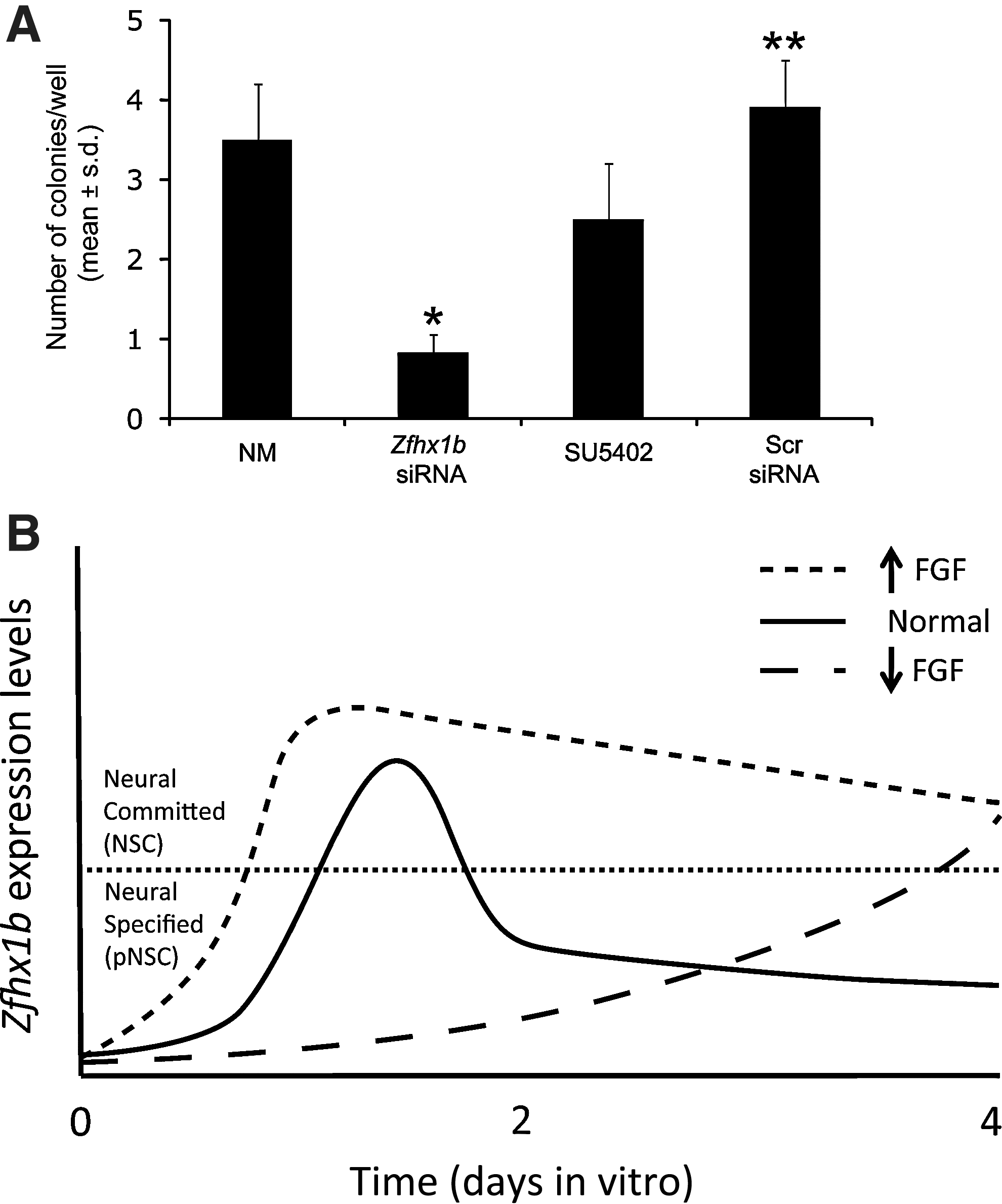
Progression toward a more committed NSC identity requires Zfhx1b.
Discussion
The initial specification of early vertebrate neural identity is modulated primarily by the inhibition of endogenous repressors of neural fate, such as BMP [17,35,36] and Wnt [37 –39] signaling. This process is largely recapitulated in ES cell models of neural induction [4,17,25] and does not require exogenous factors [34]. Our findings here extend these observations by highlighting a functional role for the transcription factor Zfhx1b, which is modulated by these pathways and was shown to be both necessary and sufficient for early neural cell development. Early onset expression of Zfhx1b coincides with the relatively rapid acquisition of a neural fate in mouse ES cells in serum-free and low-density cultures. Neither endogenous sources nor exogenous sources of FGF signaling are required for this initial neural fate transition [32,34] as shown in the present results, but FGF signaling does facilitate the further development of these initially specified cells toward a more committed neural identity—a step that we propose requires Zfhx1b function.
Increased expression of Zfhx1b during the initial stages of neural specification may allow Zfhx1b to regulate or inhibit the BMP-signaling pathway through its interaction with the BMP effector, Smad1 [40]. However, the Smad-binding domain is not required for the neural inductive capability of Xenopus SIP1/Zfhx1b [12], and it is possible that Zfhx1b may be able to directly upregulate the early neural gene Sox1 to induce a neural fate, as there are several conserved putative Zfhx1b-binding sites on the mouse Sox1 upstream regulatory region (unpublished observations). Moreover, we have shown that overexpression of Zfhx1b in mouse ES cells, which have not been subjected to neural differentiation conditions, is sufficient to induce Sox1:GFP expression. However, as both Sox1 and Zfhx1b are rapidly induced during neural specification, it is possible that Zfhx1b may regulate the maintenance of Sox1 expression. Studies in Xenopus [14,41] as well as chick [42] indicate functional regulation of Group B1 Sox genes by Zfhx1b. Therefore, the identification of direct downstream targets of Zfhx1b, such as Sox1, and the role of this transcriptional circuitry in the context of neural specification remains to be fully investigated.
Our present findings allow us to draw some parallels on the functional role of Zfhx1b in neural specification in vivo and in ES cell models. For example, Zfhx1b function in the mouse ectoderm occurs later during the transition from a pNSC to a definitive NSC, but not during the initial specification of a neural fate [23]. This transition is facilitated in the embryo by FGF-dependent activation of Zfhx1b expression, and the overexpression of this gene is sufficient to promote a definitive NSC identity [23]. In mouse ES cells, inhibition of FGF signaling through the use of the FGFR inhibitor, SU5402 [33], decreased Zfhx1b expression, but did not alter the ability of ES cells to transit to a Sox1:GFP+ neural identity within 48 h, consistent with previous findings [34]. Furthermore, gain-of-function or loss-of-function experiments revealed an increase or decrease, respectively, of definitive NSC colony formation from ES cell cultures. These observations strongly suggest that, similar to ectoderm cells in vivo, Zfhx1b plays a fundamental role in ES cells in vitro to promote the transition from a pNSC to an NSC identity. We propose a model to explain the regulation of Zfhx1b by FGF signaling and its impact of NSC development (Fig. 6B). First, Zfhx1b is rapidly upregulated under permissive in vitro conditions, and this effect can be enhanced with additional FGF signaling or suppressed by blocking endogenous FGF signaling. Other factors, such as BMP agonists/antagonists, are likely to be modulated during this transition as well. During this early phase, Zfhx1b expression continues to increase, but is not required for ES cells to transit to a neural-specified pNSC identity (Oct4+/Sox1+). Second, Zfhx1b expression levels then peak as pNSC transition to a neural-committed NSC identity (Sox1+), and Zfhx1b function is required for this transition. Levels of Zfhx1b likely decrease thereafter. Additional FGF signaling may transiently accelerate the pNSC-to-NSC transition, but the effect is quickly saturated. Extra FGF signaling may also sustain high levels of Zfhx1b for longer in these cells that are already committed. In contrast, blocking endogenous FGF signaling prevents the pNSC-to-NSC transition and allows for the accumulation of pNSCs and the delayed recovery of Zfhx1b expression, which would then be upregulated in the expanded pNSC population.
Nonetheless, there are differences that require further attention. For example, initial inhibition of FGF signaling promotes self-renewal in mouse ES cells via the Erk1/2 MAPK pathway [43], but subsequent exposure to Erk-mediated FGF signaling is required for differentiation [29,32], especially within the first 24 h [29]. A recent report suggests that RA signaling controls the switch from the pluripotent state to differentiation by first suppressing Oct4 expression as well as mediating Erk signaling to induce Fgf8 expression [44]. Commitment toward a neural lineage might therefore require endogenous FGF signaling, and we observed in the present study that inhibition of FGF signaling with SU5402 over 4 days resulted in a substantial number of ES cells (Oct4+ only) and cells expressing both Oct4 and the Sox1:GFP transgene—the presumptive pNSC population. Whether RA signaling can directly induce expression of Zfhx1b or whether the increase in endogenous Fgf8 can regulate Zfhx1b expression remains to be discerned.
In human ES cells, ZFHX1B has been shown to be regulated through the Activin-Nodal signaling pathway [18,45] and shown to be required for neurectoderm differentiation, but not its initial specification [18]. Activin-Nodal signaling has also been shown to play an important role in cell fate decisions between the mesoderm and the ectoderm in the chick embryo [46], as well as the Xenopus embryo [47,48]. Mouse Nodal mutant embryos exhibit a premature neurectoderm fate within the epiblast, indicating that Nodal signaling serves to inhibit neural development, and thus restricts development of the neurectoderm to the anterior [49]. In addition, a recent study using mouse ES cells showed that inhibition of Nodal signaling promotes neural development through inhibition of the Wnt-signaling pathway [24]. Interestingly, we previously demonstrated that simultaneously blocking both FGFR signaling (with SU5402) and Wnt signaling (with DKK1) ex vivo resulted in normal numbers of FGF-dependent NSC colonies subsequently generated from primary neurectoderm cultures [23]. We also reported that the expression of Zfhx1b is highly induced in the presence of the Wnt inhibitor, DKK1 [23], suggesting that Wnt signaling can also regulate Zfhx1b expression. Nonetheless, we could not identify a role for Wnt-regulated Zfhx1b expression in neural specification of ES cells. Further analysis to determine the role of Zfhx1b with respect to its regulation or interaction with the Nodal-signaling pathway will determine whether Zfhx1b can be a mediator between FGF-, Wnt-, and BMP/Nodal-signaling pathways to modulate NSC development.
Although Zfhx1b is sufficient to promote neural lineage commitment directly from ES cells or embryonic ectoderm, other genes have also been identified to play a role in this process. The zinc-finger nuclear protein Zfp521 has recently been shown to be essential for the induction of neural gene expression and intrinsic differentiation from mouse ES cell to neurectodermal identity [50]. However, unlike Zfhx1b, overexpression of Zfp521 could not induce neural differentiation of mouse ES cells in the presence of LIF, BMP4, or SU5402 [50]. In Xenopus embryos, Zfp521 is expressed much later than the onset of neural induction and thus plays a relatively late role in neural lineage development [50]. It seems that Zfp521 can exert its neural-inducing effects on cells already primed for a neural fate, but not on pluripotent cells. Our results indicate that transient overexpression of Zfhx1b is sufficient to drive neural specification of ES cells in the presence of LIF and serum. It could be that Zfhx1b and Zfp521 function in parallel or that Zfhx1b may be upstream of Zfp521, but future experiments will be required to determine the relative contribution of these 2 transcription factors.
Human ES cells, as well as human induced pluripotent stem (iPS) cells, hold tremendous potential for both basic research as well as practical applications, such as regenerative medicine. Like their human ES cell counterpart, human iPS cells can also be directed to differentiate along the neural lineage, however, with variable efficiencies depending on the cell type and the starting cell line [51]. Mowat-Wilson syndrome (MWS), which includes a spectrum of neurodevelopmental deficits, is caused by haplo-insufficient deletions in the human ZFHX1B gene [52]. Aspects of this neurodevelopmental disorder may be possibly attributed to defects in early NSC development. Functional analyses of ZFHX1B from iPS cells derived from MWS patients (providing that the same patient-specific iPS cells could be engineered to regain 2 normal alleles of ZFHX1B to be used as isogenic cells for comparison [53]) would be a fruitful approach toward delineating the disease mechanisms of this devastating childhood syndrome and to provide a broader insight into the relationship between defective NSC development and patient-specific neurodevelopmental disorders.
Footnotes
Acknowledgments
We thank Dr. A. Nagy, Dr. A. Smith, Dr. L. Grabel, Dr. L. Bonfanti, Dr. W. Stanford, and Dr. P. Tylzanowski for reagents. We also thank Boyang Zhang for assistance with FACS. Research funding was provided by OGS (L.T.H.D.) and NSERC (V.T.).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.